Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative, dopamine deficiency disorder. The main therapeutic strategies for PD treatment relies on dopamine precursors (levodopa), inhibition of dopamine metabolism (monoamine oxidase [MAO] B and catechol-O-methyl transferase inhibitors), and dopamine receptor agonists. Recently, a novel selective and irreversible MAO B propargylamine inhibitor rasagiline (N-propargyl-1-R-aminoindan, Azilect®) was approved for PD therapy. In contrast to selegiline, the prototype of MAO B inhibitors, rasagiline is not metabolized to potentially toxic amphetamine metabolites. The oral bioavailability of rasagiline is 35%, it reaches Tmax after 0.5–1 hours and its half-life is 1.5–3.5 hours. Rasagiline undergoes extensive hepatic metabolism primarily by cytochrome P450 type 1A2 (CYP1A2). Rasagiline is initiated at 1 mg once-daily dosage as monotherapy in early PD patients and at 0.5–1 mg once-daily as adjunctive to levodopa in advanced PD patients. Rasagiline treatment was not associated with “cheese effect” and up to 20 mg per day was well tolerated. In PD patients with hepatic impairment, rasagiline dosage should be carefully adjusted. Rasagiline should not be administered with other MAO inhibitors and co-administration with certain antidepressants and opioids should be avoided. Although further clinical evidence is needed on the neuroprotective effects of rasagiline in PD patients, this drug provides an additional tool for PD therapy.
Parkinson’s disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder clinically characterized as tremor at rest, muscular rigidity, bradykinesia, and postural instability. Clinical symptoms of the disease appear after 60% of the dopaminergic neurons population in the substantia nigra pars compacta (SNpc) already degenerated. One to two per cent of the adult population over the age of 60 is clinically diagnosed with PD; however, the number of affected people is much higher because of the initial asymptomatic progression of the disease (CitationTabakman et al 2004). Recent pathological attempts to classify the pathophysiological processes responsible for PD suggest a 6- stage evaluation scale of progression in PD-related symptoms (CitationBraak et al 2003). Stages 1–2 appear as lesions confined to the brain medulla oblongata, while PD-related clinical symptoms are absent. Stage 3 is defined as additional progression of the brain lesions towards midbrain, in particular in the SNpc monitored by PD-associated motor disorders. Stages 4–6 are characterized as further progression of the brain lesions towards the cortex with some changes in the sensory areas of the brain (CitationBraak et al 2003). The therapeutic approaches available today are symptomatic: they address mainly stages 3–6, although even with treatment, the longevity of these PD patients is short and their quality of life is poor. Since PD is generally regarded as a dopamine deficiency disorder it is not surprising that most drugs available to treat PD attempt to correct dopamine deficiency. However, none of these agents retards the progressive neurodegeneration associated with PD. Therefore, there is a need for novel therapeutic approaches to ameliorate the pathophysiological process of the disease.
PD cellular pathological mechanisms
PD is generally considered to be idiopathic (of unknown origin) and the precise cellular and molecular pathological processes causing the disease are not fully understood. In a minor subset of PD patients several genetic alterations have been identified and related to the expression and/or aggregation of two major proteins such as α-synuclein and parkin involved in part in the disease process (CitationDawson and Dawson 2003). In other sporadic forms of PD, additional biochemical abnormalities such as nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (CitationTatton et al 2003) and activation of proapoptotic proteins (eg, bax, caspases) (CitationHartmann et al 2000; CitationTatton 2000) have been identified. These genetic and biochemical changes are assumed to cause the apoptotic and/or necrotic cell death of the dopaminergic neurons of the SNpc by mitochondrial (CitationSzeto 2006) and proteasomal dysfunctions (CitationDawson and Dawson 2003), as well as oxidative stress (CitationSzeto 2006). Excitotoxicity (CitationRodriguez et al 1998), environmental toxins (CitationLandrigan et al 2005), infiammation (CitationMcGeer and McGeer 2004), reduced levels of neurotrophins (CitationLevy et al 2005), and changes in cerebral blood flow (CitationThanvi et al 2005) may also contribute to the development of PD. The complexity of pathological mechanisms is a significant obstacle in effective treatment of the disease.
PD therapeutic strategies
The first strategy for PD treatment relies on the use of dopamine precursors, such as levodopa (L-dopa) which is able to cross the blood–brain barrier (BBB) and is the most accepted and a powerful symptomatic drug available today for the treatment of PD. It increases the amount of dopamine in SNpc, thus compensating for the loss of dopaminergic neurons (, step 1). Most PD patients respond to L-dopa monotherapy. Nevertheless, the chronic treatment with L-dopa has several significant limitations and side-effects which include motor fluctuations, dyskinesias, and neuropsychiatric problems due to the increased dopaminergic activity of all five dopaminergic pathways in the brain, as well as up and down regulations of dopaminergic receptors (CitationOlanow and Jankovic 2005). After 3–5 years of L-dopa therapy, patients may exhibit an “on-off” phenomenon characterized by marked fluctuations in patient response to L-dopa. Such a person may be symptom free (“on” effect) and abruptly experience marked hypokinesia (“off” effect). The “on-off” phenomenon may be in part resolved by a novel pharmacokinetic approach. Taking into account that L-dopa has a narrow absorption window, an expandable gastroretentive dosage form of L-dopa delivery enables a significant extension of the absorption phase, thus providing more consistent blood levels of the drug (CitationKlausner et al 2003; CitationHoffman et al 2004). Unfortunately, in the latest stages of the disease, when the number of surviving dopaminergic neurons in the patients is extremely low, this therapeutic strategy is not effective.
Figure 1 Schematic of the major dopaminergic therapeutic strategies for Parkinson’s treatment. Step 1, dopamine precursor L-dopa; Step 2, dopamine receptor agonists; Step 3, dopamine metabolizing enzymes (COMT and MAO B) inhibitors; Step 4, neuroprotective compounds. +, increase in the synaptic dopamine or stimulation of dopamine receptors and survival pathways; −, inhibition of dopamine metabolizing enzymes or apoptotic pathways; ▲, dopamine neurotransmitter;V, dopamine receptors; DA, dopamine; MAO B, monoamine oxidase type B; COMT, cathechol-O-methyl transferase.
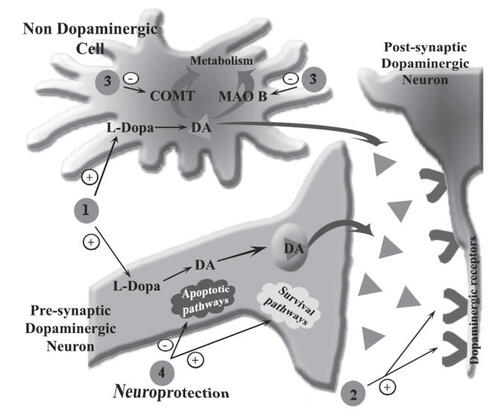
The second strategy for PD treatment is based on dopamine agonists that compensate for the lack of endogenous neurotransmitter by activating postsynaptic dopamine receptors (, step 2). Several dopamine agonists such as bromocriptine and cabergoline are widely used for symptomatic monotherapy of PD. Recently developed pramipexole and ropinirole are effective in treating symptoms of PD and data suggest that they also exert neuroprotective effects (CitationRadad et al 2005).
The third strategy for PD treatment uses inhibitors of dopamine-metabolizing enzymes: catechol-O-methyl transferase (COMT) and monoamnine oxidase B (MAO B) (Figure 3, step 3). Using this therapeutic approach the endogenous content of dopamine is increased, causing the reduction of PD symptoms. COMT inhibitors such as tolcapone and entacapone are not effective as monotherapy, but provide symptomatic relief when added to L-dopa treatment (CitationLeegwater-Kim and Waters 2006). Selegiline (deprenyl), however, is the most common inhibitor of MAO B used as monotherapy. Recently, neurologists have preferred to initiate selegiline monotherapy on patients in the early stage of PD, therefore postponing L-dopa treatment for a later stage, to avoid the side-effects associated with L-dopa (CitationOlanow and Jankovic 2005).
Several emerging therapeutic approaches are on the horizon to attenuates symptoms and/or neurodegeneration associated with PD with the hope of avoiding the motor complications seen with dopaminergic therapies: i) adenosine receptor type A2A antagonist (such as istradefylline) (CitationWu and Frucht 2005); ii) glutamate AMPA receptor antagonist (such as talampanel) (CitationWu and Frucht 2005); iii) opioid-like neuropeptide mociceptin/orphanin FQ (N/OFQ)/receptor NOP antagonist (CitationMarti et al 2005). These and other therapies currently under investigation represent a new phase of treatment strategies for PD.
Rasagiline, a novel MAO B inhibitor for PD therapy
In January 2005 the propargylamine-based, irreversible MAO B inhibitor rasagiline (Azilect®, Teva Pharmaceutical Industries Ltd., Israel) was first approved in Israel for the treatment of idiopathic PD as monotherapy or as adjunct therapy with L-dopa in patients with end-of-dose fluctuations. In February 2005 it was approved by the European Agency for the Evaluation of Medicinal Products (EMEA) for the same indication, and later on, in May 2006, rasagiline was approved by the US Federal Drug Administration (FDA). From mid 1970, selegiline (deprenyl), a selective and irreversible propargylamine drug, was the main selective MAO B inhibitor used in the clinic. Rasagiline is also a selective and irreversible propargylamine, with a slight chemical difference in the side chain structure (CitationTabakman et al 2004). Rasagiline, in contrast to selegiline, is not metabolized to potentially toxic amphetamines and its major metabolite 1-R-aminoindan has demonstrated therapeutic effects in neuronal cultures (CitationAbu-Raya et al 2002) and animal models of PD (CitationSpeiser et al 1998).
Rasagiline – pharmacokinetic characteristics
Rasagiline is well absorbed after oral administration (CitationThebault et al 2004) and readily crosses the BBB. The bioavailability of rasagiline is about 35% (CitationChen and Ly 2006) and it exerts linear absorption at doses of 1–10 mg/day (CitationFDA 2006). Maximal plasma concentration (Cmax) obtained after 1 mg and 2 mg oral dose is 2.5 ng/mL and 4.9 ng/mL, respectively. The time to reach maximal concentration (Tmax) is 0.5–1 hours and is unaffected by food intake (CitationParkinson Study Group 2005; CitationChen and Ly 2006; CitationFDA 2006). High-fat meal decreases the area under the curve (AUC) of rasagiline by 20%, which is considered clinically insignificant; therefore, rasagiline can be administered independently of food intake (CitationChen and Ly 2006; CitationFDA 2006). The volume of dis-tribution (Vd) of rasagiline varies between 87 to 243 L (CitationChen and Ly 2006; CitationFDA 2006), according to different reports. Plasma albumin binding is considered to be 60%–70% (CitationChen and Ly 2006), although a higher plasma protein binding value was reported (CitationFDA 2006). The half-life of rasagiline is 1.5–3.5 h and may be dose-dependent (CitationChen and Ly 2006; CitationFDA 2006). It is interesting to note that there is no linear pharmacokinetic-pharmacodynamic (PK-PD) correlation that can be explained by the prolonged irreversible inhibition of MAO B up to 40 days (pharmacodynamic t½). PK profile of rasagiline is similar in men and women. Rasagiline undergoes extensive hepatic metabolism and almost complete biotransformation in the liver, primarily by cytochrome P450 1A2 (CYP 1A2) to form 1-R-aminoindane (major metabolite), 3-hydroxy-N-propargyl-1-aminoindan, and 3-hydroxy-aminoindan. The major metabolite 1-R-aminoindan has a Tmax of about 2 hours (CitationStern et al 2004). Oral clearance of the drug is 94.3 L/hour, and since the liver blood flow is about 90 L/hour, this finding implies that extra-hepatic processes are not involved in elimination of rasagiline.
Rasagiline therapy in PD patients as evident from clinical trials
The recommended dose of rasagiline in treatment of PD is 1 mg once daily as monotherapy or 0.5 mg once daily as adjunctive to L-dopa. If satisfactory clinical response is not achieved, the dose may be increased to 1 mg administered once daily. The effectiveness of rasagiline was concluded from several clinical trials as monotherapy in early PD or as adjunct therapy with L-dopa in advanced PD. Rasagiline monotherapy efficacy was demonstrated by the accepted Unified Parkinson Disease Rating Scale (UPDRS) scores compared with placebo (). Significant benefits were observed in the reduction in “off” time of L-dopa treated patients, improvement in motor activity (rigidity and tremor) comparable to the efficacy of selegiline and lazabemide, a 2-aminoethyl carboxamide derivative which is an antioxidant with reversible, and a highly selective MAO B inhibition property. Evaluation of lazabemide discontinued in phase III clinical trials (CitationParkinson Study Group 2002). Upon treatment with rasagiline in combination with L-dopa, a decrease in L-dopa dosage may be expected. Indeed in the clinical trials performed, up to 16% of patients received a 13% reduced dosage of L-dopa (CitationFDA 2006).
Table 1 Clinical trials to evaluate rasagiline efficacy, safety, and side-effects
In a subanalysis of delayed-start TEMPO study, quality of life (QOL) was measured by Parkinson’s Disease Quality of Life questionnaire (PDQUALIF) at 0, 14, 26, and 52 weeks. Rasagiline treatment improved QOL scores in PD patients compared with placebo after 6 months of initial TEMPO study. But no difference in QOL scores was observed after 12 months, when all the patients had received rasagiline for at least 26 weeks (CitationBiglan et al 2006).
Clinical risk management of rasagiline therapy
Rasagiline was well tolerated in volunteers and PD patients, with treatment discontinuation rates similar to placebo (). A subanalysis of TEMPO and PRESTO clinical trials revealed that no cognitive and behavioral adverse events in either rasagiline 1 mg or placebo groups exceeded 10%, and the differences between rasagiline and placebo never exceeded 3%. This safety profile was achieved along with improved PD symptoms and better control of motorfluctuations (CitationElmer et al 2006). Most adverse effects were defined as “uncomfortable” rather than “serious”. Rasagiline was well tolerated at doses up to 20 mg/day and no cases of overdose have been reported. The most common adverse effects seen in volunteers included headache, insomnia, xerostomia, abdominal discomfort, nausea, and diarrhea. Other adverse effects observed in clinical trials, although not different from placebo, included postural hypotension, dyskinesias, arthralgia, weight loss, anorexia, depression, vomiting, balance difficulty, and hallucinations (CitationChen and Ly 2006). The long-term safety profile of rasagiline is similar to that of short-term treatment (CitationFDA 2006).
As a theoretical risk of hypertensive crisis exists with all MAO inhibitors, restriction in dietary tyramine (eg, cheese and wine) and sympatomimetic amines (eg, phenylephrine) is recommended in patients treated with rasagiline. Patients should be advised about symptoms and signs of hypertensive crisis that include severe headache, nausea and vomiting, blurred vision, chest pain, difficulty in thinking, stupor, coma, and seizures. However, the clinical trials indicated that rasa-giline, like selegiline, is not associated with “cheese effect” at clinically relevant MAO B inhibition dosages. Specifically, oral challenge with 75 mg (CitationParkinson Study Group 2002) or 50 mg (CitationParkinson Study Group 2005) tyramine did not significantly change blood pressure or heart rate of rasagiline-treated patients (CitationBiglan et al 2006).
In patients with mild hepatic impairment the AUC of rasagiline increases by 80% and Cmax increases by 38% (CitationChen and Ly 2006). Therefore, the dose of rasagiline recommended in patients with mild hepatic impairment is 0.5 mg/day (CitationFDA 2006). In moderate hepatic impairment the AUC increases by 568% and Cmax by 83%; therefore, therapy with rasagiline is not recommended in patients with moderate or severe hepatic impairment. Less than 1% of rasagiline is excreted unchanged in urine; therefore no dose adjustment is required in patients with renal insufficiency.
Rasagiline should not be administered along with other MAO inhibitors. Ciprofloxacin, a CYP1A2 inhibitor, at 500 mg twice daily dose increased rasagiline AUC by 83%. Therefore, the dose of rasagiline should be reduced by 50% in patients receiving ciprofloxacin or other CYP1A2 inhibitors like cimetidine and fluvoxamine (CitationChen and Ly 2006). Omeprazole, a CYP1A2 inducer, may reduce AUC of rasagiline; therefore, an increased dosage may be required to achieve the same clinical efficacy.
In vitro studies indicated that rasagiline at a very high con-centration (160 times higher than Cmax) did not inhibit CYP 1A2, 2A6, 2C9, 2C19, 2D6, 2E1, 3A4 (CitationChen and Ly 2006; CitationFDA 2006). Therefore, we can predict that polypharmacy with rasagiline and other drugs which are substrates of the above CYPs will not result in metabolic drug-drug interactions although we cannot exclude other pharmacokinetic and/or pharmacodynamic interactions.
MAO inhibitors co-administered with antidepressants have been associated with serotonin syndrome which is a medical emergency clinically manifested by anxiety, mental status changes, hypertension, diarrhea, hyperreflexia, myoclonus, loss of consciousness and death. In LARGO, TEMPO, and PRESTO clinical trials several patients were treated with antidepressants, including amitriptyline ≤50 mg/daily, trazodone ≤100 mg/daily, citalopram ≤20 mg/daily, sertraline ≤100 mg/daily, and paroxetine ≤30 mg/daily. Fluoxetine and fluvoxamine were not allowed. Although there are insufficient data to determine safety upon its co-administration with serotonin selective reuptake inhibitors, there have been no reports connecting serotonin syndrome to rasagiline.
We would like to stress that co-administration of an opioid receptor agonist meperidine (pethidine) with rasagi-line is contraindicated due to possible serotonin syndrome. At least 14 days should elapse between discontinuation of rasagiline and initiation of meperidine treatment. Moreover, interaction is theoretically expected with tramadol, metha-done, and propoxyphene as these opioid receptor agonists have been shown to inhibit reuptake of serotonin in vitro (CitationGillman 2005).
Relevance of rasagiline – neuroprotective effects for PD treatment
Today’s interest in PD therapy is in the development of drugs with multiple beneficial effects. In addition to the compensation of the reduction of dopamine in SNpc the drug to be developed is required to ameliorate the progression of dopaminergic neuron degeneration, ie, to induce neuroprotection of the degenerating neuron. Over the last decade a large body of evidence indicates that selegiline and rasagiline are neuroprotective in a variety of pharmacological models in vitro and in vivo (CitationAbu-Raya et al 2000; CitationTabakman et al 2004). For example, selegiline and rasagiline (10 mg/kg body weight) markedly attenuated the neurotoxic effect of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) in a non-human primate PD model at a behavioral, histological, and biochemical level in parallel to significant inhibition of MAOs activity (CitationKupsch et al 2001). Paradoxally, the anti-apoptotic and prosurvival properties of rasagiline appear to be independent of MAO B inhibition (, step 5) but mediated by the propargyl moety (as reviewed CitationTabakman et al 2004). The pharmacological mechanism of rasagiline-mediated neuroprotection was studied in neuronal cell cultures using different types of oxidative and trophic with-drawal stress models. The antiapoptotic effects of rasagiline are proposed to be mediated by activation of antiapoptotic proteins as a result of drug binding to the fl avin adenine dinucleotide (FAD) binding site in GAPDH and other anti-apoptotic proteins resulting in neuroprotection (CitationTabakman et al 2004). Based on the proof of concept of neuroprotective effects of rasagiline in in vitro and in vivo pharmacological models, the potential neuroprotective effect of rasagiline was also considered in the clinical trials.
The delayed start TEMPO study is the first attempt to examine the neuroprotective effects of rasagiline in the framework of a double-blind, randomized, placebo-controlled clinical trial (CitationParkinson Study Group 2004). During the randomization of the patients two groups were created, one which received the drug during the whole period and another one that started the treatment at the middle of the study in order to separate between immediate symptomatic effects of the drug from a delayed, disease-modifying contribution (neuroprotection). In this clinical trial design, in the second part of the study the placebo-controlled group received rasagiline; therefore at the end of the trial the symptomatic effects are presumably equalized between the two groups. The results revealed improvement in motor performance in patients that received rasagiline during the whole trial period compared with patients treated with rasagiline only during late phase. This finding may indicate a neuroprotective effect of rasagiline in the patients, but this aspect needs further exploration (CitationParkinson Study Group 2004).
Future directions
Current treatments of PD deal well with the symptomatic clinical presentation. The available therapeutic strategies mainly elevate the reduced levels of dopamine by utilizing different pharmacological mechanisms as schematically presented in . However, none of the available thera-peutic options can slow the progression of the disease. Unmet needs create opportunities for developing and evaluating neuroprotective agents. In theory, these drugs will modify the course of the disease by preventing or delaying the death of dopaminergic neurons and, moreover, other types of neuronal cells, thus being beneficial in the first stages of PD (CitationBonuccelli and Del Dotto 2006). MAO B inhibitors, such as selegiline and to a greater extent rasagiline, have been shown to have disease-modifying potential (CitationStocchi 2006). The neuroprotective effect of selegiline is compro-mised by its many amphetamine neurotoxic metabolites, and introduction of rasagiline circumvents this problem. It is important to remember that the neuroprotective mechanism of rasagiline and other propargylamines derivatives in dif-ferent neuronal models appear to be independent of MAO B inhibition. Therefore, it is essential to further elucidate the pharmacological mechanism of action of propargylamines in order to get a better insight into the neuroprotective path-ways and to apply them for new pharmacological targets for the development of novel anti-PD drugs. It is tempting to speculate that in future the novel drugs to be developed for PD treatment will be multi functional by correcting both the lack of dopamine as well as providing neuroprotection to the degenerating neurons (CitationWu and Frucht 2005). To opti-mize fully their therapeutic efficacy it would be important to identify the proper mode of administration (pharmaceutical compounding) that takes into account their pharmacokinetic properties (CitationHoffman and Stepensky 1999).
References
- Abu-RayaSBlaugrundETrembovlerVRasagiline, a novel MAO B inhibitor with neuroprotective effects under ischemic conditions in PC12 cellsDrug Dev Res20005028590
- Abu-RayaSTabakmanRBlaugrundENeuroprotective and neurotoxic effects of monoamine oxidase-B inhibitors and derived metabolites under ischemia in PC12 cellsEur J Pharmacol20024341091611779573
- BiglanKMSchwidSEberlySRasagiline improves quality of life in patients with early Parkinson’s diseaseMov Disord2006216162316450340
- BonuccelliUDel DottoPNew pharmacologic horizons in the treatment of Parkinson diseaseNeurology200667S30817030738
- BraakHDel TrediciKRubUStaging of brain pathology related to sporadic Parkinson’s diseaseNeurobiol Aging20032419721112498954
- ChenJJLyAVRasagiline: A second-generation monoamine oxidase type-B inhibitor for the treatment of Parkinson’s diseaseAm J Health Syst Pharm2006639152816675649
- DawsonTMDawsonVLMolecular pathways of neurodegeneration in Parkinson’s diseaseScience20033028192214593166
- ElmerLSchwidSEberlySRasagiline-associated motor improvement in PD occurs without worsening of cognitive and behavioral symptomsJ Neurol Sci2006248788316828804
- FDACenter for Drug Evaluation and Research [online]2006 Accessed 18 December 2006. URL: http://www.fda.gov/cder/foi/label/2006/021641lbl.pdf.
- GillmanPKMonoamine oxidase inhibitors, opioid analgesics and serotonin toxicityBr J Anaesth2005954344116051647
- HartmannAHunotSMichelPPCaspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s diseaseProc Natl Acad Sci U S A20009728758010688892
- HoffmanAStepenskyDPharmacodynamic aspects of modes of drug administration for optimization of drug therapyCrit Rev Ther Drug Carrier Syst19991657163910677802
- HoffmanAStepenskyDLavyEPharmacokinetic and pharmacodynamic aspects of gastroretentive dosage formsInt J Pharm20042771415315158977
- KlausnerEALavyEBartaMNovel gastroretentive dosage forms: evaluation of gastroretentivity and its effect on levodopa absorption in humansPharm Res20032014667314567643
- KupschASautterJGotzMEMonoamine oxidase-inhibition and MPTP-induced neurotoxicity in the non-human primate: comparison of rasagiline (TVP 1012) with selegilineJ Neural Transm2001108985100911716151
- LandriganPJSonawaneBButlerRNEarly environmental origins of neurodegenerative disease in later lifeEnviron Health Perspect20051131230316140633
- Leegwater-KimJWatersCTolcapone in the management of Parkinson’s diseaseExpert Opin Pharmacother2006722637017059382
- LevyYSGilgun-SherkiYMelamedETherapeutic potential of neurotrophic factors in neurodegenerative diseasesBioDrugs2005199712715807629
- MartiMMelaFFantinMBlockade of nociceptin/orphanin FQ transmission attenuates symptoms and neurodegeneration associated with Parkinson’s diseaseJ Neurosci200525959160116237164
- McGeerPLMcGeerEGInflammation and neurodegeneration in Parkinson’s diseaseParkinsonism Relat Disord200410Suppl 1S3715109580
- OlanowCWJankovicJNeuroprotective therapy in Parkinson’s disease and motor complications: a search for a pathogenesis-targeted, disease-modifying strategyMov Disord200520Suppl 11S31015822111
- Parkinson study groupA controlled trial of rasagiline in early Parkinson disease: the TEMPO StudyArch Neurol20025919374312470183
- Parkinson study groupA controlled, randomized, delayed-start study of rasagiline in early Parkinson diseaseArch Neurol200461561615096406
- Parkinson study groupA randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO studyArch Neurol200562241815710852
- RabeyJMSagiIHubermanMRasagiline mesylate, a new MAO-B inhibitor for the treatment of Parkinson’s disease: a double-blind study as adjunctive therapy to levodopaClin Neuropharmacol2000233243011575866
- RadadKGilleGRauschWDShort review on dopamine agonists: insight into clinical and research studies relevant to Parkinson’s diseasePharmacol Rep2005577011216382188
- RascolOBrooksDJMelamedERasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomized, double-blind, parallel-group trialLancet20053659475415766996
- RodriguezMCObesoJAOlanowCWSubthalamic nucleus-mediated excitotoxicity in Parkinson’s disease: a target for neuroprotectionAnn Neurol199844S175889749591
- SpeiserZLevyRCohenSEffects of N-propargyl-1-(R)aminoindan (rasagiline) in models of motor and cognition disordersJ Neural Transm Suppl1998522873009564629
- SternMBMarekKLFriedmanJDouble-blind, randomized, controlled trial of rasagiline as monotherapy in early Parkinson’s disease patientsMov Disord2004199162315300656
- StocchiFRasagiline: defining the role of a novel therapy in the treatment of Parkinson’s diseaseInt J Clin Pract2006602152116451296
- SzetoHHMitochondria-targeted peptide antioxidants: novel neuro-protective agentsAAPS J20068E5213117025271
- TabakmanRLechtSLazaroviciPNeuroprotection by monoamine oxidase B inhibitors: a therapeutic strategy for Parkinson’s disease?Bioessays200426809014696044
- TattonNAIncreased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson’s diseaseExp Neurol2000166294311031081
- TattonWGChalmers-RedmanRBrownDApoptosis in Parkinson’s disease: signals for neuronal degradationAnn Neurol200353Suppl 3S6170 discussion S70–212666099
- ThanviBLoNRobinsonTVascular parkinsonism – an important cause of parkinsonism in older peopleAge Ageing200534114915713855
- ThebaultJJGuillaumeMLevyRTolerability, safety, pharmacodynamics, and pharmacokinetics of rasagiline: a potent, selective, and irreversible monoamine oxidase type B inhibitorPharmacotherapy200424129530515628826
- WuSSFruchtSJTreatment of Parkinson’s disease: what’s on the horizon?CNS Drugs2005197234316142989