Abstract
Over recent decades, basic research has yielded a large volume of data on many potentially clinically relevant genetic determinants of drug efficacy and toxicity. Until recently, most examples involved genes encoding drug-metabolizing enzymes, particularly the cytochromes P450. More recently, rapid advances in genomic technologies have enabled broader, genome-wide searches for determinants of drug response. In parallel with these pharmacogenetic studies, a new drug discovery platform, termed pharmacogenomics, has emerged which utilises genetic information to guide the selection of new drugs most likely to survive increasingly demanding safety and efficacy assessments. Together, these advances are widely promoted as the basis of a new era of drug-based therapeutics tailored to the individual. The extent to which individualized or personalized medicine will emerge as a sustainable new therapeutic paradigm is, however, the topic of much debate. It is clear that an increasingly complex series of barriers must be overcome if we are to successfully harness genomic advances in the clinical setting. Potential barriers may include cost-effectiveness of the test, ethical concerns over the use of DNA, and required educational and equipment infrastructure. Although long overdue, many of these potential barriers are now being subjected to closer examination and as a result, a framework for successful clinical uptake of pharmacogenomics is emerging.
In recent years, clinical pharmacogenomics has emerged as an umbrella term which in, its simplest terms, can be defined as the use of genetic information to improve the clinical outcomes of pharmacotherapy. As new technologies, including microarray technology, proteomics (profiling protein expression) and metabonomics (profiling small molecule levels), find their way into the clinical setting, we can anticipate a broadening of this definition to the use of biological information to improve the clinical outcomes of drug therapy. The aim of pharmacogenomics in the clinical setting is simple: to maximize the chances of effective treatment of a specific indication and minimise the likelihood of adverse drug reactions. The potential clinical benefits of pharmacogenomics are immense and these have been widely articulated and speculated in the remarkable number of pharmacogenomics reviews that have been published in recent years. These benefits are perhaps most clear with respect to adverse drug reactions. Adverse drug reactions are the fifth leading cause of death in the United States (CitationLazarou 1998) and represent a major social and financial burden. Should pharmacogenomics be able to make even small inroads into this area, it would clearly represent a great financial and ethical benefit to the community at large.
In the substantial literature that has emerged over recent years, pharmacogenomics has been widely hyped as having the potential to greatly accelerate the implementation of personalized medicine, using therapeutic regimens tailored to each individual’s genetic profile. Indeed, in many review papers on pharmacogenomics, the general impression has been that it is just around the corner. However, despite the constant press, pharmacogenomics has had little utilization in clinical practice (CitationGardiner and Begg 2005). Of late, however, there have been a number of indications that a more pragmatic view may be emerging (CitationPhillips et al 2003; CitationTucker 2004).
While acknowledging the relatively limited clinical uptake of pharmacogenomics, it is important to note that poor clinical implementation of basic pharmacogenomic research does not necessarily indicate a failure. Pharmacogenomic research can result in other useful outcomes, such as deeper insight into the mechanisms and population distributions of variance in pharmacotherapy. A better understanding of the expected clinical variation can be useful, even in the absence of a pharmacogenomic test to predict the variance. Furthermore, mechanistic understanding of the source of drug variability facilitates the development of improved new drugs. As discussed later, pharmaceutical companies generally prefer to use pharmacogenomics to develop new drugs with improved pharmacology, rather than developing pharmacogenomic tests to limit use of their drugs in the clinic. While pharmacogenomic research may lead to greater understanding of the mechanism leading to therapeutic failure or adverse events, testing for genotype may not be the best method to use clinically. For example, rather than genotyping a particular CYP enzyme, it may be preferable to phenotype with a probe substrate or undertake therapeutic drug monitoring. A recent survey of clinical use of pharmacogenomics in Australia and New Zealand found that the most frequently performed genetic test was for thiopurine methyltransferase - to predict patients at high risk of myelosuppression following standard doses of azothiopurine or mercaptopurine (CitationGardiner and Begg 2005). However, the frequency of phenotyping exceeded genotyping by five-fold (CitationGardiner and Begg 2005).
This paper will seek to understand the barriers to the clinical implementation of pharmacogenomics. While it is relatively simple to enumerate the potential barriers, it is very difficult to determine which barriers present the most significant problems. Many of these barriers are sufficient in isolation to stall implementation of a pharmacogenomic test. More commonly, clinical implementation of pharmacogenomics is inhibited by multiple barriers that all need to be addressed before the tests can be put into practice. It is difficult to delineate the optimal path forward under these circumstances and many questions arise. Unfortunately, for the most part, there is insufficient objective research to answer these questions fully. While further research on specific pharmacogenomic research problems is important, so is research into the ethical, political, economic and social issues associated with the implementation of clinical pharmacogenomics.
Unraveling the complexities of the factors influencing the eventual clinical implementation of pharmacogenomics may be best handled by following the general development pathway of pharmacogenomic tests from initial conception to broad clinical use (). At each stage on this pathway, there are factors that directly and indirectly influence the clinical implementation of pharmacogenomics. The flow of topics covered in this review will generally mirror the flow of . Hence we will initially focus on potential barriers encountered early in the development of a pharmacogenomic test, and progressively lead into barriers faced during the attempt to clinically utilize the pharmacogenomic protocol.
Figure 1 Idealized flow chart of the processes undertaken to bring pharmacogenomic testing from concept to clinical use.
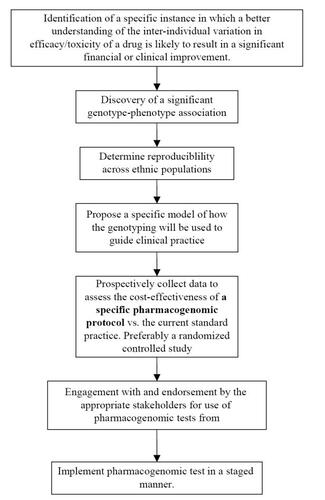
Identification of specific drug phenotypes most amenable to pharmacogenomic testing
The more cost-effective a pharmacogenomic test is, the more likely it will be taken up in the clinical setting. Thus, in order to improve clinical implementation of pharmacogenomics, it is strategic to focus research on those situations that are likely to result in the most cost effective tests. In general, it is thought that the cost-effectiveness of health care technologies is primarily dependent on the cost and effectiveness of the technology, the morbidity and mortality associated with the phenotype, and the cost of treating the phenotype (CitationVeenstra et al 2000). One study has attempted to identify a number of primary characteristics that will enhance the cost-effectiveness of pharmacogenomics (CitationVeenstra et al 2000). The most appropriate situations are those in which the clinical or economic consequences are severe and common, and in which current methods for monitoring drug response are suboptimal (CitationVeenstra et al 2000). Examples of these situations may include chronic illnesses requiring extended therapy, therapy requiring an extended period before the efficacy can be assessed, situations in which inappropriate therapy can have irreversible consequences and finally, treatments associated with severe adverse events that have the potential for significant morbidity (CitationAlmuete 2000).
Discovery of a significant genotype-phenotype association
Once a particular clinical pharmacotherapy-related phenotype has been selected as important, it is necessary to find an association between one or more single nucleotide polymorphisms (SNPs) and the phenotype. It is known that cost-effectiveness of a pharmacogenomics test is generally enhanced in situations in which the association between genotype and clinical phenotype is strong, and the variant allele frequency is relatively high (CitationVeenstra et al 2000).
Scoring the usefulness of a SNP-phenotype association
There are many ways to score the usefulness of the association. Commonly used statistics include positive and negative predictive power, specificity and sensitivity, odds ratio and relative risk. Perfect associations, resulting in no false positives and negatives, are uncommon. Some SNPs may allow few false positives at the expense of more false negatives, and vice versa. It is necessary to understand the clinical consequences of false positives and false negatives to judge the utility of any particular SNP. For example, if it is imperative to ensure that no one has a serious adverse effect from a drug, a test with very high negative predictive power (ie, if test is negative, a very high likelihood that the person will not have the adverse effect) is called for. The consequence of this may be a lower positive predictive power. Thus, there will be some patients that could have received the drug, but did not get the drug due to a false positive pharmacogenomic test. The availability of a good alternative treatment affects the relative importance of positive and negative predictive power (CitationTate and Goldstein 2004). It is obvious that assessing the utility of various SNPs is complex and requires extensive consultation with clinicians.
Experimentally determining genotypes
Setting aside the difficulty in assessing the absolute and relative utility of SNPs for predicting the phenotype, there are also many experimental factors that can influence finding a good SNP as the basis of a pharmacogenomic test. It is estimated that there are approximately 6 million common SNPs (greater than 10% frequency) among all human populations and many fold more rare SNPs (CitationCarlson et al 2004). The most sensible initial approach is to assess SNPs in genes that are known to be associated with the pharmacology of the drug. These are typically genes governing the pharmacokinetics of the drug but may also include those influencing the pharmacodynamics of the drug or pathogenesis of the disease being treated. This approach is commonly known as the candidate gene approach and has been the cornerstone of the discipline for many years. However, it is highly plausible that the observed drug response phenotype is due to a gene that has not been previously linked with the pharmacology of the drug. This is especially likely for adverse events which may be due to trace drug metabolites produced by unknown metabolic pathways. Even limiting the search for candidate genes to the known pharmacology of a drug can result in a very large number of candidate genes when associated gene pathways are taken into account, particularly in the case of drugs with complex metabolic pathways.
Should analysis of these initial candidate genes prove unsuccessful, it may be necessary to turn to highly multiplexed technologies for assessing larger numbers of SNPs across the genome. Affymetrix’s new Genome-Wide Human SNP Array 6.0, which contains 906,600 SNPs and additional probes to facilitate gene copy number determination. Although costs of the chip-based SNP technologies are falling, these technologies are still expensive and currently out of reach of most researchers and diagnostic settings. There are also many issues with data analysis of the rich data generated by these approaches. Methods for calling genotypes are still in their infancy and it is not clear how accurate the calls are. More importantly, there are the issues associated with multiple hypothesis testing. P values associated with individual tests need to be adjusted to take into account the many hypotheses (usually one for each SNP in this case) that have been tested (CitationStorey and Tibshirani 2003). A failure to do so will give misleading results on which SNPs are statistically significantly associated with the phenotype in question.
Some of the problems related to SNP genotyping may be alleviated through the use of haplotype approaches which aim to limit the number of SNPs warranting analysis. The International HapMap project aims to find tag SNPs that identify unique haplotype blocks - DNA sequences that are inherited together (CitationInternational Haplotype Consortium 2003). With this knowledge, it is thought that the identification of a few alleles of a haplotype block can unambiguously identify all other polymorphic sites in the region. Recently, the first haplotype map of the human genome emerging from the HapMap project was published (CitationInternational Haplotype Consortium 2005). This map provides accurate and complete genotypes for more than a million SNPs from four populations. Recent research suggests that between 65% and 85% of the human genome may be organized into haplotype blocks of at least 10,000 bases. As a result, researchers need to study only about 300,000 to 600,000 tag SNPs to identify the haplotypes in the human genome. These tag SNPs should hold information on the associated SNPs, thereby reducing the number of SNPs to be assessed. The next generation of SNP arrays is likely to use the tag SNPs.
Biological limitations of SNPs
It is uncertain whether SNPs in isolation will be able to predict most drug response phenotypes sufficiently well to prove cost-effective. The majority of well known pharmacogenomic associations represent predominantly monogenic traits with high penetrance and large, discrete functional consequence (CitationNebert et al 2003; CitationMeyer 2004). However, drug treatment generally represents a complex phenotype, influenced by many genes and environmental factors (Nebert 2000; CitationMeyer 2004). As a result, the phenotype is commonly a continuous gradient (CitationNebert and Vesell 2004). In these cases, variation in the activity of multiple major genes, each potentially being insignificant in isolation, must be considered to understand the variation in drug response.
Moreover, the activity of these genes may not be predicted sufficiently by SNPs, in isolation or as haplotypes. Each gene may potentially be influenced by endogenous and exogenous modifiers (CitationMeyer 2004). Phenomena such as gene silencing (CitationSchramke and Allshire 2003), epistasis (CitationMcGovern et al 2003), genomic imprinting (CitationLewis et al 2003), and RNA interference may also influence drug response phenotypes and need to be considered in addition to SNPs (CitationNebert et al 2003). The dynamic nature of the genome, for example, alterations in gene expression patterns to compensate for perturbation of a gene product or presence of environmental stimulus, adds additional complexity to the situation (CitationNebert et al 2003).
There is another clinically important issue likely to be associated with using SNPs for predicting drug response. Most human genes have approximately 3 to 10 major variant alleles, and potentially hundreds of rare variant alleles (CitationCarlson et al 2003). Thus, subjects having rare alleles would probably not be discovered prior to receiving the drug and would potentially be at risk of receiving suboptimal therapy (CitationNebert et al 2003). The clinical implications of this situation require further consideration.
In light of these caveats, methodologies based on genotyping may be limited in situations where the environmental influences on drug efficacy and toxicity are significant. In cases where multiple genes contribute significantly to the phenotypes, there will be additional complications. Firstly, the pharmacogenomic tests will be more complicated than those currently used, both in terms of genotyping and interpretation. The interpretation will be further complicated if there are interactions between the genotypes, which affect the efficacy/toxicity (ie, if the genes do not have independent influences on the phenotype). Secondly, the sample sizes to identify and validate the complement of genetic variations affecting efficacy/toxicity would be much greater.
In the future, it is likely that alternative technologies such as metabonomics will be used to complement genotyping strategies for the prediction of drug response. Metabonomics is a rapidly emerging science, involving the analysis of biofluids including urine and plasma, in order to reflect whole organism biochemical profiles and regulation of function (Nicholson 2002). The technique combines the use of multivariate statistics with an analytical platform, usually nuclear magnetic resonance (NMR) or liquid-chromatography-mass spectrometry. Its advantages include highly sensitive real time monitoring of the physiological and biochemical status, relative economy after an initial outlay for the analytical infrastructure and the ability to incorporate environmental influences. Besides being able to predict drug response a priori, this technology may be used for early detection of drug response and technology.
Determine reproducible association across ethnic populations
If the initial association between a SNP and drug phenotype is detected in a single ethnic group, it is important to assert that this association is also valid in other ethnic groups. If the association is valid only in a single ethnic group, this will cause difficulties in implementation of the pharmacogenomic test. In these cases, it will be imperative to ensure that the test is only applied to the specific ethnic group, a difficult undertaking given the dramatic increase in interbreeding between people of differing ethnic backgrounds (CitationNebert and Vesell 2004; CitationShah 2004). Implementation of such a restrictive approach may also raise significant ethical and regulatory challenges in some countries (CitationLee 2005; CitationBuckley and McKinnon 2004). For example, in Australia where medicines are highly subsidized by the Federal Government on the basis of a rigorous assessment of cost-effectiveness, it is possible to envisage a scenario where subsidy could potentially be restricted along ethnic lines. The social acceptability of such a scenario is unclear and warrants further consideration.
There are a number of reasons why the pharmacogenomic value of a SNP may vary between ethnic groups. Firstly, it is known that most genes have large differences in allelic frequencies across ethnic groups (CitationNebert and Menon 2001; CitationSalsbury et al 2003). This can significantly affect the cost-effectiveness of the test. Furthermore, for most genotype-phenotype associations, the SNP has not been proven to actually cause the variation in phenotype. The SNP may simply be in linkage disequilibrium with the causal genotype. This situation will become even more common with increasing use of genome-wide scanning of associated SNPs, using technologies such as SNP chips. The problem arises due to the variation in haplotype blocks between ethnic groups (CitationCarlson et al 2003; CitationCrawford et al 2004). Therefore if the SNP is not causal, there is a good chance that the SNP may not be associated with other ethnic groups. Finally, it is possible that the underlying physiology may differ among ethnic groups due to different gene-gene and gene-environment interactions (CitationTate and Goldstein 2004). Thus it is important to study the SNP-phenotype association in multiple ethnic groups.
Propose a specific protocol of how the genotyping will be used to guide clinical practice
It is not sufficient to have an association between a SNP and the drug response. Widespread clinical usage of the association will require a well-defined protocol on exactly how the genotype information will be used to guide dosage and/or drug selection. It is important to emphasise that it is a particular pharmacogenomic protocol that must be validated prior to clinical implementation, not simply the association between the SNP and a given drug response. Collecting data and retrospectively optimizing a protocol gives a biased representation of the pharmacogenomic test’s utility. The hypothesis (ie, the pharmacogenomic protocol) should be completely defined prior to testing its validity. This is standard practice in clinical medicine and it is imperative that pharmacogenomics also adhere to good scientific and clinical practice.
Prospectively collect data to assess the cost-effectiveness of a specific pharmacogenomic protocol vs. the current standard practice: preferably a randomized controlled study
Cost-effectiveness of a pharmacogenomics test is generally enhanced in situations in which the association between genotype and clinical phenotype is strong and well established (CitationVeenstra et al 2000). Many reviews on clinical pharmacogenomics point to the lack of good evidence as a major factor underlying slow acceptance of many pharmacogenomic associations (CitationEnsom 2001; CitationMeyer 2004; CitationGardiner and Begg 2005).
In many cases the evidence for a genotype-phenotype association comes from a retrospective study in which multiple hypotheses are tested. Prior to clinical implementation, large prospective hypothesis-driven studies in multiple ethnic groups are required. If possible, it is highly preferable to randomize patients to either (i) pharmacogenomic guided treatment or (ii) the current standard method of drug and dose selection. This will allow for the most valid assessment of whether the particular pharmacogenomic protocol should supersede current clinical practice.
Analysis of these prospective studies is complicated. Systematic methods for assessing the costs and benefits associated with pharmacogenomic tests are available, but generally underused (CitationPhillips et al 2003). There are strong economical pressures in health care and as a minimum a pharmacogenomic protocol must be shown to have greater overall benefit than cost. The costs and benefits associated with different health alternatives can be complex and difficult to compare. Cost-benefit analysis and cost-effectiveness analysis are frameworks that are widely used to aid decisions regarding the use of health technologies (CitationVeenstra et al 2000; CitationPhillips et al 2003). Should the developers of pharmacogenomic tests wish to compete for finite healthcare funding, it will be necessary to demonstrate the worth of a specific pharmacogenomic protocol using these standard frameworks.
Engagement with and endorsement by the appropriate stakeholders for use of pharmacogenomic tests
Pharmacogenomics is unlikely to flourish unless all stakeholders are involved and their concerns acknowledged. As a general rule, products of new technology tend to progress through a technology adoption life cycle, during which they sequentially penetrate different segments of the required uptake chain. These segments can differ dramatically in their preparedness to adopt and therefore require unique strategies in order to ensure uptake. The delivery of optimal drug therapy involves many groups, which makes this issue particularly complicated. Patients and their families, medical practitioners, allied health groups, pathology providers, government regulatory agencies, groups providing therapeutic advice, ethicists and health funding agencies can all be identified as stakeholders in pharmacogenomics. Each of these groups is likely to have different concerns and barriers to their involvement in pharmacogenomics. Furthermore, the concerns of different groups may not coincide.
The lay view of pharmacogenomics indicates that there is great concern that drugs developed within a pharmacoge-nomic framework would be more expensive (CitationAlmarsdottir et al 2005). There is also general concern that pharmacoge-nomics would lead to further inequalities between rich and poor countries and within societies (CitationAlmarsdottir et al 2005). A failure to address these identified lay concerns will inevitably limit uptake of pharmacogenomic tests.
Food and drug administration (FDA)
There is clear evidence that over recent times, the FDA is working to ensure a regulatory framework that is cognizant of pharmacogenomics. Clearly, pharmacogenomic tests with FDA approval are likely to generate confidence in the utility of the test and as a result, this is likely to put positive pressure on their clinical uptake. There are also indications that the FDA is encouraging pharmaceutical companies to conduct pharmacogenomic research during the development of new drugs and voluntarily submit the resulting data (CitationHampton 2005). This is more likely for adverse events. The current financial models for drug development ensure that the developers of new drug products will generally prefer to maximize the population of people approved to use a given drug. As a result, it can be assumed that drug developers will be concerned that pharmacogenomic information could be used to keep a drug off the market or limit its use to a specific patient sub-population (CitationHampton 2005). The reduction in gross sales may be further exacerbated if the cost of the pharmacogenomic test becomes closely linked with the cost of the drug (CitationDanzon and Towse 2002).
It is clear that the FDA plans to play an important role in regulating pharmacogenomic tests. Indeed, recently the FDA delayed the sale of a pharmacogenomics test for genetic variations influencing drug metabolism, citing the need for appropriate pre-market determination by the FDA (CitationHampton 2005).
Implement pharmacogenomic test in a staged manner
Staged implementation
Successful development of a widely used pharmacogenomic test is most likely to proceed in a staged manner. Such an approach relies on moving from the easiest site of implementation to the most difficult and also, from the smallest target group to the largest. Thus, it is likely that successful pharmacogenomic development will begin in the setting of a major teaching hospital, progress through other hospital environments and then to broader medical environments.
Pharmacogenomic infrastructure
A number of the barriers to clinical pharmacogenomics are related to infrastructure. This includes the ethical and political frameworks, the re-education of many different groups in the health sector, and the physical infrastructure for the pharmacogenomic tests. These particular barriers are not specific to particular pharmacogenomic tests. Once the general infrastructure is set up for a single pharmacogenomic test, further tests will require only relatively minor adjustments to the infrastructure. The problem is making the case for the initial investment.
Education
Education of health professionals is likely to pose a considerable barrier to the widespread uptake of pharmacogenomics. For example, surveys of tertiary pharmacy institutions in the US (CitationBrock et al 2002) and community pharmacists (CitationSansgiry and Kulkarni 2003) show that contemporary genomic-based education is generally lacking across the pharmacy profession. In light of the perceived lack of pharmacogenomic education among various health care professionals, the International Society of Pharmacogenomics (ISP) has recently issued a call for pharmacogenomics to be incorporated immediately into the core curricula of medical, pharmaceutical and health science programs (CitationGurwitz et al 2005).
It can however be argued that pharmacogenomics is intuitively very similar to traditional therapeutic drug monitoring (TDM) with which most medical practitioners and pharmacists feel comfortable. In the case of TDM, drug measurements are used to guide optimal dosage selection. In the case of pharmacogenomics, biological measurements are used to guide optimal drug selection and dosage. It is the nature of the measurements that pose the educational challenges. It is true that genotype data is different from most other test results with which clinicians are familiar. Those without experience in dealing with genotypes and haplotypes can easily find this information intimidating, confusing and frustrating. There are also some idiosyncrasies of this data that need to be understood for proper interpretation. However, for the most part, a basic understanding of the concepts can be quickly attained. While the facility that performs the pharmacogenomic tests would give some interpretation of the results, it is imperative to have someone physically present in the clinical environment with the depth of knowledge to ensure the data is used properly. This person could also serve in an educational role. The clinical pharmacist seems best suited to this role. A clinical pharmacist well versed in the interpretation of pharmacogenomic tests would take a large burden off the physicians and nurses, and facilitate a smooth transition to using these tests. In addition, it is important that the clinical pharmacist be able to integrate genotype information into existing knowledge of other factors that can significantly affect pharmacotherapy (eg, food, concomitant medications, pathological conditions, gender, age, weight, environment) in order to develop the optimal treatment plan for each individual. It is unlikely that current training during pharmacy degrees will be sufficient for this purpose, and post-graduate pharmacogenomics courses will need to be developed to enable efficient and accurate interpretation of the test results.
Ethical issues
There are many expressed concerns about the ethical issues surrounding use of genetic information in medicine. In the context of pharmacogenomics, this is probably of greater importance to genetic tests that are predictive of efficacy rather than adverse events. As ethical issues in pharma-cogenomics have begun to be dissected, there is recognition that the ethical issues related to pharmacogenomics are similar in character to ethical issues raised by other clinical circumstances. However, the prospect of individualised or personalised medicine brings many of these issues into clear focus. These ethical issues generally fall into three broad categories: the equitable provision of healthcare, the possibility that genetic variants may track with race or ethnicity, and the questions of consent, access and privacy surrounding pharmacogenomic information.
It is true that in many clinical settings, there is a general stigma associated with genetic testing. Even when the genetic test will give no more information than an equivalent phenotyping assay, most people will probably feel more comfortable with a phenotyping alternative. Alternative technologies such as metabonomics will not face this stigma. Metabonomic research will result in associations between (generally endogenous) chemical concentrations in biofluids (most commonly blood or urine) and the drug phenotype. Once the chemical is identified, a simple chemical assay for the specific chemical can be developed using technologies that are standard in pathology labs. Although not discovered using metabonomics, the measurement of serum creatinine concentrations to adjust the dosage of many drugs is a prime example of the type of tests this approach will result in. Tests for serum creatinine are undertaken routinely throughout the world without many of the concerns that surround genetic based tests.
Physical infrastructure
This mainly consists of instrumentation for genotyping. As costs for genotyping decrease, the greater the chance that pharmacogenomic tests with small to moderate utility will prove cost-effective. In the future it may be possible to undertake genotyping at point of care which will reduce costs associated with requiring a follow-up consultation (CitationPhillips et al 2001). A current concern is the wide variation in the cost of genotyping (CitationGardiner and Begg 2005), which is likely to be partially responsible for variable uptake of some well studied pharmacogenomic tests such as that for thiopurine methyltransferase (TMPT). It has been suggested that widespread clinical uptake of trastuzumab was enabled by the approval of a standardized, simple and commercially available method. Whether this extrapolates to other phar-macogenomics test is uncertain due to the abundance of differing technologies for genotyping.
Synergy with information technology
As information technology is further integrated into the day-to-day working of the health professions, new opportunities for pharmacogenomics will arise. Integrating complex pharmacogenomic data with conventional factors places an increased burned on health professions involved with optimizing the pharmacotherapy of individual patients. It is expected that decision support software will be developed to aid in this process. This will become especially important for the multi-gene pharmacogenomic protocols (potentially incorporating other technologies) expected in the future.
Conclusion
The greater the barriers to the clinical adoption of pharma-cogenomics, the greater the evidence and size of the improvement in clinical outcome required. The problem to date is that the evidence and importance of most pharmacogenomic associations are not sufficient to overcome the barriers to the clinical implementation. Fortunately, a number of barriers to clinical utilization are not specific to particular pharma-cogenomic tests and are likely to be significantly reduced with time. For example, technological advances are likely to improve the availability and reproducibility and decrease the difficulty and cost of genotyping. In addition, some barriers are one-off costs. Predominant among these are pharmacoge-nomic education of clinicians and the general public, and the development of general ethical protocols and financial frameworks for clinical pharmacogenomics.
Most pharmacogenomic associations do not proceed past the second step on the road to clinical implementation. Why this is the case is not clear. Perhaps it is indifference following discovery of the initial SNP-phenotype association. Perhaps it is a lack of knowledge of how to proceed. Perhaps, because the SNP-association studies are not strong enough to be cost-effective.
It is obvious that much room remains to improve the use of pharmacotherapy. However, it is not apparent that pharmacogenomics as it currently stands will be able to significantly improve pharmacotherapy for the majority of drugs. There are many critical factors in to which pharma-cogenomics can provide little insight. Chief among these are environmental influences (including interactions with foods, natural therapies and other drugs) and behavioral factors that influence compliance with treatment regimens (CitationEnsom 2001). Together, these are probably responsible for a significant proportion of the detrimental inter-individual variability resulting in poor efficacy and toxicity. It is likely that complementary technologies, such as metabonomics will be able to compensate for some limitations of genotype-phenotype associations.
As infrastructure becomes better established and phar-macogenomics becomes thought of as simply another test that can guide therapy, rather than a radical technology that raises concern it is likely that the balance between costs and benefits will shift in favor of clinically implementing pharmacogenomic protocols.
References
- AlmarsdottirABBjornsdottirITraulsenJMA lay prescription for tailor-made drugs - focus group reflections on pharmacogenomicsHealth Policy2005712334115607385
- AlmueteVIDrug therapy and pharmacogenomics. AphA 2000 – American Pharmaceutical Association Annual Meeting Available from URL: http://www.medscape.com/viewarticle/419158
- BrockTPFaulknerCMWilliamsDMContinuing-education programs in pharmacogenomics for pharmacistsAm J Health-Syst Pharm200259722511977857
- DanzonPTowseAThe economics of gene therapy and of pharmacogeneticsValue Health2002551311873384
- BuckleyPMcKinnonRAPharmacogenomics, ethics and the communityAustralian Pharmacist200423224
- CarlsonCSEberleMARiederMJSNP and haplotype variation in the human genomeMutat Res2003526536112714183
- CarlsonCSEberleMARiederMJAdditional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humansNature Genet2003335182112652300
- CarlsonCSEberleMARiederMJSelecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibriumAm J Hum Genet2004741062014681826
- CrawfordDCCarlsonCSRiederMJHaplotype diversity across 100 candidate genes for inflammation, lipid metabolism, and blood pressure regulation in two populationsAm J Hum Genet2004746102215015130
- EnsomMHChangTKPatelPPharmacogenetics: the therapeutic drug monitoring of the future?Clin Pharmacokinet20014078380211735602
- GardinerSJBeggEJPharmacogenetic testing for drug metabolizing enzymes: is it happening in practice?Pharmacogenet Genomics200515365915864139
- GurwitzDLunshofJEDedoussisGPharmacogenomics education: international society of pharmacogenomics recommendations for medical, pharmaceutical, and health schools deans of educationPharmacogenomics J200554221515852053
- HamptonTFDA seeks genome-based drug dataJAMA-J Am Med Assoc2005291323
- LazarouJPomeranzBHCoreyPNIncidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studiesJAMA-J Am Med Assoc199827912005
- LeeSSRacializing drug design: implications of pharmacogenomics for health disparitiesAm J Public Health2005952133816257939
- LewisBPShihIHJones-RhoadesMWPrediction of mammalian microRNA targetsCell20031157879814697198
- McGovernDPvan HeelDANegoroKFurther evidence of IBD5/CARD15 (NOD2) epistasis in the susceptibility to ulcerative colitisAm J Hum Genet2003731465614655097
- MeyerUAPharmacogenetics - five decades of therapeutic lessons from genetic diversityNature Rev Genet200456697615372089
- NebertDWJorge-NebertLVesellESPharmacogenomics and “individualized drug therapy”: high expectations and disappointing achievementsAm J Pharmacogenomics200333617014672516
- NebertDWMenonAGPharmacogenomics, ethnicity, and susceptibility genesPharmacogenomics J20011192211913722
- NebertDWVesellESAdvances in pharmacogenomics and individualized drug therapy: exciting challenges that lie aheadEur J Pharmacol20045002678015464039
- PhillipsKAVeenstraDLOrenEPotential role of pharma-cogenomics in reducing adverse drug reactions: a systematic reviewJAMA-J Am Med Assoc200128622709
- SalisburyBAPungliyaMChoiJYSNP and haplotype variation in the human genomeMutat Res2003526536112714183
- SansgirySSKulkarniASThe human genome project: assessing confidence in knowledge and training requirements for community pharmacistsAm J Pharm Edu200367219
- ShahJCriteria influencing the clinical uptake of pharmacogenomic strategiesBr Med J20043281482615205293
- SchmeddersMFeuersteinGKollekRProspects and limits of pharmacogenetics: The thiopurine methyl transferase (TMPT) experienceAm J Pharmacogenomics200331495512814323
- SchramkeVAllshireRHairpin RNAs and retrotransposon LTRs effect RNAi and chromatin-based gene silencingScience200330110697412869699
- StoreyJDTibshiraniRStatistical significance for genomewide studiesProc Natl Acad Sci USA20031009440512883005
- TateSKGoldsteinDBWill tomorrow’s medicines work for everyone?Nature Genet20043611 SupplS344215508001
- TuckerGPharmacogenetics-expectations and realityBr Med J20043294615231589
- Van AkenJPhillipsKAVeenstraDAn introduction to cost-effectiveness and cost-benefit analysis of pharmacogenomicsPharmacogenomics20034231912718713
- The International Hap Map ConsortiumThe international HapMap projectNature20034267899514685227
- The International Hap Map ConsortiumA haplotype map of the human genomeNature2005437129932016255080
- VeenstraDLHigashiMKPhillipsKAAssessing the cost-effectiveness of pharmacogenomicsAAPS Pharmsci20002E2911741245