Abstract
Focal adhesion kinase (FAK) and steroid receptor coactivator (Src) are intracellular (nonreceptor) tyrosine kinases that physically and functionally interact to promote a variety of cellular responses. Plenty of reports have already suggested an additional central role for this complex in cancer through its ability to promote proliferation and anoikis resistance in tumor cells. An important role for the FAK/Src complex in tumor angiogenesis has also been established. Furthermore, FAK and Src have been associated with solid tumor metastasis through their ability to promote the epithelial mesenchymal transition. In fact, a strong correlation between increased FAK/Src expression/phosphorylation and the invasive phenotype in human tumors has been found. Additionally, an association for FAK/Src with resistances to the current anticancer therapies has already been established. Currently, novel anticancer agents that target FAK or Src are under development in a broad variety of solid tumors. In this article we will review the normal cellular functions of the FAK/Src complex as an effector of integrin and/or tyrosine kinase receptor signaling. We will also collect data about their role in cancer and we will summarize the most recent data from the FAK and Src inhibitors under clinical and preclinical development. Furthermore, the association of both these proteins with chemotherapy and hormonal therapy resistances, as a rationale for new combined therapeutic approaches with these novel agents, to abrogate treatment associated resistances, will also be reviewed.
The nonreceptor tyrosine kinases FAK and SRC
The FAK-SRC complex in the integrin and tyrosine kinase receptor setting
Integrins are a family of transmembrane receptors that link the extracellular matrix (ECM) and the intracellular actin-cytoskeleton. These cell-matrix areas of adhesion are known as focal adhesion (FA) contacts/areas. Integrins cluster when they bind to ECM. Integrin clustering has a structural role but also induces the activation of intracellular signaling pathways that lead to important cellular responses such as proliferation, survival, migration and invasion in both normal and tumor cells.Citation1 In this setting, the linked activities of two nonreceptor intracellular tyrosine kinases, focal adhesion kinase (FAK) and steroid receptor coactivator (Src), is a common intracellular point of convergence in the signaling initiated by this integrin-ECM interaction. In response to the clustering, FAK associates to the cytoplasmic tail of the integrin and in response to this association FAK phosphorylates at its tyrosine residue 397 (Y397). Although this Y397 phosphorylation is mainly due to autophosphorylation; transphosphorylation by growth factors might also occur. This phosphorylated tyrosine provides a docking site for Src ().Citation2 The tyrosine kinase Src, then, phosphorylates additional sites on FAK, leading to further increased activity of FAK and allowing the recruitment of proteins that contain Src homology 2 (SH2) domains such as Grb2 and PI3K.Citation3 The mutually activated FAK/Src complex then initiates a cascade of phosphorylation events of new protein–protein interactions to trigger several signaling pathways that eventually leads to different cellular responses. For instance, Grb2, once bound to FAK can recruit SOS into the complex and activates the downstream Ras-MAPK pathway and/or may also transduces the signal through the activation of the PI3K-Akt cascade.Citation3–Citation5
Figure 1 FAK/Src complex mediated signaling pathway.
Abbreviations: ERK, extracellular signal regulated kinase; FA, focal adhesions; MEK, mitogen-activated protein; FAK, focal adhesion kinase; VEGF, vascular endothelial growth factor; MMPs, matrix metalloproteinases.
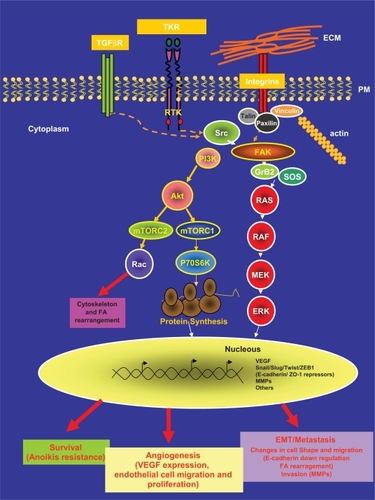
Adhesion is a hallmark of solid cancer cells and integrins are a nexus between intracellular signaling and cytoskeletal dynamics; besides, integrin-mediated signaling also intersects with growth factor-mediated signaling through various levels of cross-talk. In this complex network, the FAK/Src complex also transduces signals from tyrosine kinase receptors (TKRs) or integrates signals from both, integrins and TKRs, (see ) in normal and tumor cells.Citation3,Citation4 As an illustration of this connection in breast cancer, transforming growth factor β (TGF-β) has recently been found to be able to induce membrane-proximal clustering of human EGF (Epidermal Growth Factor) receptor 2, (HER2) and integrins by activating Src-FAK and receptor association to the cytoskeleton.Citation6
Platelet-derived growth factor receptor (PDGF-R), epidermal growth factor receptor (EGF-R), fibroblast growth factor receptor (FGF-R), insulin-like growth factor-1 receptor (IGF-1R), hepatocyte growth factor (HGF-R), colony-stimulating factor-1 receptor (CSF-1R) or, stem cell factor receptor (SCF-R) are also TKRs that may transduce their signal by FAK/Src activation. As a consequence, the FAK/Src complex is potentially involved in different steps of tumorogenesis and further growth and metastatic progression of malignant tumors.Citation7,Citation8
The increased expression or activity of FAK and/or Src in tumors is associated with a more invasive and aggressive phenotype and has lead to the development of Src and FAK inhibitors as new anticancer drugs.Citation9–Citation12 These drugs are able to block proliferation, survival, angiogenesis and/or migration/invasion in preclinical tumor modelsCitation13 and some of them have already shown preliminary antitumor activity in clinical trials with cancer patients.Citation14–Citation19
FAK and Src structure
c-Src was the first characterized human oncogene. In 1909 Peyton Rous identified the Rous Sarcoma’s Virus (RSV).Citation20,Citation21 Later, in 1958, the v-Src gene was identified as the cause that allowed RSV to produce the sarcoma when the virus infected healthy chickens. The v-Src gene was taken up by RSV and incorporated into its genome conferring the virus the advantage of being able to stimulate uncontrolled proliferation in host chicken cells. Finally the human homologue of the v-Src gene, c-Src, was characterized as the first oncogene in humans.Citation22
c-Src is a nonreceptor tyrosine kinase. The Src family comprises of eight members in humans (Src, Fyn, Yes, Lyn, Lck, Hck, Blk and Fgr) with a molecular weight between 52–62 KDa.Citation22,Citation23 Each Src kinase family member is comprised of six domains. A SH4 domain placed at the N-terminal tail is involved in targeting Src to the plasmatic membrane. Adjacent to the SH4 domain, a region that is specific to each Src family member followed by a SH3 and a SH2 domain, both of them involved in the interaction of Src with other intracellular proteins ().Citation23 Additionally, in the C-terminal tail, there is an SH1 domain involved in adenosine tri-phosphate (ATP) and substrate binding. This SH1 domain shows tyrosine kinase activity. The phosphorylation in the Y419 residue of the SH1 domain is required for maximum kinase activity. Immediately adjacent to the SH1 domain, there is another C-terminal region that acts as a negative regulatory domain that is itself regulated by phosphorylation. After phosphorylation of the Y530 residue, placed in this negative regulatory domain, Src undergoes conformational changes and becomes inactive.Citation23 Src activation is regulated at many different levels. In response to a signal stimuli Src translocates from the cytosol to the membrane where it will be activated by phosphorylation, the intracellular localization of Src is therefore one of the key regulatory mechanisms that control Src activation.Citation24 In addition the binding of FAK to the SH2 domain of Src relieves its autoinhibitory interaction that leads to the activation of Src. Once activated, Src phosphorylates FAK on a number of additional tyrosine residues, leading to further increased activity of FAK.Citation2,Citation23,Citation25 In this sense, FAK acts as a molecular scaffold protein to activate and recruit Src to its substrates.
Figure 2 FAK and Src structure.
Notes: A) FAK structure. FAK harbors a central region with kinase activity that is flanked by a N-terminal region that contains the FERM domain and by a C-terminal region that contains the FAT domain. The autophosphorylation in its Y397 residue increases its catalytic kinase activity and allows the binding of specific intracellular proteins. FAK is phosphorylated by Src in its Y576 and Y577 residues allowing its full catalytic activity. In absence of stimulus, the FERM domain works as a negative regulator of FAK activity through its interaction with the kinase domain preventing the phosphorylation in Y397. In response of stimuli, the FERM domain interacts with the cytoplasmic tail of the integrins. Allowing the autophosphorylation of FAK in this tyrosine. The FAT domain, placed in the C-terminal region, mediates the co-localization of FAK with the FA areas through the interaction of FAK with the FA associated proteins talin and paxillin. Two proline-rich domains (PR) mediate the interaction of FAK with SH3 containing proteins such as p130 CAS. B) Src structure. Src is comprised of six domains: a SH4 involved in targeting Src to the plasmatic membrane, a region U that is specific of each Src family member, a SH3 and a SH2 domain involved in the interaction of Src with other intracellular proteins and a SH1 domain involved in ATP and substrate binding. The phosphorylation in Y419 residue of the SH1 domain is required for maximum kinase activity. Placed immediately adjacent to the SH1 domain there is a negative regulatory domain. After phosphorylation of the Y530 residue, in the negative regulatory domain, Src becomes inactive.
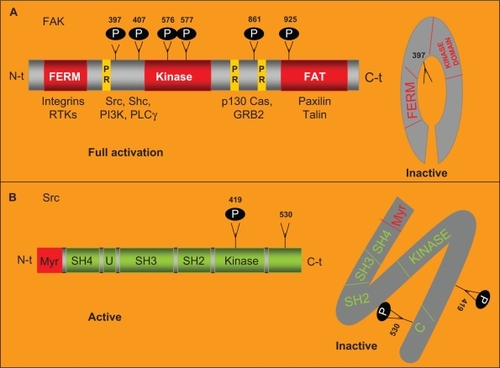
FAK was first identified in the search for proteins that where tyrosine phosphorylated in an integrin dependent manner and also in Src transformed fibroblasts as a key substrate of Src oncoprotein.Citation26 It was described as a focal adhesion-associated nonreceptor protein tyrosine kinaseCitation27,Citation28 ubiquitously expressed and encoded by an evolutionarily highly conserved gene.
FAK harbors a central region with kinase activity that is flanked by a large N-terminal region that contains the erythrocyte band four 1-ezrin-radixin-moesin (FERM) domain and by a C-terminal region that contains the focal adhesion targeting (FAT) domain. The Y397 residue, immediately adjacent to the kinase domain, is autophosphorylated in response to the clustering of integrins. This autophosphorylation increases the catalytic activity of FAK and creates a high affinity binding site for the SH2 domain of Src. This interaction recruits and activates Src. The formation of the complex with Src is the most critical event in FAK-associated signaling. Src binds the Y397 residue and phosphorylates other FAK residues including Y576 and Y577 placed on the catalytic loop of the kinase and Y861 that are important for full catalytic activity of FAK. Y925 has been also identified as an important site for phosphorylation by Src, which involves Src in induced epithelial mesenchymal transitions (EMTs).Citation29,Citation30 In the absence of stimuli from integrins and/or TKRs, the FERM domain works as a negative regulator of FAK activity. This domain interacts with the kinase domain preventing Y397 phosphorylation. Conversely, in response to co-clusters of integrins and TKRs, the FERM domain interacts with the cytoplasmic tail of the integrin allowing FAK-autophosphorylation in this Y397 residue. The FAT domain, placed in the C-terminal region, mediates the colocalization of FAK with the FA areas through the interaction of FAK with the FA associated proteins talin and paxillin. Furthermore, two proline rich domains, placed adjacent to the FAT domain, mediate the interaction of FAK with SH3 containing proteins such as p130CAS.Citation9,Citation11 The ability of FAK to promote the activation of p130CAS depends on Src activity. Although the scaffold function of FAK to recruit Src to its substrates is has already been well established, however less is known regarding FAK kinase activity.
Besides integrins, the N-terminal region of FAK also interacts with TKRs, and this interaction may be involved in the regulation of FAK activity and in addition in cross-talks between integrins and TKRs.Citation31 Phosphorylation of FAK. Y861 promotes association of FAK with the αyβ5 integrin following vascular endothelial growth factor (VEGF) stimulation.Citation32
Different FAK isoforms from alternative splicing have been found: FAK, proline rich tyrosine kinase 2 (PYK2) and FAK related nonkinase (FRNK).Citation9 FRNK lacks the catalytic domain working as a FAK inhibitor competing with nontruncated endogenous FAK for the localization at FA areas.Citation33
FAK activity is under strict regulation by a variety of kinases and phosphatases such as glycogen synthetase kinase 3 type β (GSK3 β), tyrosine phosphatase SHP-2, serine/threonine protein phosphatase type 1 as well as by Src.Citation9,Citation34,Citation35
FAK and Src and their function in normal and tumor cells
Although, most available articles in the literature show FAK and Src as independent proteins, the current idea that both proteins work as a protein complex in the cellular signaling networks is emerging. FAK would form a binary complex with Src family kinases which can phosphorylate other substrates and trigger multiple intracellular signaling pathways that would induce different cellular responses.
FAK and Src are not only critical modulators of signaling pathways mediated by TKRs and integrins, they also respond to stimuli from G protein-coupled receptors, cell–cell adhesion proteins (ie, cadherins) and steroid hormone receptors to control a variety of normal and oncogenic cellular responses such as cell survival, proliferation and migration/invasion.Citation4,Citation22,Citation23,Citation34,Citation36,Citation37
FAK-null embryos exhibit an early embryonic lethal phenotype.Citation38 These embryos show multiple defects, including a disorganized cardiovascular system due to extensive defects in angiogenesis and vasculogenesis.Citation38 Accordingly, over expression of FAK in vascular endothelial cells promotes angiogenesis.Citation39 In addition, conditional deletion of FAK in adult mouse epithelium was not lethal, and probably due to a functional compensatory effect mediated by its related family member PYK2.Citation40 Although PYK2 knock out mice develop normally except they do exhibit defective macrophage migration.Citation41,Citation42 The above data suggested that endothelial cells may posses an adaptive capacity to switch to PYK2 dependant signaling after deletion or inhibition of FAK. Thus, FAK and PYK2 inhibition may result in an antiangiogenic effect.
An interesting interaction has also been reported between FAK and the tumor suppressor protein p53, via the FERM domain that triggers p53 degradation, so that loss of FAK results in activation of p53 which could eventually suggest new approaches to trigger cytotoxic drug induced apoptosis.Citation43
Although, Src-null mice were viable, the analysis of homozygous mutants showed that they were deficient in bone remodeling (they had an impaired osteoclast function) and also developed osteopetrosis. This phenotype demonstrated that Src is not required for general cell viability possibly due to a Src functional overlap with other related tyrosine kinases such as FAK. Therefore, Src may play an essential role in bone formation.Citation44 Accordingly, cancer patients treated with a Src inhibitor showed reduced serum levels of bone resorption markers suggesting Src inhibitors as a possible effective treatment for established bone metastasis.Citation45,Citation46
In normal adult tissues and tumor cells, FAK and Src control many important biological processes within the cell. They have been associated with responses of cell growth and survival.Citation9,Citation10 In addition, they have also been involved in anoikis (apoptosis induced when anchorage-dependent cells detach from the surrounding ECM) resistance in tumor cells.Citation47–Citation50 Hence, FAK/Src activation may promote the anchorage-independent growth/transformation of tumor cells through the inhibition of the apoptotic response.Citation48,Citation49 FAK activation, in response to integrins, has an important role in FA turnover/rearrangement. This process is crucial for cell spreading and migration in physiological and pathological processes (ie, FA turnover is essential for EMTs during the metastatic behavior of tumor cells and in endothelial cell migration during tumor angiogenesis).Citation9 Phosphorylation of FAK-Y925 is the major Src-specific phosphorylation event that is associated with integrin adhesion dynamics and E-cadherin deregulation during Src-induced EMT.Citation29,Citation30
Furthermore, FAK has already been found, at elevated levels, in the majority of human cancers (head and neck, colon, breast, prostate, liver, thyroid, and others), particularly in highly invasive metastases.Citation51 High levels of Phospho-FAK Y397 has already been found in: ovarian invasive tumors;Citation52 acute myeloid leukemia;Citation53 squamous cell carcinoma of the laryn;Citation54 invasive cervical carcinoma;Citation55 invasive human colon cancer cells;Citation56 medullary thyroid cancer cell lines;Citation57,Citation58 human pancreatic cancer cells;Citation59 glioma cells;Citation60 and other tumor types. Furthermore, high levels of FAK phosphorylated in other tyrosine residues have already been found in specific tumor types. Papillary thyroid cancer samples show high phosphor-Y861-FAK levels and high levels of phopho-Y861-FAK have also been correlated with sensitivity to the Src inhibitor AZD0530 in papillary and also in anaplastic thyroid cancer models.Citation61 Although, the relationship between Src and cancer progression is best documented in colon and breast cancer.Citation10,Citation62 Src over expression or over activation has also been shown in a variety of human biopsies from primary tumors and their metastases ().Citation23
Figure 3 Role of the FAK-Src complex in the malignant progression of solid tumors.
Abbreviations: ECM, extracellular matrix; EMT, epithelial mesenchynal transitions; FAK, focal adhesion kinase; Src, steroid receptor coactivator.
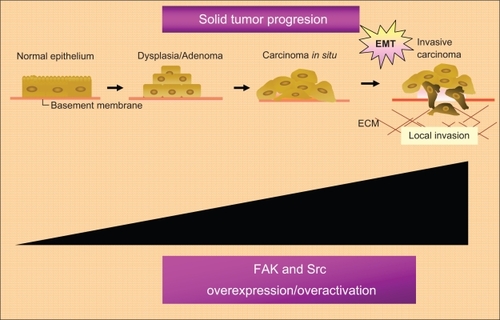
An additional pro-angiogenic role for FAK and Src signaling in tumors has also been suggested.Citation63–Citation69 FAK expression has been found in tumor endothelial cells from grade III and IV astrocytoma biopsies; whereas FAK expression was absent in endothelial cells of normal brain biopsies.Citation63 Accordingly, tumor endothelial cells transfected with FRNK (a negative FAK regulator) showed less migration in vitro than control cells; suggesting that FAK is involved in tumor-angiogenesis, at least in part, through the induction of endothelial cell migration.Citation63 Preclinical data from prostate cancer cell lines have also suggested a role for FAK signaling in the induction of VEGF expression in tumor cells.Citation70 Additionally, an intracellular cross-talk between the Ang-1 TKR (Tie-2) involved in angiogenesis in tumors and integrin pathways has also been shown.Citation71 The binding of integrin α1β5 to ECM-glycoproteins may lead to the association of Tie-2 with integrin α1β. The stimulation of Tie-2 by Ang-1 may promote the recruitment of FAK to the TKR-integrin areas inducing an endothelial cell response (sprouting and stabilization of the new tumor vessels).Citation71 Src has also been associated with VEGF production in tumor cells.Citation66 In fact, Src inhibition decreases angiogenesis in vivo.Citation68,Citation69
Immunohistological data about the expression/correlation between active FAK/Src on primary tumors and on their metastases is still awaited, to explore the value of FAK/Src as predictors of tumor outcome.
In fact, as we will review below, a link for FAK/Src with chemoresistance has already been reported in tumor models. In fact, FAK downregulation enhances docetaxel cytotoxicity in ovarian cancer cells.Citation72 Moreover, FAK downregulation also increases gemcitabine chemosensitivity in pancreatic cancer cells.Citation73 Accordingly, a variety of reports show a role for Src in the promotion of chemoresistance. Citation74–Citation77 Src inhibitors have already shown single agent activity in cancer patients after their progression to chemotherapy.Citation78 Src inhibition promotes chemosensitivity in pancreatic cancer cells.Citation74 In addition, a combination of 5-fluorouracil (5-FU) and a Src inhibitor in 5-FU-resistant human pancreatic cancer cell lines restored 5-FU-induced apoptosis.Citation74 The potential mechanism for 5-FU chemosensitivity induced by Src inhibitors might be associated with the inhibition of the epidermal growth factor receptor-AKT (EGFR–AKT) pro-survival pathway induced by 5-FU. Furthermore, a role for Src in mediating acquired endocrine resistance is also well established.Citation10
Other FAK family members have also been associated with cancer. In fact, Pyk2 also shows a high expression and an association with tumor progression in a variety of tumor types such as: astrocytomas; breast; glioma; prostate; hepatocarcinoma; and nonsmall cell lung cancer.Citation79–Citation83 In addition, other Src family members have also been associated with solid and hematological tumors; and inhibitors against Src family members are under development as new anticancer drugs.Citation84
FAK-Src and tumor associated epithelial mesenchymal transition
Epithelial mesenchymal transition (EMT) is a complex of cellular and molecular processes by which epithelial cells acquire mesenchymal and migratory properties.Citation85 EMT takes place during critical phases of embryonic development and is also a crucial step in the infiltration and progression in solid tumors. Hallmarks of EMT include loss of cell–cell contacts, induction of FA turnover and increased expression of mesenchymal (fibronectin, vimentin, N-cadherin, α-smooth muscle actin, and others) and invasiveness (ie, metalloproteinases) markers.Citation86 The EMT is at the convergence of different molecular pathways involving cell survival and resistance to apoptosis, invasion and tumor angiogenesis, metastasis and drug resistance in advanced tumors.Citation87
A critical molecular feature in the loss of cell–cell contacts during EMT is the downregulation of the adhesion molecule E-cadherin (delocalization/loss of E-cadherin expression). A variety of membrane receptors such as integrins, TKRs, serine-threonine kinase receptors are able to induce E-cadherin downregulation during development and tumor progression through the activation of specific intracellular signaling cascades such as Ras-MAPK and PI3K-Akt-mTOR. In fact, transcriptional repressors (Snail, Slug, Twist, or ZEB1/2) involved in EMTs during development are also induced in response to EMT stimuli to repress E-cadherin expression during tumor progression.Citation86 Plenty of evidence suggests that FAK and Src, through its ability to integrate signals from numerous signaling receptors, plays a critical role in tumor-associated EMTs promoting intracellular signaling pathways that lead to the induction of E-cadherin repressors and to the subsequent E-cadherin downregulation as well as that promote FA turnover to allow tumor cell migration/invasion ().Citation88–Citation91
New anticancer drugs that target FAK and Src
Based on evidence that supports FAK as a molecular scaffold protein, activated by Src to recruit its substrates; and that Src, as a tyrosine kinase is involved in the catalytic activation of FAK, and triggers FAK kinase activity to promote a variety of cellular responses during tumor progression, preclinical and clinical studies with new agents that employ different mechanisms for the blockade of FAK or Src kinases are currently underway.Citation9,Citation10 We show below a summary of the most advanced FAK and Src inhibitors under development (see ).
Table 1 Summary of FAK and Src inhibitors under clinical and preclinical development
Historically the first drugs synthesized with the aim of inhibiting T-cell activation via the Src family kinases Lck and Fyn were PP1 and PP2. The latest one is very selective for Src family kinases (SFKs). After PD173955 and PD173956 emerged with a lower selectivity than that of PP2, since these compounds were inhibitors of; Abl, Csk, platelet derived growth factor receptor (PDGFR) and EGFR. CGP76030 and CGP77675 were also multi-targeted agents against SFKs, Abl, EGFR and VEGF receptor (VEGFR) and SFKs, EGFR, FAK, and VEGFR, respectively. A third generation of molecules characterized by their higher potency in enzyme assays and their dual inhibition of c-Abl in a variety of imatinib-resistant c-Abl mutations appeared. The strong activity against c-Abl together with the potent anti-Src activity is explained by the strong structural similarity of the ATP binding domains in both kinases. The dual selectivity speeded up the development of these compounds.
At present, researchers are strongly focused on the study of the potential therapeutic benefits from the use of ATP-competitive kinase inhibitors against FAK and Src. These inhibitors interact with the ATP-binding pocket of FAK or Src and subsequently prevent FAK and Src autophosphorylation and therefore their activation.Citation11
Dasatinib
Dasatinib (BMS-354825) is an orally active small multiselective inhibitor, which inhibits the kinases Src and Abl with IC50 values of 0.55 and 3.0 nM, respectively.Citation92 Dasatinib also inhibits other Src family members such as Fyn (half maximal inhibitory concentration [IC50] of 0.2 nM), LcK (IC50 of 1.1 nM) and Yes (IC50 of 0.4 nM).Citation92 Dasatinib blocks the wild type chimeric Bcr-Abl protein which arise from Philadelphia chromosome.Citation93 Dasatinib is a 20-fold more potent inhibitor than imatinib in cells expressing wild-type Bcr–Abl hybrid protein and it also has an antitumoral effect in those tumor cells expressing Bcr–Abl imatinib-resistant mutants.Citation93 Dasatinib is also able to inhibit the tyrosine kinase receptors c-KIT, PDGFR-β and ephrins (EPHA2).Citation92 Dasatinib has already shown to have broad preclinical activity in solid and hematological tumor models.Citation14,Citation92 It has already been approved by the Food and Drug Administration (FDA) and by the European Medicines Agency (EMEA) for the second line treatment of imatinib-refractory chronic myelogenous leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL).Citation14 Currently, phase I/II clinical trials with dasatinib as a single agent or in combination are underway in a variety of solid and hematological tumors, based on the capability of dasatinib for blocking the activity Src family members.Citation94 Besides effectiveness in hematologic malignancies, dasatinib can suppress tumor growth in human breast cancer, human prostate, colon, ovarian, and lung cancer lines.Citation95–Citation100
AZD0530
AZD0530 is another novel, orally administered, potent, and highly selective inhibitor of Src (IC50 value ≤ 4 nM), other Src family members like LcK and Yes (both with IC50 values < 4 nM) and Abl as well.Citation101 Preclinical activity has been shown in: skin; breast; prostate; and pancreatic tumor modelsCitation101 as well as estrogen receptor-positive breast cancer modelsCitation102 where the combination of AZD0530 with tamoxifenCitation103 and aromatase inhibitorsCitation104 prevented hormonal therapy resistance. This combination also shows an additive effect of delaying the growth of breast cancer cells.Citation105
A variety of phase I and II clinical trials are currently underway with AZD0530 in monotherapy or in combination, in patients with solid tumors based on its capability to inhibit Src.Citation106 Another possible use of AZD0530 has been recently reported. Based upon the hypothesis that Src could be involved in the development of metastases in xenograft modes where the growth of the primary tumor was controlled by radiation; this report shows that those tumors from mice receiving AZD0530 regress more quickly following radiotherapy than their control counterparts.Citation107
Bosutinib
Bosutinib (SKI-606) is a potent, orally administered, bioavailable, dual Src (IC50 value of 3.8 nM) and Abl inhibitor which has already shown to have an antitumoral effect in chronic myelocytic leukemia (CML), colon, prostate and breast cancer models.Citation108,Citation109 Preclinical breast cancer models showed a decrease in in vitro cell motility and invasion and in vivo metastases after bosutinib treatment.Citation110 A phase I clinical trial with bosutinib has been published showing; drug-related dose-limiting toxicity of grade 3 diarrhea and grade 3 rash (1 pt) with 400 mg being selected as the maximum tolerated dose.Citation111 Currently, phase II, proof of concept clinical trials, in patients with CML who had failed to improve with Imatinib, and in patients with solid tumors, are underway.Citation112
PF-562,271
PF-562,271 is a potent ATP-competitive, small molecule inhibitor of both FAK and the related kinase Pyk2 (IC50 values of 1.5 nM (0.7 ng/mL) and 14 nM (7 ng/mL), respectively). PF-00562271 also inhibits other kinases such as c-Src and insulin growth factor 1 receptor (IGF1R) with less selectivity (IC50 value of 797 nM and IC50 > 500 nM respectively). This inhibitor has shown a broad preclinical activity.Citation113 In PC3 human prostate tumor cells, PF-00562271 treatment blocks, anchorage independent tumor cell growth and tumor cell migration in vitro, has shown antitumoral effects in vivo.Citation11 PF-00562271 decreases FAK phosphorylation-status in vitro and shows antitumor efficacy in vivo, in xenografts from: human colon; breast; prostate; pancreatic; and hepatocellular carcinoma tumor cell lines.Citation113–Citation115 No weight loss, or increase in morbidity and mortality were observed in any in vivo experiment and tumor growth inhibition was dose and drug exposure dependent. Furthermore, PF-562271 also showed an additional antiangiogenic effect over tumors.Citation113 PF-562271 through the inhibition of FAK and PYK2 kinases may interfere with the ability of endothelial tumor cells to migrate, thus blocking the sprouting and stabilization of the new tumor vessels.Citation63,Citation116 PF-00562271 treatment has also led to the blockade of the expression of pro-angiogenic growth factors in tumor cells, such as VEGF.Citation70 Recently, Bagi and colleagues showed that PF-00562271 synergized with antiangiogenic agents that directly block VEGF signaling, through its ability to target different aspects of angiogenesis and tumor aggressiveness.Citation115 In addition, these authors show that the combination of these agents not only led to the blockade of tumor growth, it also impacted upon the ability of the tumor to recover on withdrawal of the therapy. PF-562,271 has also showed an in vivo effect in preventing the loss of bone, suggesting its potential activity in patients with bone metastases and cancer-associated osteoporosis.Citation117 Therefore, PF-562,271 may comprise of a combined action over tumors: antiproliferative; proapoptotic; antiangiogenic; and antimetastatic action. Based on the preclinical data, a dose escalation phase 1 clinical trial with PF-562,271, administered orally as a single agent, in patients with solid tumors is currently underway. Tumor responses with PF-562,271 have been already reached in ovarian, colon together with head and neck cancer patients. Preliminary results showed a manageable safety profile with PF-562,271. In addition, continuous oral dosing is feasible and may be extended over 6 to 12 months in the majority of patients. The most common adverse events in the 32 evaluated patients with monitored safety data was: nausea in 14 patients (<Grade 3); vomiting in 12 patients (only 1 patient with Grade ≥3); fatigue in 8 patients (<Grade 3); and diarrhea in 6 patients (<Grade 3). Prolonged disease stabilization has already been observed in a variety of solid tumors. However, the maximum tolerated dose (MTD) and recommended Phase 2 dose have still to be published.Citation15,Citation16 In addition to PF 573,228, a closely related, early prototype FAK inhibitorCitation118 has been reported to have appealing activity in combating ovarian cancer metastases,Citation119,Citation120 demonstrating the growing body of evidence that supporting research of Src/FAK inhibitors in epithelial carcinoma.
TAE 226
TAE 226 is a low molecular weight, ATP-competitive tyrosine kinase inhibitor of FAK and IGF1R with an IC50 range of 100 to 300 nM/L.Citation121 TAE 226 is still under preclinical development. Flow cytometry analysis of human glioma cell lines under TAE 226 treatment have shown an increase in the apoptotic and G0 (quiescent/nonproliferative) fractions after treatment, when these cells were compared with control/nontreated cells.Citation60 TAE 226 induced-apoptosis in the glioma tumor model is mediated by caspases and is correlated with the p53 status. In fact, apoptosis was only induced in the subset of glioma cell lines containing the mutant p53 gene.Citation60
Additionally, TAE 226 treatment also prevented the in vitro attachment of these glioma cell lines. Furthermore, an in vivo intracranial glioma xenograft model showed a significantly higher median survival in the group of mice treated with TAE 226, at concentrations of 50–75 mg/kg. The treatment of ovarian cancer cell lines with TAE 226 inhibited cell growth in both a time- and dose-dependent manner; and enhanced docetaxel-mediated growth inhibition by 10 and 20 fold in the taxane-sensitive and taxane-resistant ovarian cell lines, respectively. In addition, TAE 226 alone and in combination with chemotherapy significantly prolonged survival in tumor-bearing mice. The efficacy of TAE 226 was related to: reduced pericyte coverage; the induction of apoptosis of tumor-associated endothelial cells; reduced microvessel density and tumor cell proliferation.Citation121 TAE 226 also displayed an antitumoral effect in human pancreatic cell lines,Citation122 esophageal cancer cell lines and xenografts, by a potent inhibition of PI3K-AKT-mTOR cell survival signaling.Citation123 Clinical trials with this new dual FAK-IGF1R inhibitor are planned. Recently, interesting experiments have suggested that the FAK–IGF–1R interaction site could be targeted; the specific disruption of this protein–protein interaction with another small molecule inhibitor (INT2-31) reinforces the potential novel role of this antineoplastic strategy.Citation124
FAK/Src and chemotherapy resistance
As we mentioned above, an association between FAK activation and resistance to chemotherapy has been broadly reported in human tumor models.Citation72,Citation73,Citation125–Citation128 Accordingly, the combination of conventional chemotherapeutic drugs with FAK-targeting agents apparently offers greater efficacy in preclinical models than chemotherapy as a single agent. Treatment with FAK antisense oligonucleotides significantly induced apoptosis in human glioblastoma cells associated with a decrease in FAK protein levels.Citation129 The in vitro cytotoxic effect achieved with the anti-FAK agent in monotherapy was almost the same as those obtained with different chemotherapeutic regimens such as cisplatin, etoposide and nimustine hydrochloride.Citation129 When FAK antisense oligonucleotides and chemotherapy were administered in combination the antitumoral effect was clearly additive.Citation129 Treatment of squamous cell carcinoma models with recombinant FRNK peptides combined with etoposide, paclitaxel or 5-FU also showed an additive antitumoral effect.Citation127
The effect of combined chemotherapy and anti-FAK agents were also explored in human HCC cells in vitro.Citation130 When TNF-α plus cycloheximide was combined with FAK-antisense, an increase in the apoptotic index was observed.Citation130 Additionally, FAK siRNA was also able to potentiate gemcitabine action in pancreatic cancer cellsCitation73 increasing the apoptotic index. The in vivo treatment with FAK siRNA, in combination with gemcitabine, induced in a statistically significant manner, a larger inhibition in the size of the tumors than gemcitabine in monotherapy.Citation73 FAK siRNA incorporated in liposomes was administered to mice bearing tumors from human ovarian cancer cells.Citation131 Mice treated with siRNA-DOPC showed a decrease in tumor weight. Docetaxel in combination with siRNA-DOPC resulted in an even greater reduction in tumor weight.Citation131 This combination also showed: antiangiogenic properties;Citation131 it decreased microvessel density; VEGF and MMP-9 secretion; and increased apoptosis in tumor cells, in addition to tumor-associated endothelial cells.Citation131 Treatment with siRNA-DOPC resulted in a decrease in the tumor weight of cisplatin-resistant xenografts as well.Citation131 These data suggest that the combination of anti-FAK agents with docetaxel or cisplatin may be a valuable therapeutic approach in the chemotherapy of resistant ovarian cancer. Smith and colleagues showed that FAK downregulation enhanced the effects of 5-FU in human melanoma cells.Citation132 FAK antisense oligonucleotides significantly increased cell detachment and apoptosis when they were administered alone or in combination with 5 FU.Citation132 This led to the decrease in FAK protein levels, an effect that was also observed with the 5-FU alone. Accordingly, FAK blockade plus 5-FU showed and additive effect.
There are also, preclinical data supporting the role of Src in chemoresistance. The inhibition of Src reversed chemoresistance toward 5-FU in human pancreatic carcinoma cells.Citation74 Furthermore, Src inhibition also impaired both inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells.Citation75 The combination of dasatinib and chemotherapy (5′-5′-DFUR or cisplatin) was synergistic in triple-negative breast cancer cells.Citation99 In addition, phase 1 clinical trials are currently ongoing, with the Src inhibitor dasatinib in combination with chemotherapy in solid tumors.Citation94,Citation133
Interestingly the combination of paclitaxel/carbolatin standard chemotherapy in ovarian cancer has shown interesting synergism from this combined approach at the cell line level, that requires further investigation.Citation134
In conclusion, current evidence shows that FAK/Src-targeting compounds enhance the action of conventional anticancer agents at least in preclinical tumor models. Nonetheless, further molecular studies testing the activation status of both kinases in tumor biopsies and clinical trials, with anti-FAK/anti-Src agents and different chemotherapeutic schedules are still required to confirm if this complex is involved in treatment resistance and if the combination can enhance the efficacy of conventional chemotherapy in the clinical setting.
The interest of targeting FAK and Src in breast cancer
The Src/FAK signaling pathway is related to multiple receptor tyrosine kinases (RTKs) and intracellular mediators with a prominent role in the biology of the different subtypes of breast cancer.Citation135
On one hand, c-Src interacts with and contributes to the signaling cascade of different RTKs; modulates their turnover by interfering in the endocytosis; and ubiquitination; in to taking part in the cytoskeleton rearrangement, migration and survival processes started at the RTKs’ level in tumor cells.Citation7 In breast cancer there is evidence of the interaction between c-Src and EGFR, (although short of a synergistic activity), connected to the crosstalk between estrogen receptor (ER) and EGFR.Citation136,Citation137 HER2 is coexpressed with c-Src in breast cancerCitation138 and their interaction seems to facilitate the migratory and metastatic phenotype of these cells.Citation139,Citation140 Likewise, HER2 is actually involved in c-Src regulation,Citation140 another mechanism of activation of HER2 such as the transactivation through G-protein receptors (ie, CXCR4).Citation141 C-Src has also been proposed to be involved in the modulation of HER2–HER3 heterocomplexes in an intracellular mediated pattern.Citation142
However, Src has also been linked to the endoplasmic reticulum (ER) nongenomic activityCitation143 and the homeostasis of the ERCitation144 in the tumor cells.
Src inhibitors have been developed in different breast cancer subtypes. Triple negative breast cancer has emerged as a potential field to be explored by this family of drugs. Two independent groups have described genetic profiles associated with a response to dasatinibCitation95,Citation145 highlighting the particular sensitivity of the basal-like phenotype to this Src inhibitor. This molecular marker approach has led to a phase 2 trial in the clinical settingCitation146 showing modest, although encouraging, results to be tested in further trials, in combination with chemotherapy.
Hormone receptor-positive breast cancer, resistant to the classic endocrine therapy strategies, has become another field actively studied. In the tamoxifen resistance setting, the upregulation of different RTKs’ signaling pathways has been involved. It has been suggested that the resistant phenotype is not just the result of an estrogen independent growth,Citation147 but is also linked to an alteration in the relationship between the cells and the extracellular matrix, so that these tumor cells acquire an invasive and migratory phenotypeCitation148,Citation102 that favors tumor dissemination. It has been demonstrated that anti-HER2 therapies are able to eliminate the agonist effect of tamoxifen, restoring its antitumoral capacityCitation149 and the blockade of both pathways, showing an increased efficacy against endocrine-resistant tumors.Citation100,Citation150,Citation151
However, these combinations have not shown a definitive effect regarding the migratory and invasive phenotype,Citation148 moreover, the tumoral cells eventually develop double resistance that results in an even more invasive behavioral pattern.Citation102,Citation152 This dual resistant phenotype is characterized by an increase Src kinase activity, that defines another opportunity to target endocrine resistant breast cancer. The in vitro utility of Src inhibitors; due to the Src/FAK relationship in the acquisition of endocrine resistance in breast cancer, has already been tested.Citation153–Citation156 However, a recent article showed an opposite role for Src in breast cancer. Campbell and colleagues analyzed 262 breast cancer specimens, before tamoxifen treatment, for active Src expression by tissue microarray. The authors showed that phosphorylated c-Src in the nucleus was significantly associated with improved patient outcome in ER-positive breast cancer.Citation157 The current findings suggest a crosstalk between ER and Src/FAK kinases, so that the addition of agents that block Src and/or FAK to hormonal therapy may improve the efficacy of the current endocrine therapies (aromatase inhibitors and tamoxifen). Clinical trials in breast cancer patients with dasatinib and aromatase inhibitors are underway in breast cancer patients.Citation94
Conclusions
As we reviewed above FAK and Src form a mutually activated complex that acts as a common intracellular point of convergence in the signaling initiated by a variety of membrane receptors (RTKs, Integrins, G-coupled receptors, ER and others) to trigger a cascade of phosphorylation events and new protein–protein interactions in tumor cells and tumor endothelial cells, that allow the angiogenic and metastatic behavior of tumors. In fact, preclinical data with anti-Src and anti-FAK agents under development show that both types of inhibitors lead to antiproliferative, antiangiogenic and antimetastatic responses in human tumor models; a synergistic effect with other anticancer agents has been also observed. Therefore the inhibition of one of these kinases appears to be a successful therapeutic approach to avoid recurrence and dissemination of the primary tumor and also the progression of metastatic lesions.
Currently, we have robust data to believe FAK and c-Src inhibitors as a novel and promising anticancer strategy to combine with current anticancer therapies. A synergistic effect has already been shown when they are combined with other antitarget agents (ie, gefitinib, imatinib and sunitinib). Furthermore, these drugs have also been shown to be good candidates in the avoidance of chemotherapy and hormonotherapy resistances.
In addition, although it remains difficult to asses the efficacy of antimetastatic agents in the clinical setting, its appears that the inhibition of Src and FAK may have a potent anti-invasive effect, to delay tumor dissemination rather than real tumor shrinkage. Furthermore, although levels have already been seen as a possible predictor of response to Src inhibitors there is still a lack of suitable biomarkers that would be able to predict a response to these agents. In addition to current clinical studies of biomarker assays, the use of more sophisticated imaging technologies and the testing of the tumor, guided by biochemical rational, will help to maximize the development of these new compounds.
We have reviewed those trials with FAK and Src inhibitors under clinical development as a single agent or in combination with other therapeutic approaches. They have already shown clinical benefits in cancer patients with solid tumors. The identification of useful biomarkers to assess target inhibition, anti-invasive efficacy and predict treatment response will be crucial for future clinical trials.
Acknowledgements
We would like to thank to Nieves Ruíz-Ayllón for the editorial assistance. The authors report no conflicts of interest relevant to this research.
References
- GuoWGiacontiFGIntegrin signaling during tumor progressionNat Rev Mol Cell Biol200451081682615459662
- CalalbMBPolteTRHanksSKTyrosine of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinasesMol Cell Biol1995159549637529876
- MitraSatyajit KSchlaepferDavid DIntegrin-regulated FAK–Src signaling in normal and cancer cellsCurrent Opinion in Cell Biology200618551652316919435
- BrutonVGMacPhersonIRJFrameMCCell adhesion receptors, tyrosine kinases and actin modulators: a complex three-way circuitryBiochim Biophys Acta2004169212114415246683
- PlayfordMPSchallerMDThe interplay between Src and integrins in normal and tumor biologyOncogene20041823487928794615489911
- WangSEXiangBZentRQuarantaVPozziAArteagaCLTransforming growth factor beta induces clustering of HER2 and integrins by activating Src-focal adhesion kinase and receptor association to the cytoskeletonCancer Res200969247547819147560
- BromannPAKorkayaHCourtneidgeSAThe interplay between Src family kinases and receptor tyrosine kinasesOncogene200423487957796815489913
- ChiangACMassaguéJMolecular basis of metastasisN Engl J Med2008359262814282319109576
- Van NimwegenMJVan DewaterBFocal adhesion kinase: a potential target in cancer therapyBiochem Pharmacol20077359760916997283
- MorganLNicholsonRIHiscoxSSRC as a therapeutic target in breast cancerEndocr Metab Immune Disord Drug Targets20088427327819075780
- ParsonsJTSlack-DavisJTilghmanRRobertsWGFocal adhesion kinase: targeting adhesion signaling pathways for therapeutic interventionClin Cancer Res200814362763218245520
- FinnRSTargeting Src in breast cancerAnn Oncol20081981379138618487549
- Slack-DavisJKMartinKHTilghmanRWIwanickiMCellular characterization of a novel focal adhesion kinase inhibitorJ Biological Chem2007282201484514852
- JarkowskiASweeneyRPNilotinib: a new tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemiaPharmacotherapy200828111374138218956997
- SiuLLPhase 1 study of a focal adhesion kinase (FAK) inhibitor PF-00562271 in patients (pts) with advanced solid tumorsAbstract 35272007 ASCO Annual MeetingChicago, IL, USA
- SiuLLA phase I clinical, pharmacokinetic (PK) and pharmacodynamic (PD) evaluation of PF-00562271 targeting focal adhesion kinase (FAK) in patients (pts) with advanced solid tumorsAbstract 35342008 ASCO Annual MeetingChicago, IL, USA
- AraujoJArmstrongAJBraudELDasatinib and docetaxel combination treatment for patients with castration-resistant progressive prostate cancer: A phase I/II study (CA180086)Abstract 50612009 ASCO Annual MeetingChicago, IL, USA
- KlugerHMDudekAMcCannCA phase II trial of dasatinib inadvanced melanomaAbstract 90102009 ASCO Annual MeetingChicago, IL, USA
- MayerEBaurainJSparanoJDasatinib in advanced HER2/neu amplified and ER/PR-positive breast cancer: Phase II study CA180088Abstract 10112009 ASCO Annual MeetingChicago, IL, USA
- RousPATransmission of a malignant new growth by means of a cell-free filtrateJAMA191156198202
- RousPAA sarcoma of the fowl transmissible by an agent separable from the tumor cellsJ Exp Med191113439741119867421
- Steven MartinGThe hunting of the SrcNature Rev Molec Cell Biol2001246747511389470
- YeatmanTJA renaissance for SRCNat Rev Cancer20044647048015170449
- IngleyESrc family kinases: regulation of their activities, levels and identification of new pathwaysBiochim Biophys Acta200817841566517905674
- CoxBDNatarajanMStettnerMRGladsonCLNew concepts regarding focal adhesion kinase promotion of cell migration and proliferationJ Cell Biochem2006991355216823799
- KannerSBMonoclonal antibodies to individual tyrosine-phosphorylated protein substrates of oncogene-encoded tyrosine kinasesProc Natl Acad Sci1990879332833322110361
- ShallerMDBorgmanCACobbBSpp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesionsProc Natl Acad Sci199289519251961594631
- SchallerMDParsonsJTFocal adhesion kinase and associated proteinsCurr Opin Cell Biol199467057717833050
- BruntonVGAvizienyteEFinchamVJIdentification of Src-specific phosphorylation site on focal adhesion kinase: dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behaviorCancer Res20056541335134215735019
- AvizienyteEWykeAWJonesRJSrc-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalingNat Cell Biol20024863263812134161
- SiegDJHauckCRIlicDFAK integrates growth-factor and integrin signals to promote cell migrationNat Cell Biol20002524925610806474
- EliceiriBPPuenteXSHoodJDSrc-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signalingJ Cell Biol2002157114916011927607
- SchallerMDBorgmanCAParsonsJTAutonomous expression of a noncatalytic domain of the focal adhesion associated protein tyrosine kinase pp.125FAKMol Cell Biol1993137857918423801
- MitraSKSchaepferDDIntegrin-regulated FAK-C-src signaling in normal and cancer cellsCurr Opin Cell Biol20061851652316919435
- ZhaoJGuanJLSignal transduction by focal adhesion kinase in cancerCancer Metastasis Rev2009281–2354919169797
- ParsonsJTMartinKHSlackJKTaylorJMFocal adhesion kinase: a regulator of focal adhesion dynamics and cell movementOncogene200019495606561311114741
- SummyJMGallickGESrc family kinases in tumor progression and metastasisCancer Metastasis Rev20032233735812884910
- IlicDFurutaYKanazawaSReduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient miceNature19953775395447566154
- PengXUedaHZhouHOverexpression of focal adhesion kinase in vascular endothelial cells promotes angiogenesis in transgenic miceCardiovascular Res200464421430
- WeisSMLimSTLutu-FugaKMCompensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell. FAKJ Cell Biol2008181435018391070
- GuinamardROkigakiMSchlessingerJRavetichJVAbsence of marginal zone B cell in Pyk2-deficient mice defines their role in the humoral responseNat Immunol20001313610881171
- OkigakiMDavisCFalascaMPyk2 regulates multiple signaling events crucial for macrophage morphology and migrationProc Nat Acad Sci2003100107401074512960403
- LimSTChenXLLimYNuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradationMol Cell200829192218206965
- SorianoPMontgomeryCGeskeRBradleyATargeted disruption of the c-src proto-oncogene leads to osteopetrosis in miceCell19916446937021997203
- RoodmanGDMechanisms of bone metastasisN Engl J Med20043501655166415084698
- RucciNTetiSusa MInhibition of protein kinase c-Src as a therapeutic approach for cancer and bone metastasesAnticancer Agents Med Chem20088334234918393792
- ZhanMZhaoHHanZCSignaling mechanisms of anoikisHistol Histopathol200419397398315168359
- DuxburyMSItoHZinnerMJAshleySWWhangEEFocal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cellsSurgery2004135555556215118593
- LiuGMengXJinYInhibitory role of focal adhesion kinase on anoikis in the lung cancer cell A549Cell Biol Int200832666367018343694
- Díaz-MonteroCMWygantJNMcIntyreBWPI3-K/Akt-mediated anoikis resistance of human osteosarcoma cells requires Src activationEur J Cancer20064210149150016759849
- Gabarra-NieckoVSchallerMDDuntyJMFAK regulates biological processes important for the pathogenesis of cancerCancer Metastasis Rev200322435937412884911
- Grisaru-GranovskySSalahZMaozMPrussDBellerUBar-ShavitRDifferential expression of protease activated receptor 1 (Par1) and pY397FAK in benign and malignant human ovarian tissue samplesInt J Cancer2005113337237815455382
- RecherCYsebaertLBeyne-RauzyOExpression of focal adhesion kinase in acute myeloid leukemia is associated with enhanced blast migration, increased cellularity, and poor prognosisCancer Res20046493191319715126359
- AronsohnMSBrownHMHauptmanGKornbergLJ Expression of focal adhesion kinase and phosphorylated focal adhesion kinase in squamous cell carcinoma of the larynx. Aronsohn MS, Brown HM, Hauptman G, Kornberg LJ.
- MoonHSParkWIChoiEAChungHWKimSCThe expression and tyrosine phosphorylation of E-cadherin/catenin adhesion complex, and focal adhesion kinase in invasive cervical carcinomasInt J Gynecol Cancer200313564064614675348
- DingJLiDWangXWangCWuTFibronectin promotes invasiveness and focal adhesion kinase tyrosine phosphorylation of human colon cancer cellHepatogastroenterology200855882072207619260479
- PantaGRNwariakuFKimLTRET signals through focal adhesion kinase in medullary thyroid cancer cellsSurgery200413661212121715657578
- PantaGRDuLNwariakuFEKimLTDirect phosphorylation of proliferative and survival pathway proteins by RETSurgery2005138226927416153436
- TongZKunnumakkaraABWangHNeutrophil gelatinase-associated lipocalin: a novel suppressor of invasion and angiogenesis in pancreatic cancerS Cancer Res2008681561006108
- LiuTJLaFortuneTHondaTInhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivoMol Cancer Ther2007641357136717431114
- SchweppeREKeregeAAFrenchJDSharmaVGrzywaRLHaugenBRInhibition of Src with AZD0530 reveals the Src-Focal Adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancerJ Clin Endocrinol Metab20099462199220319293266
- ChenJIs Src the key to understanding metastasis and developing new treatments for colon cancer?Nat Clin Pract Gastroenterol Hepatol20085630630718477987
- HaskellHNatarajanMHeckerTPFocal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cellsClin Cancer Res2003962157216512796381
- AngelucciABolognaMCurr. Targeting vascular cell migration as a strategy for blocking angiogenesis: the central role of focal adhesion protein tyrosine kinase familyPharm Des2007132121292145
- EarleySPlopperEDisruption of focal adhesion kinase slows transendothelial migration of AU-565 breast cancer cellsBiochem Biophys Res Commun200635040541217010315
- EliceiriBPPaulRSchwartzbergPLHoodJDLengJChereshDASelective requirement for C-src kinases during VEGF-induced angiogenesis and vascular permeabilityMol Cell1999491592410635317
- NiuGConstitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesisOncogene2002212000200811960372
- KilarskiWWJuraNGerwinsPInactivation of Src family kinases inhibits angiogenesis in vivo: implications for a mechanism involving organization of the actin cytoskeletonExp Cell Res2003291708214597409
- LairdADSrc family kinase activity is required for signal tranducer and activator of transcription 3 and focal adhesion kinase phosphorylation and vascular endothelial growth factor signaling in vivo and for anchorage-dependent and-independent growth of human tumor cellsMol Cancer Ther2003246146912748308
- ShetaEAHardingMAConawayMRTheodorescuDFocal adhesion kinase, Rap1, and transcriptional induction of vascular endothelial growth FactorJ Natl Cancer Inst200092131065107310880549
- CasconeINapioneLManieroFSeriniGBussolinoFJStable interaction between alpha5beta1 integrin and Tie2 tyrosine kinase receptor regulates endothelial cell response to Ang-1Cell Biol200517069931004
- HalderJLandenCNJrLutgendorfSKFocal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cellsClin Cancer Res20051124 Pt 18829883616361572
- DuxburyMSItoHBenoitEZinnerMJAshleySWWhangEERNA interference targeting focal adhesion kinase enhances pancreatic adenocarcinoma gemcitabine chemosensitivityBiochem Biophys Res Commun2003311378679214623342
- IschenkoICamajPSeeligerHInhibition of Src tyrosine kinase reverts chemoresistance toward 5-fluorouracil in human pancreatic carcinoma cells: an involvement of epidermal growth factor receptor signalingOncogene200827577212722218794807
- DuxburyMSItoHZinnerMJAshleySWWhangEEInhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cellsClin Cancer Res20041072307231815073106
- DuxburyMSItoHZinnerMJAshleySWWhangEEsiRNA directed against c-Src enhances pancreatic adenocarcinoma cell gemcitabine chemosensitivityJ Am Coll Surg2004198695395915194078
- ShahANGallickGESrc, chemoresistance and epithelial to mesenchymal transition: are they related?Anticancer Drugs200718437137517351389
- MayerEBaurainJSparanoJDasatinib in advanced HER2/neu amplified and ER/PR-positive breast cancer: Phase II study CA180088Abstract: 10112009 ASCO Annual MeetingChicago IL, USA
- GutenbergABrückWBuchfelderMLudwigHCExpression of tyrosine kinases FAK and Pyk2 in 331 human astrocytomasActa Neuropathol2004108322423015221336
- BehmoaramEBijianKJieSFocal adhesion kinase-related proline-rich tyrosine kinase 2 and focal adhesion kinase are co-overexpressed in early-stage and invasive ErbB-2-positive breast cancer and cooperate for breast cancer cell tumorigenesis and invasivenessAm J Pathol200817351540155018832579
- SunCKNgKTSunBSThe significance of proline-rich tyrosine kinase2 (Pyk2) on hepatocellular carcinoma progression and recurrenceBr J Cancer2007971505717551499
- IiizumiMBandyopadhyaySPaiSKRhoC promotes metastasis via activation of the Pyk2 pathway in prostate cancerCancer Res200868187613762018794150
- ZhangSQiuXGuYWangEUp-regulation of proline-rich tyrosine kinase 2 in non-small cell lung cancerLung Cancer200862329530118571765
- JohnsonFMGallickGESRC family nonreceptor tyrosine kinases as molecular targets for cancer therapyAnticancer Agents Med Chem20077665165918045060
- ThieryJPEpithelial-mesenchymal transitions in development and pathologiesCurr Opin Cell Biol200315674074614644200
- PeinadoHOlmedaDCanoASnail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?Nat Rev Cancer20077641542817508028
- SabbahMEmamiSRedeuilhGMolecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancersDrug Resist Updat2008114–512315118718806
- BaileyKMLiuJJCaveolin-1 up-regulation during epithelial to mesenchymal transition is mediated by focal adhesion kinaseBiol Chem2008283201371413724
- CicchiniCLaudadioICitarellaFTGFbeta-induced EMT requires focal adhesion kinase (FAK) signalingExp Cell Res2008314114315217949712
- MandalMMyersJNLippmanSMEpithelial to mesenchymal transition in head and neck squamous carcinoma: association of Src activation with E-cadherin down-regulation, vimentin expression, and aggressive tumor featuresCancer200811292088210018327819
- WeiJXuGWuMOverexpression of vimentin contributes to prostate cancer invasion and metastasis via src regulationAnticancer Res2008281A32733418383865
- LombardoLJLeeFYChenPDiscovery of N-(2-chloro-6methylphneyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5 carboxamide (BMS-354825),a dual Src/Abl. kinase inhibitor with potent antitumor activityin pre-clinical assaysJ Med Chem200447276658666115615512
- TokarskiJSNewittJAChangCYThe structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutantsCancer Res2006 1;66115790579716740718
- KopetzSShahANGallickGESrc continues aging: current and future clinical directionsClin Cancer Res200713247232723618094400
- FinnRSDeringJGintherCDasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“triple-negative” breast cancer cell lines growing in vitroBreast Cancer Res Treat2007105331932617268817
- HuangFReevesKHanXIdentification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selectionCancer Res20076752226223817332353
- ParkSIZhangJPhillipsKATargeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse modeCancer Res20086893323333318451159
- LeXiao-FengMaoWeiqunLuZhenBastRobert CJrDasatinib induces autophagy as well as growth arrest in human ovarian cancer cellsAbstract 388 AACR 2009
- TryfonopoulosDO’DonovanNCorkeryBClynesMCrownJActivity of dasatinib with chemotherapy in triple-negative breast cancer cellsAbstract: e146052009 ASCO Annual MeetingChicago, IL, USA
- SeoaneSMonteroJCPandiellaAOcanaAEffect of dasatinib on the activity of trastuzumab in HER2- overexpressing breast cancer cellsAbstract: 10842009 ASCO Annual MeetingChicago, IL, USA
- HennequinLFAllenJBreedJN-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitorMed Chem2006492264656488
- HiscoxSMorganLGreenTPBarrowDGeeJNicholsonRIElevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cellsBreast Cancer Res Treat20069726327416333527
- HiscoxSJordanNJSmithCDual targeting of Src and ER prevent acquired antihormone resistance in breast cellsBreast Cancer Res Treat20091151576718493848
- ChenYGuggisbergNJordaMCombined Src and Aromatase Inhibition Impairs Human Breast Cancer Growth In vivo and Bypass Pathways Are Activated in AZD0530-Resistant TumorsClin Cancer Res200915103396340519451593
- HerynkMHBeyerARCuiYCooperative action of tamoxifen and c-Src inhibition in preventing the growth of estrogen receptor-positive human breast cancer cellsMol Cancer Ther20065123023303117172405
- Clinical Trials.gov Clinical Trials AZDO530 at: http://www.clinicaltrial.gov/ct2/results?term=AZD0530+ accessed March 2010.
- StratfordIJTelferBGreenTPInhibition of metastatic dissemination following loco-regional control of a primary tumor: examining a novel treatment paradigm with the Src kinase inhibitor AZD0530Abstract 4946 AACR 2009
- BoschelliDHWuBYeFSynthesis and Src kinase inhibitory activity of a series of 4-[(2,4-dichloro-5-methoxyphenyl)amino]-7-furyl-3-quinolinecarbonitrilesJ Med Chem200649267868787617181170
- RabbaniSAValentinoMLAliSBoschelliFInhibitor of Src kinase SKI-606 (Bosutinb) blocks prostate cancer growth, invasion and experimental skeletal metastases in vitro and in vivo by regulating the expression of key intracellular signaling molecules and genes involved in prostate cancer progressionAbstract 2325 AACR 2009
- JallalHValentinoMLChenGBoschelliFAliSRabbaniSAA Src/Abl kinase inhibitor, SKI-606, blocks breast cancer invasion, growth, and metastasis in vitro and in vivoCancer Res20076741580158817308097
- MessersmithWAKrishnamurthiSHewesBABosutinib (SKI-606), a dual Src/Abl tyrosine kinase inhibitor: Preliminary results from a phase 1 study in patients with advanced malignant solid tumorsAbstract 35522007 ASCO Annual MeetingChicago, IL, USA
- Clinical Trials.gov Clinical Trials Bosutinib at: http://www.clinicaltrial.gov/ct2/results?term=Bosutinib+ accessed March 2010.
- RobertsWGUngEWhalenPAntitumor Activity and Pharmacology of a Selective Focal Adhesion Kinase Inhibitor, PF-562,271Cancer Res20086861935194418339875
- JaymeBStokesSJAdairJKTargeting FAK in pancreatic cancer: a novel therapeutic approachAbstract 843, AACR 2209
- BagiCMChristensenJCohenDPSunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft modelCancer Biology and Therapy20098985686519458500
- CasconeINapioneLManieroFSeriniGBussolinoFJStable interaction between alpha5beta1 integrin and Tie2 tyrosine kinase receptor regulates endothelial cell response to Ang-1Cell Biol200517069931004
- BagiCMRobertsGWAndresenCJDual focal adhesion kinase/Pyk2 inhibitor has positive effects on bone tumors: implications for bone metastasesCancer2008112102313232118348298
- Slack-DavisJKMartinKHTilghmanRWCellular characterization of a novel focal adhesion kinase inhibitorJ Biol Chem200728220148451485217395594
- StoneRLNickAMSpannuthWThe clinical and biological significance of focal adhesion kinase activation in ovarian carcinomaAbstract 3636 AACR 2009
- Dos SantosLAThe FAK Inhibitor PF573228 Demonstrates Anti-proliferative and anti-invasive activity in MUC16/CA125 ovarian carcinoma cellsAbstract 3519 AACR 2009
- HalderJLinYGMerrittWMTherapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinomaCancer Res20076722109761098318006843
- LiuWBloomDACanceWGKurenovaEVGolubovskayaVMHochwaldSNFAK and IGF-IR interact to provide survival signals in human pancreatic adenocarcinoma cellsCarcinogenesis20082961096110718263593
- HaoHWangZBaoXDual tyrosine kinase inhibitor for focal adhesion kinase and insulin-like growth factor-I receptor (TAE226) leads to apoptosis in esophageal cancer by inhibiting AKT-mTOR survival signalingAbstract 1840 AACR 2009
- UcarDKurenovaEZhengDA novel small molecule that targets the FAK and IGF-1R site of interaction inhibits growth of human cancerAbstract 2008 AACR 2009
- VilledieuMDeslandesEDuvalMHéronJFGauduchonPPoulainLAcquisition of chemoresistance following discontinuous exposures to cisplatin is associated in ovarian carcinoma cells with progressive alteration of FAK, ERK and p38 activation in response to treatmentGynecol Oncol2006101350751916387351
- Van NimwegenMJHuigslootMCamierATijdensIBvan de WaterBFocal adhesion kinase and protein kinase B cooperate to suppress doxorubicin-induced apoptosis of breast tumor cellsMol Pharmacol20067041330133916825486
- KornbergLJAdenovirus-mediated transfer of FRNK augments drug-induced cytotoxicity in cultured SCCHN cellsAnticancer Res2005254349435616309239
- FangYWangLJinJRef Focal adhesion kinase affects the sensitivity of human hepatocellular carcinoma cell line SMMC-7721 to TNF- α/cycloheximide-induced apoptosis by regulating protein kinase B levelsEur J Biochem20012684513451911502212
- WuZMYuanXHJiangPCAntisens oligonucleodes targeting the focal adhesion kinase inhibit proliferation, induce apoptosis and cooperate with cytotoxic drugs in human glioma cellsJ Neurooncol20067711712316314954
- FangYWangLJinJZhaXFocal adhesion kinase affects the sensitivity of human hepatocellular carcinoma cell line SMMC-7721 to TNF-α/cycloheximide-induced apoptosis by regulating protein kinase B levelsEur J Biochem20012684513451911502212
- HalderJKamatAALandenCNJrFocal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapyClin Cancer Res200612164916492416914580
- SmithCSGolubovskayaVMPeckEEffect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell linesMelanoma Res200515535736216179862
- SomloGAtzoriFStraussLDasatinib plus capecitabine (Cap) for progressive advanced breast cancer (ABC): Phase I study CA180004Abstract: 10122009 ASCO Annual MeetingChicago IL, USA
- TeohDAyeniTARubattJMIn vitrocharacterization of the antitumor effects of dasatinib (BMS-354825) in combination with paclitaxel and carboplatin in human ovarian cancer cell linesAbstract 828 AACR 2009
- SummyJMGallickGETreatment for advanced tumors: SRC reclaims center stageClin Cancer Res20061251398140116533761
- BiscardiJSBelschesAPParsonsSJCharacterization of human epidermal growth factor receptor and c-Src interactions in human breast tumor cellsMol Carcinog19982142612729585256
- HitosugiTSasakiKSatoMSuzukiYUmezawaYEpidermal growth factor directs sex-specific steroid signaling through Src activationJ Biol Chem200728214106971070617284441
- MuthuswamySKSiegelPMDankortDLWebsterMAMullerWJMammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activityMol Cell Biol19941417357437903421
- Elsches-JablonskiAPBiscardiJSPeavyDRTiceDARomneyDAParsonsSJSrc family kinases and HER2 interactions in human breast cancer cell growth and survivalOncogene200120121465147511313890
- VadlamudiRKSahinAAAdamLWangRAKumarRHeregulin and HER2 signaling selectively activates c-Src phosphorylation at tyrosine 215FEBS Lett20035431–3768012753909
- CabiogluNSummyJMillerCCXCL-12/stromal cell-derived factor-1alpha transactivates HER2-neu in breast cancer cells by a novel pathway involving Src kinase activationCancer Res200565156493649716061624
- IshizawarRCMiyakeTParsonsSJc-Src modulates ErbB2 and ErbB3 heterocomplex formation and functionOncogene200726243503351017173075
- SongRXZhangZSantenRJEstrogen rapid action via protein complex formation involving ERalpha and SrcTrends Endocrinol Metab200516834735316126407
- ChuIArnaoutALoiseauSSrc promotes estrogen-dependent estrogen receptor alpha proteolysis in human breast cancerJ Clin Invest200711782205221517627304
- HuangFReevesKHanXIdentification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selectionCancer Res20076752226223817332353
- FinnRSBengalaCIbrahimNPhase II Trial of Dasatinib in Triple-negative Breast Cancer: Results of Study CA180059SABCS2008
- KnowldenJMHutchesonIRJonesHEElevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cellsEndocrinology200314431032104412586780
- HiscoxSMorganLBarrowDDutkowskilCWakelingANicholsonRITamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype: inhibition by gefitinib (‘Iressa’, ZD1839)Clin Exp Metastasis200421320121215387370
- NicholsonRIHutchesonIRHarperMEModulation of epidermal growth factor receptor in endocrine-resistant, estrogen-receptor-positive breast cancerAnn N Y Acad Sci200296310411512095935
- MassarwehSOsborneCKJiangSMechanisms of Tumor Regression and Resistance to Estrogen Deprivation and Fulvestrant in a Model of Estrogen Receptor-Positive, HER-2/neu-PositiveBreast Cancer Cancer Res2006661682668273
- ShouJMassarwehSOsborneCKMechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancerJ Natl Cancer Inst2004961292693515199112
- HiscoxSJordanNJMorganLGreenTPNicholsonRISrc kinase promotes adhesion-independent activation of FAK and enhances cellular migration in tamoxifen-resistant breast cancer cellsClin Exp Metastasis200724315716717394086
- Planas-SilvaMDBruggemanRDGrenkoRTStanley SmithJRole of c-Src and focal adhesion kinase in progression and metastasis of estrogen receptor-positive breast cancerBiochem Biophys Res Commun20063411738116412380
- Planas-SilvaMDHamiltonKNTargeting c-Src kinase enhances tamoxifen’s inhibitory effect on cell growth by modulating expression of cell cycle and survival proteinsCancer Chemother Pharmacol200760453554317186241
- Planas-SilvaMDWaltzPKEstrogen promotes reversible epithelial-to-mesenchymal-like transition and collective motility in MCF-7 breast cancer cellsJ Steroid Biochem Mol Biol20071041–2112117197171
- ChenYGuggisbergNJordaMCombined Src and Aromatase Inhibition Impairs Human Breast Cancer Growth In vivo and Bypass Pathways Are Activated in AZD0530-Resistant TumorsClin Cancer Res200915103396340519451593
- CampbellEJMcDuffETatarovOPhosphorylated c-Src in the nucleus is associated with improved patient outcome in ER-positive breast cancerBr J Cancer200812299111769177419018258