Abstract
Malaria is of continuing concern in nonimmune traveling populations. Traditionally, antimalarial drugs have been developed as agents for dual indications (treatment and prophylaxis). However, since 2000, when the 5th Amendment to the Declaration of Helsinki (DH2000) was adopted, development of new malaria prophylaxis drugs in this manner has ceased. As a consequence, there may not be any new drugs licensed for this indication in the foreseeable future. Major pharmaceutical companies have interpreted DH2000 to mean that the traditional development paradigm may be considered unethical because of doubt over the likelihood of benefit to endemic populations participating in clinical studies, the use of placebo, and the sustainability of post-trial access to study medications. In this article, we explore the basis of these concerns and suggest that the traditional development paradigm remains ethical under certain circumstances. We also consider alternative approaches that may be more attractive to sponsors as they either do not use placebo, or utilize populations in endemic countries who may unambiguously benefit. These approaches represent the way forward in the future, but are at present unproven in clinical practice, and face numerous regulatory, logistical and technical challenges. Consequently, in the short term, we argue that the traditional clinical development paradigm remains the most feasible approach and is ethical and consistent with the spirit of DH2000 under the appropriate circumstances.
Introduction
Malaria is caused by four parasitic protozoa of the genus Plasmodium (P. falciparum, P. vivax, P. ovale, and P. malariae). The disease is prevalent in most tropical regions, causing up to 500 million clinical cases, and over one million deaths per year, mostly in children (CitationWHO 2005). In areas of high transmission, continuous exposure to malaria confers partial or ‘semi-immunity’. There are also smaller but growing populations of nonimmunes in endemic countries (CitationUNFPA 2007). Each year, 25–30 million nonimmune people travel from nonendemic to endemic countries for holidays, business, military deployments, or to visit friends and relatives (CitationChen and Keystone 2005). Malaria imported by this population upon their return is a significant public health concern, with approximately 30000 cases a year and fatality rates ranging from 1%–4% (CitationMuentener et al 1999; CitationMagill 2004; CitationNewman et al 2004; CitationChen and Keystone 2005; CitationChristen et al 2006; CitationLegros et al 2007). There are, in traditional thinking, two distinct populations that may benefit from the development of new antimalarial drugs. The populations of endemic countries benefit from drugs to treat symptomatic malaria, while the nonimmune traveling population in nonendemic countries (the traditional target population) benefit from drugs for malaria prevention. However, there is a growing realization that drugs with malaria prophylaxis properties can also potentially benefit nonimmune travelers from endemic countries, infants, children, and pregnant women through intermittent presumptive treatment, and everyone living in endemic countries in the context of malaria control (CitationUSAID 2007) and potential elimination (CitationBill and Melinda Gates Foundation 2007).
Prophylactic antimalarials work by either disrupting the initial development of malaria parasites in the liver (causal activity), suppressing the emergent asexual blood stages of the parasite (suppressive activity) or preventing the relapses induced by the latent liver forms (hypnozoites) of relapsing P. vivax and P. ovale malaria (presumptive antirelapse therapy or PART; see reviews by CitationHill et al 2006; CitationUSCDC 2008). At least three prophylaxis strategies of administration have been utilized to exploit these properties. The most common approach is to administer causal or suppressive drugs at efficacious prophylaxis (subtherapeutic) doses continuously throughout the period of exposure to malaria (CitationBaird et al 2007). For prophylactic drugs, with the exception of primaquine, a post-exposure PART regime is required to prevent subsequent relapse of P. vivax (CitationHill et al 2006). An alternative approach is so called ‘fire and forget’ prophylaxis, or ‘pre-exposure prophylaxis’, in which travelers are given a single dose or short course regime of a long half-life drug at a treatment dose that will protect throughout the duration of exposure (CitationShanks et al 2007). This approach is currently unproven in clinical practice, although some drugs have the potential to be used in this manner (tafenoquine, atovaquone-proguanil and piperaquine). As we shall see, no drug for malaria prevention is adequate in all respects. There is therefore a need to develop new drugs for this indication. However, no new drugs for malaria prophylaxis have been approved in the United States since 2000, despite three (mefloquine, atovaquone-proguanil, and doxycycline) having been licensed in the preceding decade. This has occurred as a direct consequence of ethical concerns raised by major pharmaceutical companies in the wake of changes to the Declaration of Helsinki in 2000 (DH2000).
The Declaration of Helsinki (CitationWMA 2004) is a statement of ethical principles adopted at the congress of the World Medical Association in 1964 (CitationCarlson et al 2004). The intent of the declaration was to formalize a set of core principles for the ethical execution of medical research (CitationCarlson et al 2004). Prior to the DH, the only similar document was the Nuremburg Code (CitationCarlson et al 2004). This was formalized during the trial of Nazi doctors for war crimes in 1947 (CitationKious 2001). The Nuremburg Code enshrined twelve ethical standards with which physicians should comply when conducting research involving human subjects, including informed consent and ensuring benefit to study participants (discussed in CitationCarlson et al 2004). The original Declaration of Helsinki embodied most of these ethical principles (CitationCarlson et al 2004). The Declaration was modified substantially in 1975, and again to a lesser extent in 1983, 1989, and 1996 as international consensus shifted, or to provide greater clarification (CitationCarlson et al 2004). The principles enshrined in the 1975 amendment were arguably the most widely recognized source of ethical guidance for two and half decades (CitationCarlson et al 2004). However the 5th Amendment, adopted in 2000 (DH2000), has resulted in controversy in many areas of medical research (CitationForster et al 2001; CitationCarlson et al 2004; CitationLie et al 2004). Ethical concerns relating to population benefit, the use of placebo and the sustainability of post-trial access to study drugs have had a profound impact on the development of new drugs for malaria prevention.
This review will describe existing and potential prophylactic drugs, outline the traditional manner in which these drugs are developed, consider the DH2000-related ethical objections to the traditional development paradigm, argue that the traditional development paradigm remains ethical and consistent with the spirit of DH2000 under some circumstances, and outline alternative approaches that, while challenging now, may, if successful, circumvent DH2000-related ethical concerns in the future. Finally, we argue that unless a pathway forward can be clearly defined, development of new prophylaxis drugs will not be feasible in the foreseeable future.
Current and future prophylactic antimalarial drugs
Currently available drugs
Five commercially available antimalarials (discussed below) have been formally registered in the United States for malaria prevention (CitationUSCDC 2008; ). The four drugs approved for continuous prophylaxis in the United States are mefloquine, atovaquone-proguanil, doxycycline, and chloroquine (CitationUSCDC 2008). Primaquine is approved for radical cure of P. vivax and P. ovale. It is recommended but not formally licensed for continuous prophylaxis (CitationHill et al 2006; CitationUSCDC 2008). Fansidar (pyrimethamine-sulfadoxine) was previously indicated for prophylaxis in the United States, but is no longer used for this purpose due to its association with fatal Stevens-Johnson syndrome (CitationMiller 1986). To our knowledge, the only other drug approved elsewhere is chloroquine-proguanil. Several recent reviews have documented the benefits and limitations of each of these (CitationMilhous 2001; CitationShanks et al 2001a; CitationPeterson 2004; CitationChen and Keystone 2005; CitationShanks and Edstein 2005; CitationHill et al 2006; CitationBaird et al 2007; CitationChen et al 2006), and the US Centers for Disease Control publishes prescribing information on a biannual basis (CitationUSCDC 2008). Summary information is outlined in . The utility of each drug can be rated in terms of cost, safety/tolerability, potential for compliance, activity against multiply-drug resistant strains of malaria, efficacy against P. vivax and regulatory status. None of the available choices are perfect, so there is a clear case for the development of alternatives.
Table 1 Characteristics of currently available drugs for malaria prevention
Prophylactic drugs in clinical development
The only compound in formal clinical development for prophylaxis is tafenoquine. Tafenoquine is a primaquine analog with a much longer half-life that exhibits activity as a suppressive, causal, and antirelapse prophylactic drug, and that in a continuous prophylaxis mode can be administered once weekly or monthly (CitationBrueckner et al 1998a, Citation1998b, Citation2001; CitationPeters 1999; CitationWalsh et al 1999; CitationLell et al 2000; CitationShanks et al 2001b; CitationNasveld et al 2002b; CitationHale et al 2003; CitationWalsh et al 2004). Tafenoquine might also potentially be administered in a ‘pre-exposure prophylaxis’ mode (CitationShanks et al 2001b; CitationHale et al 2003). Like primaquine, tafenoquine also has the disadvantage of causing hemolysis in G6PD deficiency (CitationShanks and Edstein 2005; CitationBrueckner et al 2001). Unfortunately the development of tafenoquine for prophylaxis has been substantially delayed in favor of a new indication: radical cure (treatment) of P. vivax malaria. This occurred directly as a result of concerns of the commercial sponsor (CitationLapierre and Duparc 2007), GlaxoSmithKline, that DH2000 makes the development of drugs for malaria prevention unethical (discussed later). Several other antimalarials might be considered as potential prophylactic agents if the DH2000-related ethical concerns are resolved. These include primaquine, weekly administered atovaquone-proguanil, azithromycin combinations, and piperaquine. We refer the reader to several recent reviews for a comprehensive outline of their development status (CitationChen and Keystone 2005; CitationBaird et al 2007) and to for a summary of their advantages and limitations. Primaquine is included in this list since it has not yet been formally approved for continuous prophylaxis.
Table 2 Characteristics of future prophylaxis drugs
The traditional development of antimalarial drugs for treatment and prophylaxis
The traditional clinical development of antimalarial drugs is outlined in and below. Atovaquone-proguanil (Malarone™) is used as an example since it was the last drug approved for malaria treatment and prophylaxis in the USA (CitationUSFDA 2000). However, it is important to first describe the general manner in which drugs are usually developed and approved. We have used the FDA-regulated process in the USA as an example below, and refer the reader elsewhere for more information (CitationUSCEDER 2007).
Figure 1 Clinical development of new drugs for malaria prevention.
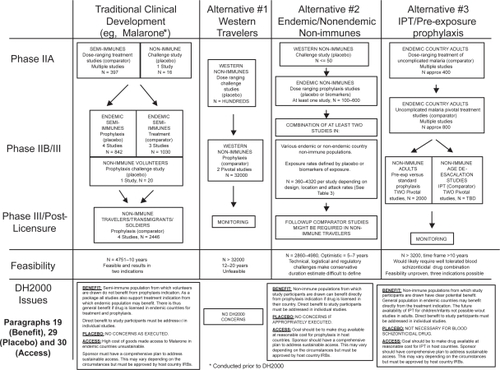
The first step is the filing of an investigational new drug (IND) application with the FDA. The absence of any safety concerns triggers initiation of the clinical program. Firstly, Phase 1 clinical studies are conducted to demonstrate the safety, establish the maximally tolerated dose, and characterize the pharmacology in normal healthy volunteers. Next, the potential efficacy and tolerability of one or more doses of the drug is evaluated in a small number of patients in Phase II studies. Thereafter, the dose of drug that will be marketed for the desired indication is selected. A series of at least two ‘pivotal Phase III’ studies are then conducted using this dose to demonstrate efficacy and safety. If the data suggest a favorable risk:benefit relationship, the sponsor then files a new drug approval (NDA) application, that, if approved, allows the drug to be marketed. Additional post-licensing or Phase IV studies may be required to further evaluate safety. Ideally, all clinical studies are conducted in the intended target population. It is normal practice to use placebo in Phase I studies. Placebo may also be used in Phase II and Phase III if no effective drugs are available, or in the interests of ensuring scientific validity.
In reality, the traditional development of antimalarial drugs differs from this idealized scenario in several respects. Firstly, pivotal Phase III prophylaxis efficacy studies historically have not been conducted in the intended target population. This is because placebo must be used in order to establish exposure to malaria, but the use of placebo is contraindicated in traveling nonimmunes because close medical supervision in most circumstances is usually not possible (discussed in more depth later). The alternative is to conduct the Phase III efficacy studies in semi-immune populations in endemic countries, as was the case with Malarone® (CitationLell et al 1998; CitationShanks et al 1998; CitationSukwa et al 1999). Close medical supervision is more feasible in these endemic area populations. A key consequence of this approach is that prophylactic efficacy cannot be proven in the traditional target population in the normal fashion. Rather, a series of additional supplementary nonpivotal studies are required. In the case of Malarone, these included two small challenge studies in nonimmune volunteers (CitationShapiro et al 1999; CitationBerman et al 2001), a placebo-controlled study in Indonesian trans-migrants (CitationLing et al 2002), and a comparator study in deployed South African soldiers (Citationvan der Berg et al 1999).
Secondly, in the case of blood schizonticidal drugs the Phase III prophylactic efficacy studies may be preceded by a series of Phase II and III studies to prove efficacy for treatment of malaria. In the case of Malarone, this was necessary to select the appropriate partner drug for atovaquone, desired dosing regimes and to prove the efficacy of the combination (see reviews by CitationLooareesuwan et al 1999; CitationMarra et al 2003; CitationBoggild et al 2007). Without these studies, the development of Malarone for prophylaxis would have been vastly more challenging. This is because the prophylaxis regimen employs the same combination partner:drug and drug:drug ratio defined during the prior treatment studies. Also, the treatment studies support the notion that the prophylaxis regimen will be effective despite limited direct evidence of this in the target population.
Finally, since the Phase III prophylactic efficacy studies we conducted in small nontarget populations without direct comparison to conventionally used prophylaxis drugs, additional safety studies were required. In the case of Malarone, two studies were conducted in nonimmune travelers using mefloquine and chloroquine-proguanil as comparators with tolerability as the primary endpoint (CitationHogh et al 2000; CitationOverbosh et al 2001). Efficacy was a secondary endpoint in these studies. Functionally, these studies represent a hybrid of traditional Phase III pivotal and Phase IV post-licensing studies. They showed that Malarone was well-tolerated in the intended target population compared to the available alternatives. This approach worked for Malarone, but might be more problematic for some drugs if tolerability and pharma-cokinetic differences exist between the South-East Asian and African populations utilized during the Phase II/III program and the intended target population (mostly Western travelers).
In the context of DH2000, it is placebo, the utilization of semi-immune populations in the Phase III prophylaxis studies, and in the case of Malarone, and the sustainability of post-trial access to study drug, that raise ethical concerns. These issues are explored in depth in the next section.
DH2000 and its relevance to malaria prophylaxis
The 5th Amendment to the Declaration of Helsinki is different from its predecessors in several respects. Firstly, it claims under Paragraph 9, to be a universal statement of research ethics with primacy over similar documents (CitationLie et al 2004). Secondly, DH2000 substantially altered the language of existing articles, and introduced new articles in an attempt to clarify issues thought to be inadequately addressed in prior amendments (CitationCarlson et al 2004). These changes have provoked great controversy and we refer the reader elsewhere to detailed discussions of these issues (CitationReynolds 2000; CitationSimon 2000; CitationEmmanuel and Miller 2001; CitationForster et al 2001; CitationCarlson et al 2004; CitationLie et al 2004). However, DH2000 has also changed the ethical context with respect to three issues of critical importance to the development of new prophylactic antimalarial drugs. These relate to social benefit (Paragraph 9), placebo controls (Paragraph 29) and post-trial access to study drug (Paragraph 30).
Primacy of the 5th Amendment:
Paragraph 9 of DH2000 (CitationWMA 2004) states the following: “Research Investigators should be aware of the ethical, legal and regulatory requirements for research on human subjects in their own countries as well as applicable international requirements. No national ethical, legal or regulatory requirement should be allowed to reduce or eliminate any of the protections for human subjects set forth in this declaration”.
It is clear from this provision that the current version of the Declaration of Helsinki is intended to have priority over national laws and regulations (CitationForster et al 2001). The text differs substantially from the 1996 version, which states that “physicians are not relieved from criminal, civil and ethical responsibilities under the laws of their own countries” and that its provisions were intended to be “recommendations” (CitationForster et al 2001). In the past, it was possible to argue that the Declaration was a source of guidance, and research studies were ethical provided that they were approved by local institutional review boards and were compliant with local laws and regulations. Now, it is possible to argue that a study is unethical because it is not ‘DH2000 compliant’, even if all local laws and regulations are followed. Consequently, individual researchers or national regulatory agencies protect themselves against uncertainty by publicly stating which version of the Declaration of Helsinki (or other ethical code) is to be followed (CitationForster et al 2001; CitationRiis 2003). For example, the US FDA requires that foreign clinical trials must be conducted to an ethical standard that is consistent with the 1989 version of DH or the laws and regulation of the host country, whichever provides the greatest protection (CitationUSFDA 2001).
In order to avoid any accusation of unethical behavior, it is almost inevitable that large multinational pharmaceutical companies would insist that their sponsorship of international research studies be conducted in accordance with both relevant national regulations and DH2000, whichever is more stringent, and to avoid such endeavors where a conflict exists.
Social benefit
Paragraph 19 states the following: “Medical research is only justified if there is a reasonable likelihood that the populations in which the research is carried out stand to benefit from the results of that research” (CitationWMA 2004). The provision is a new addition to the Declaration, and was intended to ensure that medical research is responsive to the needs of the population in which it is conducted.
In the context of Paragraph 19, the appropriateness of the three Malarone semi-immune prophylaxis studies, and, by extension, the traditional development paradigm itself have been challenged on the basis that the communities involved would be very unlikely to benefit from this indication (CitationNosten 2000). Prophylactic antimalarial drugs are not in widespread use in endemic countries (CitationNosten 2000). This is due to several factors, including the impracticality of their deployment on a long term basis to entire populations, concerns over development of resistance, potential unknown risks of adverse events with long term usage, and the specter of a decline in the development of semi-immunity that may result if malaria prevention could not be sustainably provided.
Since prophylactic drugs are not usually indicated in semi-immunes for the reasons stated above, some pharmaceutical companies have concluded that there is no reasonable likelihood of benefit and such studies are unethical (CitationLapierre and Duparc 2007).
Placebo controls
Paragraph 29 states that: “The benefits, risks, burdens and effectiveness of a new method should be tested against those of the best prophylactic, diagnostic, and therapeutic methods. This does not exclude the use of placebo, or no treatment, in studies where no proven prophylactic, diagnostic or therapeutic method exists” (CitationWMA 2004).
The intention of this provision was to prevent a reoccurrence of controversial placebo-controlled studies. An oft-cited example is the use of a placebo control in studies designed to determine the utility of HIV drugs for prevention of vertical transmission of HIV, despite the fact that proven treatments were available (CitationLa Vaque and Rossiter 2001). This provision sparked such controversy and great debate that the WMA issued a clarification in 2002, stating the circumstances under which a placebo control may be acceptable even if a proven therapeutic method exists:
“Where for compelling and scientifically sound methodological reasons its use is necessary to determine the efficacy or safety of a prophylactic, diagnostic method; or where a prophylactic, diagnostic or therapeutic method is being investigated for a minor condition and the patients who receive placebo will not be subject to any additional risk of serious or irreversible harm” (CitationWMA 2004).
Despite this clarification, the pharmaceutical industry have adopted the view point that the use of placebo is unethical in the context of Phase III malaria prophylaxis studies since effective drugs are available (CitationLapierre and DuParc 2007).
Post-trial access to prophylactic drugs
Paragraph 30 states that “at the conclusion of the study, every patient entered into the study should be assured of access to the best proven prophylactic, diagnostic and therapeutic methods identified by the study” (CitationWMA 2004).
At its meeting in France 2004, the WMA issued the following clarification to Article 30:
“The WMA hereby reaffirms its position that it is necessary during the study planning process to identify post-trial access by study participants to prophylactic, diagnostic and therapeutic procedures identified in the study or access to appropriate care. Post-trial access arrangements or other care must be described in detail in the study protocol so the ethical review committee may consider such arrangements during its review” (CitationWMA 2004).
The intention of Article 30 was to prevent the withdrawal of study drug from subjects participating in clinical trials who would normally not have access to the study medication due to either expense or unavailability (see CitationBlackmer and Haddad 2005). From the perspective of a potential sponsor, many aspects of this provision are problematic. As CitationBlackmer and Haddad (2005) and CitationRiis (2003) have outlined, the issues of scope and responsibility are not adequately defined. It is unclear whether the burden of providing post-trial access to treatment falls on the sponsor or the public health system of the country (CitationBlackmer and Haddad, 2005). As pointed out by CitationRiis (2003), the time frame of provision of access is also unclear: should this be until the product reaches the market, or does this mean indefinitely? It is also unclear how Paragraph 30 should apply in a non-Phase III setting, where the study may prove nothing in terms of a new prophylactic method. Finally, it can also be argued that the requirement to provide access (Paragraph 30) also applies to the general population (Paragraph 19), since how can one reasonably benefit if provisions are not made to ensure sustainable access? Indeed, if the principle of distributive justice is followed to its logical conclusion, it may be reasonable to argue that failure to ensure in advance the reasonable availability of a future product in a timely manner to study participants and the populations from which they are drawn is sufficient reason for a study to not be executed at all (discussed by CitationMacklin 2001).
Once again, the Malarone example is insightful. The sponsor attempted to make Malarone available on a limited compassionate basis through the Malaria Donation Project (CitationShretta et al 2000, Citation2001). However, it is clear in retrospect that the high cost of making Malarone was always going to pose challenges in terms of ensuring sustained access (CitationShretta et al 2000, Citation2001). Post-trial availability was not discussed in the publications reporting the results of the prophylaxis studies (CitationShanks et al 1998; CitationLell et al 1998; CitationSukwa et al 1999), presumably because they were conducted prior to DH2000. Therefore, it is unclear to what degree local institutional review boards were fully informed as to their implications. This underscores the point that a plan for sustainable access must be addressed a priori.
The most conservative strategy for a large, publicly-owned pharmaceutical company to discharge the risk associated with the above ethical uncertainties would be to ensure that Phase III clinical development programs are not executed in endemic countries unless there is an a priori commitment from senior management to market the drug for the intended indication at a reasonable price.
Impact of DH2000 on clinical development of drugs for malaria prevention
As a consequence of the uncertainties raised by DH2000, many large pharmaceutical firms have adopted the following mantra in order to manage ‘ethical’ risk: Compliance with the spirit of DH2000 can only be assured if a commitment exists a priori, to register and make available at a reasonable cost, a specific compound, for the specific indication, in those countries in which the clinical program is executed and placebo cannot be used since effective drugs for prophylaxis exist. As a consequence, they have reached the conclusion that this burden cannot be met in the context of prophylactic drugs. They have ceased active involvement in development of drugs for this indication as a result.
Most companies active in the development of antimalarial drugs today are doing so on a noncommercial basis. In this context, their focus is on the treatment indication, and many companies do provide the fruits of that research effort at or below cost to endemic countries. Coartem™ is an example of this (CitationNovartis 2006). The value to a sponsor of such engagement is primarily the positive public profile generated by tackling a neglected disease. There is thus very little incentive to risk being considered unethical by developing prophylaxis drugs with a small commercial market in nonendemic countries.
This is the central reality that must be engaged if one is to conceive of a pathway forward for the development of new prophylactic drugs. Whilst one can argue, as we will do, that alternative interpretations of the DH2000 can allow prophylactic drug development to occur in an ethical and DH2000 compliant manner, one must also reconsider the traditional development paradigm in light of this reality. These two issues are the central matters of interest in the remainder of this review.
Is the traditional development paradigm ethical and DH2000-compliant?
Previously, we outlined the ethical case against the traditional development paradigm from a DH2000 perspective. However, it is reasonable to consider why it is necessary to comply with DH2000 at all. The authors of DH2000 intended it to be the primary source of ethical guidance. However, the basis of this claim can be challenged. Firstly, the legal basis of WMA’s claim of primacy is debatable (CitationForster et al 2001). DH2000 is not a treaty, has not been ratified by national governments and so is not legally binding. Secondly, the moral and practical necessity for such a claim is now arguably defunct (CitationForster et al 2001). This is because the Nuremburg Code and the original Declaration of Helsinki came into existence when no recognized code of international research ethics was in place. This is no longer the case, with the advent of strict regulatory guidelines in most jurisdictions. CitationRiis (2003), one of the original authors of the DH, has argued that DH2000 should serve as a subordinate code of ethics, and that national and regional statutes should take precedence. From our perspective, it seems logical that national regulations should be thus be considered the source of ethical guidance where a conflict exists with DH2000, but that DH2000 could usefully be considered the primary source of guidance in jurisdictions that choose to adopt it as national policy, or where no source of ethical guidance exists. If you accept this argument, a clinical program can be considered ‘ethical’, if not DH2000 compliant, provided that all appropriate local and international laws are followed. An important caveat is that the interests of vulnerable populations cannot be protected in jurisdictions with weak national regulations, or in those that lack the institutional capability to allow the appropriate constitution of ethical review boards and oversight of medical research. This is certainly the case in some malaria-endemic countries.
Nevertheless, the fact remains that some national governments may elect to adopt DH2000 as national policy. Therefore, DH2000 concerns must be addressed in a substantive way. The key issues that must be addressed are those of ‘placebo’, ‘social benefit’ ‘access/sustainability’. With respect to the use of placebo, it is clear that the 2004 clarification to Paragraph 19 suggests that the use of a placebo is appropriate, even if effective treatments exist, if there is a compelling methodological reason or the risk of severe or irreversible harm is low. Both qualifications apply in the context of malaria. A placebo controlled arm in Phase II challenge and Phase III field studies is necessary for scientific and methodological reasons for the development of drugs for malaria prevention. In the context of a challenge study, a placebo arm is essential for demonstrating the infectivity of mosquitoes (CitationShapiro et al 1999; CitationBerman et al 2001; CitationBejon et al 2005; CitationHeppner et al 2005a; CitationWang et al 2005; CitationDunachie et al 2006; CitationWalther et al 2006). In Phase III prophylaxis studies (eg, those that supported the licensure of Malarone) the use of placebo reduces the sample size required by several orders of magnitude. The effect of this is that the clinical development of a drug is potentially more logistically feasible, less expensive, and may result in more rapid approval and availability to populations who may benefit. Placebo-controlled malaria challenge studies in nonimmunes are considered ethical (Miller and Grady 2003). Malaria, if rapidly diagnosed and treated, has minimal risk of severe or irreversible harm (CitationChurch et al 1997; CitationVerhage et al 2005). Thus, placebo is appropriate in controlled settings where regular blood smears are examined to detect malaria, and the ensuing infection can be effectively treated with conventional antimalarial drugs. Therefore, we would argue that the use of placebo controlled arms in clinical malaria studies is DH2000-compliant and should not be a concern from an ethical perspective, provided the studies are conducted in appropriate settings.
Paragraph 19 clearly states that a reasonable likelihood must exist that the populations in which the research is carried out stand to benefit from the results of that research. A reasonable interpretation of this language is that ‘populations’ does not refer only to study participants and that ‘reasonable likelihood’ does not simply mean an abstract promise of benefit in the future. The pharmaceutical industry have interpreted this to mean that prior to execution of a Phase III program, a sponsor must have the intent to license and market the drug in the country of interest for the specific indication being investigated. From our perspective this argument seems unnecessarily restrictive because (i) the concept of the ‘results of the research’ is not strictly defined under DH2000, (ii) sponsors make go/no go decisions at the level of the program rather than a specific study, and (iii) under the traditional paradigm, the clinical development of many drugs for the treatment and prophylaxis indications are linked. It is therefore reasonable, and consistent with the spirit of DH2000, to extend the concept of ‘results of the research’ to include the licensure of a drug for treatment as well as prophylaxis. Thus, development of a drug for malaria prophylaxis is consistent with the principles of DH2000, provided that the intent of the sponsor, a priori, is to develop and market a drug for prophylaxis and treatment. In this ideal scenario, the potential benefit of the research for all (in endemic and nonendemic countries) can be maximized.
However, many aspects of clinical research fall outside this ideal. Specific examples are Phase I malaria challenge studies (eg, Shapiro et al 2002), the Phase II studies that identified the appropriate combination partner for atovaquone (reviewed by CitationLooareesuwan et al 1999), or the Phase III studies that demonstrated the prophylactic efficacy of primaquine (CitationBaird et al 2001). In these instances, DH2000 does not prescribe the appropriate benefits due the study population and participants. Indeed, in the context of a Phase I study, there is no benefit to the study participants, and the possibility of general future benefit remains theoretical. There are also no provisions under DH2000 to resolve ethical dilemmas created by uncertainties in the language of the document itself (CitationForster et al 2001). As pointed out by CitationMacklin (2001), many of the ambiguities in DH2000 reflect substantive ethical dilemmas that may never be resolved. There are two reasonable approaches that a sponsor may take and remain consistent with the spirit of DH2000. In the context of sustainable access (Paragraph 30, discussed later in this section), DH2000 mandates that an appropriate (ideally local) ethical review committee be presented with a comprehensive plan a priori. It would be logical to extend the same argument to an assessment of potential benefit. Alternatively, as suggested by CitationRiis (2003), one may use national or regional statutes or regulations as a source of guidance. Both approaches seem logical, ethical and consistent with the spirit of DH2000. An important caveat here is that many endemic countries do not have the institutional capability to execute robust independent ethical reviews. However, this is not true in many cases.
When considering benefit, one must remember that DH2000 is not the sole source of guidance. Shortly after the adoption of DH2000, a prestigious group of North American bioethicists and African clinician scientists proposed the ‘fair benefits’ framework as an alternative to DH2000 (CitationAnon 2002). Both DH2000 and the fair benefits philosophy aim to avoid the exploitation of vulnerable populations, but address the issue of benefit differently. Proponents of fair benefit argue that benefit to a community of study participants should be in proportion to the risks of the proposed research (CitationAnon 2002). Furthermore, the assessment of what constitutes a fair benefit is a decision best left to those individuals or communities participating directly in the research (CitationAnon 2002). For example, provision of needed healthcare for a community may be an acceptable benefit for participation in a clinical trial. Conceptually, this approach sounds reasonable, but is unlikely to resolve the ethical risks for sponsors. In the context of a Phase III study involving a new antimalarial drug, the fair benefits approach is not consistent with the spirit of DH2000, since it leaves open the possibility that the study population will not benefit from the study drug. Furthermore, whilst community consent to any program of research is desirable, this must also be accompanied by review and approval by an appropriately constituted ethical board in order to remain within the spirit of DH2000. Finally, proponents of the fair benefits philosophy leave unaddressed the concepts of equality and distributive justice. Thus, in our view, a sponsor is best able to remain consistent with the spirit of DH2000 if (i) the intent a priori is to market an antimalarial drug for as many indications of interest to host endemic countries as possible, and/or (ii) to provide an appropriate institutional review board the opportunity to review and assess the likelihood that benefit would result from the research.
In terms of access, the pharmaceutical industry has interpreted collectively the provisions of Paragraph 19 and 30 to mean that a sponsor, when planning a Phase III program, must commit, a priori, to licensing and making available the study drug at reasonable cost, for the relevant indication, in the countries in which the study subjects were recruited. In the context of the traditional development paradigm, this would in most cases translate into provision of the study drug for malaria treatment and prophylaxis at cost. This seems reasonable in the context of an over-arching plan to develop a new drug, but may be inappropriate in some circumstances. It leaves unaddressed the issue of access for trial participants in the gap between trial termination and licensure. Furthermore, the development of antimalarial drugs is often not organized systematically by a single sponsor. Many antimalarial drugs have been shown to be effective for treatment or prophylaxis in ad hoc studies sponsored by different universities, travel clinics or other research institutions. Such institutions may not have the resources to commit to a donation or compassionate use program. It may be reasonable to argue in such cases that the drugs concerned are already affordable, or, with the possible exception of atovaquone-proguanil, will become affordable once patent protection has lapsed and generic manufacturers enter the market. It is clear from the clarification to Paragraph 30 that the decision about what constitutes a reasonable ‘access’ plan is a decision intentionally left to local review boards, with the implication that not all boards will arrive at the same consensus on what is appropriate. Therefore, regardless of the circumstances, a sponsor can remain consistent with the spirit of DH2000, provided a plan to address sustainable access is approved by an appropriately constituted local review board.
Alternative clinical development paradigms
In the previous section, we argued that the traditional development paradigm remains ethical under certain circumstances. Whilst we hope this case is persuasive, it is prudent to also consider alternatives. Alternative 1 would be to execute the entire clinical program in the traditional target population (see ). This approach would resolve all the ethical issues, but was rejected by the Malarone clinical development team. The stated reason was that the sample size (n = 16000 per study) required for each pivotal Phase III study was unfeasible. This is because this approach would have depended on measuring the superiority of the investigational drug compared to a standard drug with efficacy (prevention of symptomatic malaria) as the primary endpoint (CitationHogh et al 2000), and the incidence of symptomatic malaria in patients taking effective prophylaxis is rare (CitationHogh et al 2000) and variable due to several factors (seasonality of malaria, travel destinations etc). In this section we explore two additional alternatives. Alternative 2 involves the use of nontraditional nonimmune populations where malaria exposure is estimated based on either placebo or through the use of biomarkers (see outline in ). Alternative 3 involves development of new drugs simultaneously for pre-exposure prophylaxis and IPT (see ). However, we must first turn our attention to the use of biomarkers of exposure as alternative to placebo.
Use of biomarkers of exposure as an alternative to placebo
Currently, there are no established alternatives other than placebo to estimate malaria exposure and to control the size of a prophylaxis study. One can argue that a noninferiority design to demonstrate efficacy might be appropriate since the concept of malaria prophylaxis is proven and effective regimes exist. However, the results of such a study are not interpretable if the expected attack rate is not observed in the comparator arm, because the study will be underpowered or it cannot be proven that exposure to malaria occurred. This is not hypothetical. A recent Phase III study conducted in Australian troops deployed to East Timor resulted in no incidences of symptomatic falciparum malaria in either the investigational drug or mefloquine (comparator) arms (CitationNasveld et al 2002a). Entomology and point prevalence studies of populations in the general area may not accurately predict exposure in transient populations in local areas because these populations behave differently. In addition, malaria epidemiology is highly localized (CitationGreenwood 1989; CitationBautista et al 2006). Where close follow-up cannot be achieved, placebo cannot be used. Therefore, its use in travelers and deployed military personnel is unethical under most circumstances. A method to measure indirectly the exposure in individuals taking effective malaria prophylaxis would be ideal.
Validated biomarkers (surrogates) would not only obviate the need for a placebo, and the statistical requirement for large sample sizes in studies with cases coming from prophylaxis failures (see ), but also facilitate execution in the target population. This would mitigate the ethical concerns with respect to both Paragraphs 19 and 29. Such a surrogate marker could be an antibody response, or a pattern of protein or gene expression. This approach has been demonstrated in principle by CitationOrlandi-Pradines et al (2006) amongst French troops deployed to Gabon and Cote d’Ivoire. The soldiers were provided with a variety of insect repellants, protective devices such as bed nets as well as chemoprophylaxis. Around 35% of soldiers demonstrated an immune response to pre-erythrocytic antigens (CSP, LSA1, STARP, SALSA, and SR11.1), and the method was sensitive enough to detect differences in exposure between soldiers reporting different levels of compliance with entomological deterrent devices. CSP has long been used as a general marker of immunologic exposure to malaria (CitationDruilhe et al 1986). Similar unpublished studies at WRAIR suggest that merozoite surface protein 1 (MSP1) may have sufficient sensitivity to justify further assessment for qualification for use in regulatory studies (CitationOhrt et al 2007). In a regulatory context, these associations probably qualify some antibodies as potential biomarkers of exposure to malaria. However, none of the biomarkers discussed currently meet the regulatory burden required for them to be considered as validated biomarkers of exposure (CitationBiomarker Definitions Working Group 2001; CitationUSFDA 2005, Citation2007; CitationWagner et al 2007), so additional studies would be required to address this.
Table 3 Possible phase III-equivalent study designs
Nonimmune populations in endemic countries
The traditional target population for drugs for malaria prevention is nonimmune travelers from nonendemic countries. However, any nonimmune individual traveling to an endemic area, including those from endemic countries, should take malaria prophylaxis if available to reduce the risk of death from malaria. They can therefore potentially benefit from investigational drugs for malaria prevention. Examples of such populations are endemic country travelers, migrants, military members and university students. With the exception of university students, malaria prevention trials have been successfully executed in these populations in the past (three of many examples include CitationOhrt et al 1997; CitationLing et al 2002; CitationWalsh et al 2004). It is also important to recognize that the demographics of endemic countries are changing. The population of potentially traveling urban residents (mostly nonimmune) is increasing while that of rural residents (mostly semi-immune) is decreasing proportionally (CitationUNFPA 2007). Thus, drugs for malaria prevention may play a more important role in endemic countries in the future.
Phase III studies could be conducted in endemic country nonimmunes with a placebo, or using a biomarker approach. The sample sizes and challenges associated with various possible trial designs are outlined in . The principal advantage of utilizing such populations is that, in some cases (eg, university students or temporary migrants), they may be resident in endemic regions for an extended period of time. This means that the sample sizes required are much lower, because per person attack rates will be much higher than in an equivalent traveling population that might reside in the same region for a month or less. However, identifying appropriate populations may not be straightforward. Firstly, the population should ideally be nonimmunes traveling temporarily to a region of higher endemicity (since permanent prophylaxis is not feasible they may not benefit). Secondly, they must be traveling for sufficiently long periods of time for the attack rate to approach 80% or more. Finally, if placebo must be used, the clinical context must allow the possibility of close follow up and rapid distribution of effective malaria treatment drugs. One other scientific concern with this approach is that the demographics of endemic nonimmunes may be different from nonendemic country nonimmunes with respect to ethnicity, comorbidities etc. This may be problematic if pharmacokinetics or tolerability are affected by these factors.
Safari studies
An alternative would be to conduct placebo controlled studies in nonimmune Western travelers or military personnel in endemic countries. One can envisage a scenario where nonendemic country volunteers would participate in a paid or subsidized fishing or safari trip to western Kenya or Tanzania and participate in a prophylaxis study. We term this approach the ‘safari study’. This approach addresses the ethical concerns raised by Paragraphs 19 and 30. This approach differs from a comparable study in nonimmune Western volunteers, since it would not utilize a travel clinic population of volunteers already intending to travel, but would instead actively recruit study subjects to travel to a specific high transmission area. Consequently, this approach raises its own ethical concerns (). Firstly, the airfares and local expenses of study participants would have to be compensated to a degree that some may consider coercive. Secondly, participation in the study would expose volunteers to infectious diseases that they would not otherwise encounter. Finally, it is reasonable to expect that study participants would be unwilling or unable to travel for prolonged periods. This would necessitate trips of shorter durations, lower per/person attack rates and possibly unfeasible sample sizes as a result.
The IPT/Pre-exposure prophylaxis paradigm
Intermittent presumptive treatment (IPT) is a strategy for using antimalarial drugs to minimize morbidity and mortality in high-risk populations living in malaria endemic areas by giving full treatment doses of drugs at defined time periods (CitationRosen et al 2004; CitationEgan et al 2005; CitationO’Meara et al 2005; CitationSchellenberg et al 2006). For example, IPT in infants (IPTi) is being implemented in Sub-Saharan Africa to provide protection to infants between ages 0–12 months who suffer the highest malaria attributable mortality. In this setting, a treatment dose of a drug, most commonly sulfadoxine-pyrimethamine (SP), is given along with the age appropriate vaccines in the Expanded Program of Immunization (EPI).
Published results of studies show a modest protective effect against clinical attacks of malaria and severe anemia and very low rates of serious adverse events (CitationSchellenberg et al 2001; CitationChandramohan et al 2005; CitationMacate et al 2006). The overall-risk-benefit and cost-benefit of the intervention appears favorable, but there likely remain differences in effectiveness related to schedule of administration, age of dosing, differing levels of background SP resistance, use of other malaria control measures in the population such as insecticide treated bed-nets, transmission intensity, and other factors yet to be clearly defined. IPT can also be used in other vulnerable, high-risk populations such as pregnant women (IPTp), children ages 1–5 (IPTc), and for use in areas with intense but marked seasonal transmission (sIPTc).
Intermittent presumptive treatment is really the use of anti-malarial drugs for a chemoprophylaxis indication in endemic populations at risk of malaria. Using chemoprophylaxis in endemic populations has historically been discouraged or not promoted because of fears of increased malaria morbidity and mortality after prophylaxis is discontinued (the “rebound” effect) and concerns over sustainability of the intervention (CitationGreenwood et al 1995; CitationMenendez et al 1997; CitationGeerligs et al 2003). The most recent experience with SP suggests that, at least with this drug, the rebound effect is not present or quite minimal and co-administering SP in the EPI vaccine schedule is effective. In any case, because of IPT, the strategy of using antimalarial drugs for a chemoprophylaxis indication in endemic populations is increasingly an accepted practice in the appropriate setting. Therefore, the argument against testing drugs in endemic populations because the at risk population will never benefit from the use of the drug with a prophylactic indication is not true.
Conceptually, IPT is analogous to pre-exposure pro-phylaxis in nonimmune travelers. Recently, pre-exposure prophylaxis in nonimmune travelers was proposed as a new strategy for using antimalarial drugs to prevent infection in short term (less than one month) travelers (CitationShanks et al 2007). Full treatment regimes of antimalarial drugs would be given to travelers prior to their trip that would offer solid protection for the duration of travel. These drugs would need to be long acting agents, such as piperaquine, atovaquone, or tafenoquine, that could give reliable protection. This could be seen as a variation of “IPT for travelers”.
Thus, the needs of the nonimmune short term traveler and the needs of the relatively nonimmune very young endemic populations have begun to converge with the emergence of the IPTi strategy in Sub-Saharan Africa, and the pre-exposure prophylaxis strategy for travelers. Both groups would receive therapeutic doses of safe and well tolerated drugs to be given to asymptomatic populations at risk of serious morbidity and death due to malaria.
A hypothetical clinical trajectory to develop a drug simultaneously for IPT and pre-exposure prophylaxis is outlined in . After appropriate dose ranging studies to establish an effective dose, pivotal comparator studies would be conducted in adults with uncomplicated malaria in endemic countries. These studies would utilize adequate clinical outcome as the primary endpoint and time to reinfection as the secondary endpoint. If safe and effective (primary and secondary endpoints), these studies would provide the rationale to execute pivotal, age de-escalation IPT studies in nonimmune children and infants. In parallel, Malarone-like tolerability end-point studies could be conducted in which an IPT drug was compared to standard prophylaxis in nonimmune travelers. The execution of these studies would be contingent on the demonstration of adequate post-treatment reinfection patterns in the prior treatment studies.
This approach has several possible advantages. First, there is the prospect that a drug could be developed for two or three indications. Second, many industry-sponsor-related ethical concerns are resolved using this approach. Placebos would not be necessary for blood schizonticidal drugs developed in this manner, provided that the dosing regimes for all the Phase III studies remained unchanged. If the intention of the sponsor is to market the investigational agent at a reasonable price in the host country for IPTi/c, there is a reasonable likelihood of benefit to the nonimmune pediatric population. The semi-immune adult population benefits because development of a potential drug for IPT for their children would not be feasible without their participation in the earlier treatment studies. Long term access to the drug will almost inevitably be more sustainable, since a sponsor might only be obligated to market the drug to the pediatric population in endemic countries.
There are several limitations to such an approach. First, as outlined, it is likely only to be applicable to a safe and very well-tolerated blood schizonticidal drug. Second, whilst rebound lack of immunity does not seem to be a problem so far for SP, this issue would need to be carefully thought through for a new drug. Third, this approach assumes that for prophylaxis, as for Malarone, regulatory agencies would accept a submission which did not include pivotal studies in the target population powered using efficacy endpoints. Finally, it is currently unproven in practice, since no commercially available antimalarials were developed for, and are currently used, for all these indications.
Clinical development of drugs for malaria prevention in a post-DH2000 world
As we have seen, the 5th Amendment to the Declaration of Helsinki has changed the ethical environment in which drugs for malaria prophylaxis are developed. The traditional paradigm, in which antimalarial drugs are development for malaria treatment and prevention, is considered by some in the pharmaceutical industry to be unethical, due to concerns regarding the use of placebo, the perceived lack of reasonable likelihood of benefit to some of the participating populations, and difficulties relating to ensuring sustainable post-trial access to the study drug. As we have shown, the required Phase III efficacy studies in nonimmune Western travelers, the logical target population, are unfeasible, due to the large sample sizes required. Alternative development plans, including those that involve the use of biomarkers instead of placebo, nontraditional nonimmune populations rather than Western travelers, and/or intermittent preventative treatment are potential alternatives, but face formidable regulatory, technical and logistical obstacles. The Walter Reed Army Institute of Research (WRAIR) and other institutions are in the process of conducting the clinical and epidemiological studies and initiating the regulatory consultations that may resolve some of these issues. However, in the interim, the traditional development paradigm represents the most feasible pathway forward.
How, then, should a sponsor proceed in a changed ethical environment? Our perspective as potential sponsors and codevelopment partners of future antimalarial drugs can be summarized as follows. Phase III prophylaxis studies in semi-immunes are clearly ethical under DH2000, provided that the intention of the sponsor is to market the drug for malaria indications of interest to host endemic countries. However, this ideal set of circumstances may not always prevail. In different situations, clinical studies may be considered DH2000 compliant on a case by case basis, if an appropriately constituted ethical review committee, in a country with the appropriate institutional capacity, is satisfied that a reasonable likelihood exists of benefit occurring as a result of the study. Furthermore, any clinical study or drug development plan is consistent with the spirit of DH2000 if an appropriately constituted local review board approves a comprehensive plan to address sustainable access a priori. Finally, the use of placebo is ethical and DH2000 compliant, since there are compelling scientific reasons for its use, and the risk of severe harm to study participants is minimal in the appropriate clinical context. The success of future efforts to develop drugs for antimalarial prophylaxis will ultimately be determined by how acceptable this view point is to important stakeholders such as the pharmaceutical industry, regulators, and host country institutional review boards. We hope that this perspective will be considered thoughtfully in the context of all the other factors that contribute to the feasibility of new antimalarial drug development.
Acknowledgements
This manuscript was reviewed by Walter Reed Army Institute of Research (WRAIR) and US Army Medical Research and Material Command (MRMC), and there is no objection to its publication or dissemination. The opinions expressed herein are those of the authors and do not reflect the views or opinions of the Department of the Army or the Department of Defense. We thank Dr Wilbur K. Milhous for helpful comments on the manuscript.
Disclosure
The authors are actively engaged in new drug development for antimalaria chemoprophylaxis at the WRAIR and have a professional interest in identifying a pathway forward for clinical development of new drugs for the prophylaxis indication. The authors are or in the past have been members of the joint GlaxoSmithKline-US Army tafenoquine clinical development team. None of the authors have a financial conflict of interest.
References
- Anon2002Fair benefits for research in developing countriesScience2982133412481120
- BairdJKBasriHSubiantoB1995Treatment of chloroquine-resistant Plasmodium vivax with chloroquine and primaquine or halofantrineJ Infect Dis1711678827769318
- BairdJKLaceyMDBasriH2001Randomized, parallel placebo-controlled trial of primaquine for malaria prophylaxis in Papua, IndonesiaClin Infect Dis221990711712091
- BairdJSchwartzEHoffmanS2007Prevention and treatment of vivax malariaCurr Infect Dis Rep9394617254503
- BautistaCTChanASRyanJR2006Epidemiology and spatial analysis of malaria in the Northern Peruvian AmazonAm J Trop Med Hyg7512162217172396
- BejonPAndrewsLAndersonRF2005Calculation of liver-to-blood inocula, parasite growth rates, and preerythrocytic vaccine efficacy, from serial quantitative polymerase chain reaction studies of volunteers challenged with malaria sporozoitesJ Infect Dis1916192615655787
- BermanJDNeilsenRChulayJD2001Causal prophylactic efficacy of atovaquone-proguanil (MalaroneTM) in a human challenge modelTrans R Soc Med Hyg9542932
- Bill and Melinda Gates Foundation2007Eradication of malaria [online]. Accessed November 28, 2007. URL: http://www.gatesfoundation.org/MediaCenter/Speeches/Co-ChairSpeeches/BillgSpeeches/BGSpeech-MalariaForum-071017.htm
- Biomarkers Definitions Working Group2001Biomarkers and surrogate endpoints: preferred definitions and conceptual frameworkClin Pharmacol Ther69899511240971
- BlackmerJHaddadH2005The Declaration of Helsinki: an update on paragraph 30CMAJ1739
- BoggildAKPariseMELewisLS2007Atovaquone-proguanil: report from the CDC expert meeting on malaria chemoprophylaxis (II)Am J Trop Med Hyg762082317297027
- BruecknerRPCosterTWescheDL1998aProphylaxis of Plasmodium falciparum infection in a human challenge model with WR 238605, a new 8-aminoquinoline antimalarialAntimicrob Agents Chemo4212934
- BruecknerRPLasseterKCLinET1998bFirst-time-in humans safety and pharmacokinetics of WR238605, a new antimalarialAm J Top Med Hyg586459
- BruecknerRPOhrtCBairdJK20018-AminoquinolinesRosenthalPJAntimalarial chemotherapy: Mechanisms of action, resistance, and new directions in drug discoveryNew JerseyThe Humana Press, Inc
- BryanJP2006Cost considerations of malaria chemoprophylaxis including use of primaquine for primary or terminal chemoprophylaxisAm J Trop Med Hyg754165016968914
- CarlsonRVBoydKMWebbDJ2004The revision of the Declaration of Helsinki: past, present and futureBr J Clin Pharmacol576957315151515
- ChandramohanDOwusu-AgyeiSCarneiroI2005Cluster ran-domised trial of intermittent preventive treatment for malaria in infants in area of high, seasonal transmission in GhanaBMJ3317273316195288
- ChenLHKeystoneJS2005New strategies for the prevention of malaria in travelersInfect Dis Clin N Am19185210
- ChenLHWilsonMESchlagenhaufP2006Prevention of malaria in long-term travelersJAMA29622344417090770
- ChristenDSteffenRSchlagenhaufP2006Deaths caused by malaria in Switzerland 1988–2002Am J Trop Med Hyg7511889417172391
- ChurchLWLeTPBryanJP1997Clinical manifestations of Plasmodium falciparum malaria experimentally induced by mosquito challengeJ Infect Dis175915209086149
- DavisTMHungTYSimIK2005Piperaquine: a resurgent antimalarial drugDrugs65758715610051
- DruilhePPradierOParentG1986Levels of antibodies to Plasmodium sporozoite surface antigens reflect malaria transmission and are persistent in the absence of reinfectionInfect Immun5339373525412
- DunachieSJWaltherMEpsteinJE2006A DNA prime-modified vaccinia virus Ankara boost vaccine encoding thrombospondin-related adhesion protein but no circumsporozoite protein partially protects healthy malaria-naïve adults against Plasmodium falciparum sporozoite challengeInfect Immun7459334216988273
- DunneMWSinghNShuklaM2005A multicenter study of azithro-mycin, alone and in combination with chloroquine, for the treatment of acute uncomplicated Plasmodium falciparum malaria in IndiaJ Infect Dis1911582815838784
- EdsteinMDKoteckaBMAndersonKL2005Lengthy antimalarial activity of atovaquone in human plasma following atovaquone-proguanil administrationAntimicr Agents Chemo4944212
- EganACrawleyJSchellenbergD2005Intermittent preventive treatment for malaria control in infants: moving towards evidence-based policy and public health actionTrop Med Int Health108151716135186
- EmmanuelEJMillerFG2001The ethics of placebo-controlled trials-a middle groundN Engl J Med3459151911565527
- ForsterHPEmanuelEGradyC2001The 2000 revision of the Declaration of Helsinki: a step forward or more confusionLancet25914495311705513
- GeerligsPDBrabinBJEggelteTA2003Analysis of the effects of malaria chemoprophylaxis in children on haematological responses, morbidity and mortalityBull World Health Organ812051612764517
- GlaxoSmithKline2004Malarone™ (atovaquone-proguanil) [prescribing information]Research Triangle Park, NCGlaxoSmith Kline
- GreenwoodBM1989The microepidemiology of malaria and its importance to malaria controlTrans R Soc Trop Med Hyg832592576161
- GreenwoodBMDavidPHOtoo-Forbes1995Mortality and morbidity from malaria after stopping malaria chemoprophylaxisTrans R Soc Trop Med Hyg89629338594677
- HaleBROwusu-AgyeiSFryauffDJ2003A randomized, double-blind, placebo-controlled, dose-ranging trial of tafenoquine for weekly prophylaxis against Plasmodium falciparumClin Infect Dis36541912594633
- HasugianARPurbaHLKenangalemE2007Dihydroartemisinin-piperaquine versus artesunate-amodiaquine: superior efficacy and post-treatment prophylaxis against multi-drug-resistant Plasmodium falciparum and Plasmodium vivax malariaClin Inf Dis44106774
- HeppnerDGKesterKEOckenhouseCF2005aToward an RTS, S-based, multi-stage, multiantigen vaccine against falciparum malaria: progress at the Walter Reed Army Institute of ResearchVaccine17–18224350
- HeppnerDGWalshDSUthaimongkolN2005bRandomized, controlled double-blind trial of daily oral azithromycin in adults for the prophylaxis of Plasmodium vivax malaria in Western ThailandAm J Trop Med Hyg73842916282291
- HillDRBairdJKPariseME2006Primaquine: Report from CDC expert meeting on malaria chemoprophylaxisAm J Trop Med Hyg7540115
- HoghBClarkePDCamusD2000Atovaquone-proguanil versus chloroquine-proguanil for malaria prophylaxis in non-immune travelers: a randomized, double blind studyLancet35618889411130385
- KiousBM2001The Nuremburg Code: Its history and implicationsPrinceton J Bioethics4719
- La VaqueTJRossiterT2001The ethical use of placebo controls in clinical research: The Declaration of HelsinkiApp Psychophysiol Biofeedback262337
- LapierreDIDuparcS2007The industry perspective on the Declaration of HelsinkiCongress of International Society of Tropical Medicine, 2007, Vancouver, Canada. Abstract SY0602.
- LegrosFBouchaudOAncelleT2007Risk factors for imported fatal Plasmodium falciparum malaria, France, 1996–2003Emerg Infect Dis13883817553228
- LellBKucknerDNdjaveM1998Randomised placebo-controlled study of atovaquone plus proguanil for malaria prophylaxis in childrenLancet351709139504515
- LellBFaucherJFMisinouMA2000Malaria chemoprophylaxis with tafenoquine: a randomized studyLancet3552041510885356
- LieRKEmanuelEGradyC2004The standard of care debate: the Declaration of Helsinki versus the international consensus opinionJ Med Ethics30190315082816
- LingJBairdJKFryauffDJ2002Randomized, placebo-controlled trial of atovaquone/proguanil for the prevention of Plasmodium falciparum or Plasmodium vivax malaria among migrants to Papua, IndonesiaClin Infect Dis358253312228819
- LooareesuwanSChulayJDCanfieldCJ1999Malarone™ (atovaquone and proguanil hydrochloride): A review of its clinical development for the treatment of malariaAm J Trop Med Hyg605334110348225
- MacateEAidePAponteJJ2006Intermittent preventive treatment for malaria control administered at the time of routine vaccinations in Mozambican infants: a randomized, placebo-controlled trialJ Infect Dis1942768516826474
- MacklinR2001After Helsinki: unresolved issues in international researchKennedy Inst Ethics J11173612166444
- MagillAJ2004Malaria: Epidemiology and risk to the travelerKeystoneJKozarskyPFreedmanDTravel medicine. Chapter 12. Edinburgh, ScotlandMosby
- MarraFSalzmanJREnsomMHH2003Atovaquone-proguanil for pro-phylaxis and treatment of malariaAnn Pharmacother3712667512921511
- MenendezCKahigwaEHirtR1997Randomised placebo-controlled trial of iron supplementation and malaria chemoprophylaxis for prevention of severe anemia and malaria in Tanzanian infantsLancet350844509310602
- MilhousWK2001Development of new drugs for chemoprophylaxis of malariaMed Trop614850
- MillerFGGradyC2001The ethical challenge of infection-inducing challenge experimentsClin Infect Dis3310283311528576
- MillerKDLobelHOSatrialeRF1986Severe cutaneous reactions among American travellers taking pyrimethamine-sulfadoxine (Fansidar) for malaria prophylaxisAm J Trop Med Hyg35451582939735
- MillerRSWongsrichanalaiCBuathongN2006Effective treatment of uncomplicated Plasmodium falciparum malaria with azithromycin-quinine combinations: a randomized, dose-ranging studyAm J Top Med Hyg734016
- MuentenerPSchlagenhaufPSteffenR1999Imported malaria (1985–1995): Trends and perspectivesBull World Health Org775606610444879
- NasveldPBrennanLKitchenerS2002aA randomised, double-blind comparative study to evaluate the safety, tolerability and effectiveness of tafenoquine and mefloquine for the prophylaxis of malaria in non-immune Australian soldiers [abstract]Annual Meeting of the Americal Society of Tropical Medicine and Hygiene2002. Abstract 326.
- NasveldPKitchenerSEdsteinM2002bComparison of tafenoquine WR238605 and primaquine in the post exposure (terminal) prophylaxis of vivax malaria in Australian Defense Forces PersonnelTrans R Soc Trop Med Hyg96683412625150
- NewmanRDPariseMEBarberAM2004Malaria-related deaths among U.S. travelers, 1963–2001Ann Intern Med1415475515466772
- NoedlHKrudsoodSChalermatranaK2006Azithromycin combination therapy with artesunate or quinine for the treatment of uncomplicated Plasmodium falciparum malaria in adults: a randomized, phase 2 clinical trial in ThailandClin Infect Dis4312647117051490
- NostenF2000Prophylactic effect of Malarone against malaria: all good news?Lancet3561864511130375
- Novartis. 2006. Novartis accelerates production of life-saving malaria treatment Coartem [online]. Accessed June 15, 2007. URL: http://hugin.info/134323/R/1030389/165274.pdf
- OhrtCDoolanDBriceG2007Plasmodium antibodies as surrogate markers of malaria exposure5th European Congress on Tropical Medicine and International HealthAmsterdam, NetherlandsMay 2007
- OhrtCRichieTRWidjajaH1997Mefloquine compared with doxycycline for the prophylaxis of malaria in Indonesian soldiers. A randomized, double-blind, placebo-controlled trialAnn Intern Med126963729182474
- O'MearaWPBremanJGMcKenzieFE2005The promise and potential challenges of intermittent preventive treatment for malaria in infants (IPTi)Malar J4315644136
- Orlandi-PradinesEPenhoatKDurandC2006Antibody responses to several malaria pre-erythrocytic antigens as a marker of malaria exposure among travelersAm J Trop Med Hyg749798516760507
- OverboschDSchilthuisHBienzleU2001Atovaquone-proguanil versus mefloquine for malaria prophylaxis in non-immune travelers: Results from a randomized double-blind studyClin Infect Dis3310152111528574
- PetersW1999The evolution of tafenoquine: antimalarial for a new millennium?J R Soc Med923455210615272
- PetersonE2004Malaria chemoprophylaxis: when should we use it and what are the options?Expert Rev Anti Infect Ther21193215482177
- ReynoldsT2000The ethics of placebo-controlled trialsAnn Intern Med13349110975985
- RiisP2003Thirty years of bioethics: the Helsinki Declaration 1964–2003New Rev Bioethics1155
- RitzhauptLBetleyBAHirsch2007Safety assessment of azithromycin plus chloroquine for the treatment and prevention of malaria [abstract]56th Annual Meeting of the American Society of Tropical Medicine and Hygiene2007Philadelphia, USA517
- RosenJBBremanJG2004Malaria intermittent preventive treatment in infants, chemoprophylaxis, and childhood vaccinationsLancet3631386815110499
- Sanofi-Synthelabo1999Primaquine brand of phosphate tablet [product information]Sanofi-SynthelaboNew York
- SchellenbergDMenendezCKahigwaE2001Intermittent treatment for malaria and anaemia control at time of routine vaccinations in Tanzanian infants: a randomised, placebo-controlled trialLancet3571471711377597
- SchellenbergDCisseBMenendezC2006The IPTi Consortium: research for policy and actionTrends Parasitol2229630016713739
- SimonR2000Are placebo-controlled clinical trials ethical or needed when alternative treatment exists?Ann Intern Med133474510975967
- ShanksGDGordonDMKlotzFW1998Efficacy and safety of atovaquone/proguanil as suppressive prophylaxis for Plasmodium falciparum malariaClin Infect Dis2749499770146
- ShanksGDRagamaBOAlooAJ1999Time to reappearance of malaria parasites following various drug treatment regimens in a holoendemic area of western KenyaTrans R Soc Trop Med Hyg93304510492766
- ShanksGDKainKCKeystoneJS2001aMalaria chemoprophylaxis in the age of drug resistance. II. Drugs that may be available in the futureClin Infect Dis33381511438908
- ShanksGDOlooAJAlemanGM2001bA new primaquine analog, tafenoquine (WR238605), for prophylaxis against Plasmodium falciparum malariaClin Infect Dis3319687411700577
- ShanksGDEdsteinMD2005Modern malaria prophylaxisDrugs6510912110
- ShanksGDMagillAJFreedmanDO2007Drug free holidays: Pre-travel versus during travel malaria prophylaxisAm J Trop Med Hyg771217620622
- ShapiroTARanasinhaCDKumarN1999Prophylactic activity of atovaquone against Plasmodium falciparum in humansAm J Trop Med Hyg60831610344660
- ShrettaRBrughaRRobbA2000Sustainability, affordability and equity of corporate drug donations: the case of MalaroneLancet35517182010905260
- ShrettaRWaltGBrughaR2001A political analysis of corporate drug donations: the example of Malarone in KenyaHealth Policy Plan161617011358917
- SukwaTYMulengaMChisdakaN1999A randomized, double-blind, placebo-controlled field trial to determine the efficacy and safety of Malarone™ (atovaquone/proguanil) for the prophylaxis of malaria in ZambiaAm J Trop Med Hyg60521510348223
- TaylorWRRichieTLFryauffDJ1999Malaria prophylaxis using azithromycin: a double-blind, placebo-controlled trial in Irian Jaya, IndonesiaClin Infect Dis28748110028075
- [UNFPA] United Nations Food Program2007State of world population 2007United Nations Population Fund, New YorkNew York
- [USAID] United States Agency for International Development2007President’s malaria initiative [online]. Accessed November 28, 2007. URL: http://www.fightingmalaria.gov/
- [USCDC] United States Center for Diseases Control and Prevention2008CDC health information for international travel 2008 [online]. Accessed July 11, 2007. URL: http://wwwn.cdc.gov/travel/contentYel-lowBook.aspx
- [USFDA] United States Food and Drug Administration2000FDA approves Malarone for prevention and treatment of malaria” [online]. Accessed June 15, 2007. URL: http://www.fda.gov/bbs/topics/ANSWERS/ANS01026.html
- [USFDA] United States Food and Drug Administration2001Guidance for industry: Acceptance of foreign clinical studies [online]. Accessed July 11, 2007. URL: http://www.fda.gov/cder/guidance/fstud.pdf
- [USFDA] United States Food and Drug Administration2005Guidance for industry: Pharmacogenomic data submissionsUnited States Department of Health and Human ServicesRockville, MD, United States
- [USFDA] United States Food and Drug Administration2007Malaria: Developing drug and non-vaccine products for treatment and prophylaxis (draft guidance for industry)United States Department of Health and Human ServicesRockville, MD
- [USCEDER] Center for Drug Evaluation and Research2007Frequently asked questions on drug development and investigational new drug applications [online]. Accessed November 14, 2007. URL: http://www.fda.gov/cder/about/smallbiz/faq.htm#Introduction
- van der BergJDDuvenageCSRoskellNS1999Safety and efficacy of atovaquone and proguanil hydrochloride for the prophylaxis of Plasmodium falciparum malaria in South AfricaClin Ther21741910363739
- VerhageDFTelgtDSBousemaJT2005Clinical outcome of experimental human malaria induced by Plasmodium falciparum-infected mosquitoesNeth J Med6352815768480
- WagnerJAWilliamsSAWebsterCJ2007Biomarkers and surrogate end points for fit-for-purpose development and regulatory evaluation of new drugsClin Pharmacol Ther81104717186007
- WalshDSLooareesuwanSWilairatanaP1999Randomized dose-ranging study of the safety and efficacy of WR 238605 (tafenoquine) in the prevention of relapse of Plasmodium vivax malaria in ThailandJ Infect Dis1801282710479159
- WalshDSWilaratanaPTangDB2004Randomized trial of 3-dose regimens of tafenoquine (WR238605) versus low-dose primaquine for preventing Plasmodium vivax malaria relapseClin Infect Dis39109510315486831
- WaltherMThompsonFMDunachieS2006Safety, immunogenicity, and efficacy of prime-boost immunization with recombinant poxvirus FP9 and modified vaccinia virus Ankara encoding the full-length Plasmodium falciparum circumsporozoite proteinInfect Immun7427061616622207
- WangRRichieTLBaracerosMF2005Boosting of DNA vaccine-elicited gamma interferon responses in humans by exposure to malaria parasitesInfect Immun7328637215845492
- WeissWROlooAJJohnsonA1995Daily primaquine is effective for prophylaxis against falciparum malaria in Kenya: comparison with mefloquine, doxycycline, and chloroquine plus proguanilJ Infect Dis171167182
- [WMA] World Medical Association2004World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects [online]. Accessed June 15, 2007. URL: http://www.wma.net/e/policy/b3.htm
- [WHO] World Health Organization2005World Malaria Report 2005World Health OrganizationGeneva