Abstract
The development of nonsteroidal anti-inflammatory drugs (NSAIDs) selective for cyclooxygenase (COX)-2 (named coxibs) has been driven by the aim of reducing the incidence of serious gastrointestinal (GI) adverse events associated with the administration of traditional (t) NSAIDs – mainly dependent on the inhibition of COX-1 in GI tract and platelets. However, their use has unravelled the important protective role of COX-2 for the cardiovascular (CV) system, mainly through the generation of prostacyclin. In a recent nested-case control study, we found that patients taking NSAIDs (both coxibs and tNSAIDs) had a 35% increase risk of myocardial infarction. The increased incidence of thrombotic events associated with profound inhibition of COX-2-dependent prostacyclin by coxibs and tNSAIDs can be mitigated, even if not obliterated, by a complete suppression of platelet COX-1 activity. However, most tNSAIDs and coxibs are functional COX-2 selective for the platelet (ie, they cause a profound suppression of COX-2 associated with insufficient inhibition of platelet COX-1 to translate into inhibition of platelet function), which explains their shared CV toxicity. The development of genetic and biochemical markers will help to identify the responders to NSAIDs or who are uniquely susceptible at developing thrombotic or GI events by COX inhibition. We will describe possible strategies to reduce the side effects of etoricoxib by using biochemical markers of COX inhibition, such as whole blood COX-2 and the assessment of prostacyclin biosynthesis in vivo.
Nonsteroidal antiinflammatory drugs (NSAIDs) are commonly used in the general population for treating pain and inflammatory conditions (CitationBurke et al 2006). They comprise traditional (t) NSAIDs and NSAIDs selective for cyclooxygenase (COX)-2 (named coxibs) which were developed to reduce the risk of serious gastrointestinal (GI) complications – dependent, at least in part, on the inhibition of COX-1 (CitationFitzGerald and Patrono 2001). The therapeutic effects (analgesic and anti-inflammatory) of NSAIDs, both traditional and coxibs, are mostly due to the inhibition of COX-2-dependent prostanoids (). In placebo-controlled randomized clinical trials (RCTs), coxibs (rofecoxib [Vioxx®], celecoxib [Celebrex®, Artilog®, Solexa®, Artrid®] and valdecoxib [Bextra®]) were associated with an increase in the relative risk (RR) of cardiovascular (CV) events by 1- to 2.7-fold (CitationOtt et al 2003; CitationBresalier et al 2005; CitationSolomon et al 2005; CitationPfizer 2005; CitationNussmeier et al 2005). However, the results of observational studies and a meta-analysis of data derived from trials with coxibs have shown that the CV hazard is not restricted to NSAIDs selective for COX-2 but also applies to some tNSAIDs, such as diclofenac (CitationHernandez-Diaz et al 2006; CitationKearney et al 2006). In a recent nested-case control study, we found that patients taking NSAIDs (both coxibs and tNSAIDs) had a 35% increased risk of myocardial infarction (CitationPatrignani et al 2008a). Clinical results suggest that the CV hazard associated with the administration of NSAIDs is dose-dependent (CitationPatrignani et al 2008a; CitationSolomon et al 2008). In addition, the genetic background of the individual may play a role in increased susceptibility to inhibition of NSAIDs (CitationArehart et al 2008). To limit the possible detrimental effects, associated with the administration of this efficacious class of drugs, is necessary to develop strategies of risk management through the identification of genetic and biochemical markers to select the responders to NSAIDs or who are uniquely susceptible to developing thrombotic or GI events by COX inhibition.
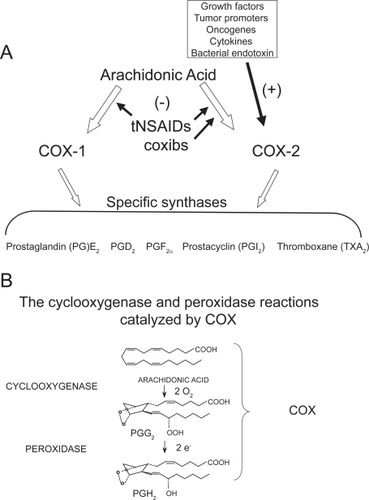
Figure 1 Pathways of prostanoid biosynthesis. (A) Prostanoids (PGE2, PGD2, PGF2α, PGI2, TXA2) are produced by COX-1 and COX-2 and specific synthases; (B) PGH2, generated by cyclooxygenase and peroxydase activity of COX, is then converted to prostanoids by the activity of different synthases.
Differential COX pathways are effective in health and disease
Biology of COX-1 and COX-2
Prostanoids are lipid autacoids – including prostaglandin (PG) E2, PGF2α, PGD2, prostacyclin (PGI2), and thromboxane(TX) A2 – that are immediately released outside the cell after intracellular biosynthesis and modulate a wide variety of physiologic and pathologic processes via the interaction with specific receptors expressed mostly on the surface of target cells (CitationNarumiya et al 1999; CitationBreyer et al 2001).
Under normal physiologic conditions, prostanoids play an essential homeostatic role in the GI cytoprotection, hemostasis, renal physiology, gestation, and parturition (CitationFunk 2001; CitationPatrono et al 2001; CitationFitzGerald 2003). Moreover, they play important roles in pathophysiologic processes such as inflammation, cancer, and thrombosis (CitationFunk 2001; CitationPatrono et al 2001; CitationFitzGerald 2003). Two isoforms of COX (COX-1 and COX-2) have been cloned and characterized (CitationSimmons et al 2004). COX-1 and COX-2 are the products of different genes. COX-1 is considered a “housekeeping gene” by virtue of constitutive low-levels of expression in most cell types. However, high levels of constitutive expression of COX-1 have been detected in the stomach, platelets, and the kidney. In addition, COX-1 can be regulated during development (CitationRocca et al 1999). In contrast, the gene for COX-2 is a primary response gene with many regulatory sites; thus, COX-2 expression can be rapidly induced by bacterial endotoxin (LPS), cytokines, such as interleukin (IL)-1β and tumor necrosis factor-α, growth factors, and the tumor promoter phorbol myristate acetate (PMA) (reviewed by CitationKang et al 2007). However, COX-2 is constitutively expressed in some cells in lung (CitationAsano et al 1996), brain (CitationYamagata et al 1993; CitationKaufmann et al 1997), kidney (CitationHarris et al 1994), pancreatic β-cells (CitationRobertson 1998), and GI carcinomas (CitationRistimaki et al 1997; CitationShao et al 2000; CitationCarlson et al 2003).
COX-1 and COX-2 share the same catalytic activities: the cyclooxygenase which oxidizes arachidonic acid (AA) to PGG2 and the peroxidase which reduces PGG2 to the unstable endoperoxide PGH2, precursor of prostanoids (CitationSmith et al 2000) (). Although the peroxidase reaction is considered the second step in the formation of PGH2, the COX reaction is absolutely dependent on peroxidase activity for its activation (CitationMalkowski et al 2000). However, the two COX-isozymes require different levels of hydroperoxides to initiate cyclooxygenase catalysis, ie, COX-2 requires considerably lower levels of hydroperoxides than COX-1 (CitationKulmacz and Wang 1995; CitationMarnett et al 1999). Elegant work performed by Funk’s group reveals that COX-1 can partially compensate for COX-2 function but that this is limited by the differential ability of these two isoforms to metabolize low concentrations of arachidonate (CitationYu et al 2007). In fact, COX-2 catalysis occurs at lower levels of free AA than COX-1 (CitationSwinney et al 1997).
These differences in the regulation of COX-1 and COX-2 catalysis contribute to the development of cell-specific pathways of prostanoid biosynthesis dominated by one or the other enzyme in vivo even in the presence of the concurrent expression of the 2 isozymes (CitationCapone et al 2007). As an example, in endothelial cells where different antioxidant defence systems are operative to maintain a reductive cytosolic environment, COX-2 is the dominant pathway turned on to generate prostacyclin, an important anti-atherogenic and anti-thrombotic mediator (CitationGrosser et al 2006) ().
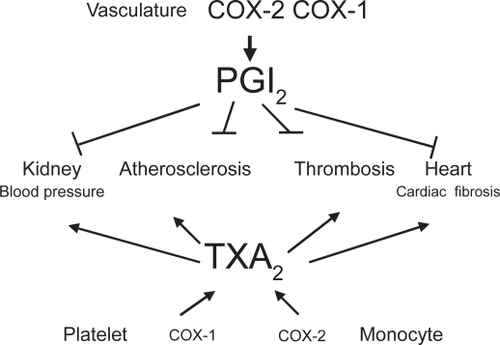
Figure 2 Vascular prostacyclin counteracts the cardiovascular effects of platelet TXA2.
COX-2 is the major pathway in pain
Other than in inflammation and pyresis, COX-2 plays an important role in nociception (CitationVane et al 1998). Noxious stimuli (eg, physical, chemical, thermal) in peripheral tissues cause damage and the inflammatory response leads to the release of pain-producing substances that activate nociceptors on the terminals of sensory nerve fibers. This pain is the hallmark of protective response to adverse stimuli and subsides with removal of the stimulus. In this context, the induction of COX-2, leading to enhanced prostanoid release, translates into sensitization of peripheral nociceptor terminals and produces localized pain hypersensitivity. In fact, prostanoids increase neuronal activity in nociceptive nerve fibers by raising cAMP levels and lowering the activation threshold for opening of tetrodotoxin-resistant sodium channels in the neuronal membrane. PGE2 and prostacyclin, produced in the peripheral terminals of sensory nerve endings, are hyperalgesic and enhance nociception produced by other mediators (such as bradykinin) (CitationMurata et al 1997; CitationNakao et al 2007). Moreover, peripheral inflammation also generates pain hypersensitivity in neighboring uninjured tissue (secondary hyperalgesia), because of increased neuronal excitability in the spinal cord (central sensation) and a syndrome comprising diffuse muscle and joint pain, fever, lethargy, and anorexia (CitationSamad et al 2001). NSAIDs play an antinociceptive action by acting both at peripheral sites and at central sites mainly through COX-2 inhibition. Both COX-1 mRNA and COX-2 mRNA as well as COX-1 and COX-2 proteins are expressed constitutively in the spinal cord but only COX-2 can be induced in response to exogenous and endogenous cytokines (CitationBeiche et al 1996; CitationInoue et al 1999; CitationTonai et al 1999). Like nociceptive pain, hyperalgesia is linked to an adverse stimulus and diminishes with healing and decreased inflammation. Prolonged acute pain and hyperalgesia, however, can evolve into chronic pain.
The results of RCTs showing a comparable pain relief by NSAIDs selective for COX-2 and tNSAIDs both in acute clinical pain models (for example, oral surgery) and chronic clinical pain models (such as osteoarthritis [OA] and inflammatory arthropaties), support a major role of inhibition of COX-2 in their clinical efficacy (CitationFitzgerald and Patrono 2001; CitationSciulli et al 2005). However, it should be pointed out that often the RCTs, comparing tNSAIDs and coxibs, had inadequate power to detect small differences possibly associated with inhibition of COX-1. Mostly, they were sized to detect equivalence, not superiority, of either (CitationPatrono et al 2001).
COX-1 is the major pathway in GI protection
Prostanoids (PGE2 and prostacyclin) are key mediators to maintain gastric mucosal blood flow and increase protective mucus as well as bicarbonate production (CitationWilson 1991). Several lines of evidence show that prostanoids produced in normal GI tissue which are required for normal physiological functioning are derived from the COX-1 isoform (CitationKargman et al 1996). However, upregulation of COX-2 in the margin of healing ulcers and evidence from the use of COX inhibitors in rodents has raised concerns that COX-2 might be involved in the healing of existing ulcers (CitationShigeta et al 1998; CitationTakahashi et al 1998). However, COX-2 up-regulation in the GI tract has been observed in cancer development (CitationRomano et al 1998; CitationPrescott and Fitzpatrick 2000; CitationNardone et al 2004; CitationWang and Dubois 2006).
COX-1 is involved in TXA2-dependent platelet function
Platelets generate TXA2 from AA through the activity of COX-1 (CitationPatrignani et al 1999). TXA2 generated in response to a variety of stimuli (for example, collagen, thrombin, and adenosine diphosphate) participates in the control of hemostasis through the induction of irreversible platelet aggregation triggered by the interaction with G-protein–coupled receptors, the TXA2 receptors (CitationAlfranca et al 2006). Recent findings show that tiny concentrations of TXA2 can cause platelet activation (CitationMinuz et al 2002; CitationPulcinelli et al 2005; CitationMinuz et al 2006). Thus, 10 nM of the TXA2 mimetic U46619 induces platelet adhesion and shape change (CitationMinuz et al 2002), and in the presence of a subthreshold concentration of collagen it causes platelet aggregation (CitationPulcinelli et al 2005). Moreover, TXA2 causes vasoconstriction and vascular proliferation; thus, it is involved in atherogenesis and increase in blood pressure () (CitationGrosser et al 2006).
COX-2-dependent prostacyclin is the major pathway in vascular protection
In vascular endothelial cells, COX-2 plays a dominant role in producing important prostanoids to regulate the functions of underlying vascular smooth muscle cells and circulating cells. Although endothelial cells may generate a different array of the prostanoids PGD2, PGE2, and prostacyclin along the vascular beds, there is robust evidence that prostacyclin is the dominant prostanoid produced in the macrocirculation (CitationMoncada et al 1977; CitationGrosser et al 2006).
Prostacyclin potently inhibits aggregation of platelets induced by TXA2 and other agonists, vascular smooth muscle cell proliferation and vascular tone, leukocyte–endo-thelial cell interactions, and cholesteryl ester hydrolase (CitationGrosser et al 2006). As shown in , most of the deleterious effects of TXA2 in the CV system are countered by prostacyclin, mainly through the activity of COX-2. Thus, inhibition of vascular COX-2 leads to loss of the protective function associated with prostacyclin. Recently, it has been reported an antioxidant role for prostacyclin through the induction of hemoxygenase-1 (CitationEgan et al 2004). For all these biological actions, prostacyclin has on the distinctive features of a cardioprotective mediator. Studies in knockout (KO) mice for prostacyclin receptor (IP) and the recent findings of acceleration of CV disease in humans by a dysfunctional IP mutation convincingly support the protective role of prostacyclin for the CV system (CitationArehart et al 2008; CitationPatrignani et al 2008b).
Biochemical biomarkers of COX inhibition
NSAIDs are distinguished on the basis of their COX-isozyme selectivity in vitro, described as the ratio of the concentrations required to inhibit the activity of the isozymes by 50% (IC50 for COX-1/IC50 for COX-2) (). The assessment of COX-1/COX-2 ratios in vitro describes an experimental COX-isozyme selectivity which mirrors the chemical features of the different NSAIDs. This is assessed using the human whole blood assays which evaluate the effects of drugs on platelet COX-1 and monocyte COX-2 activities (CitationPatrono et al 1980; CitationPatrignani et al 1994). They are capacity indexes of COX-isozyme activities to generate prostanoids from endogenous sources of AA and their pharmacological inhibition is not influenced by different pathological conditions. These assays are based on the measurement of PGE2 production, in response to lipopolysaccharide (LPS) added to heparinized human blood samples for 24 h which reflects the time-dependent induction of COX-2 in circulating monocytes (CitationPatrignani et al 1994). The parallel measurement of TXB2 production during whole blood clotting is used as an index of platelet COX-1 activity (CitationPatrono et al 1980). When the COX-1/COX-2 IC50 ratio is higher than 1 the drug is more potent in inhibiting COX-2. When the ratio is approximately 1, the drug is a non-selective inhibitor of both COX-1 and COX-2. When the ratio is lower than 1, the drug is more potent towards COX-1. We assessed COX-1/COX-2 IC50 values of several COX inhibitors. Interestingly, we have found that the biochemical selectivity is a continuous variable preventing the separation of tNSAIDs from coxibs (CitationPatrignani et al 2008c). However, we can identify some drugs more selective for COX-1 than COX-2 (such as aspirin, naproxen and ibuprofen) from the wide cluster of agents more selective for COX-2 than COX-1. This includes: i) piroxicam and indomethacin which are approximately 3-fold more selective for COX-2; ii) etodolac, meloxican, diclofenac, celecoxib, and valdecoxib which are 6- to 60-fold more selective for COX-2; iii) the highly selective COX-2 inhibitors such as, etoricoxib, rofecoxib, and lumiracoxib which have a COX-1/COX-2 IC50 ratio higher than 100 ().
Figure 3 Assessment of COX-isoform selectivity in whole blood in vitro (A) and ex vivo (B).
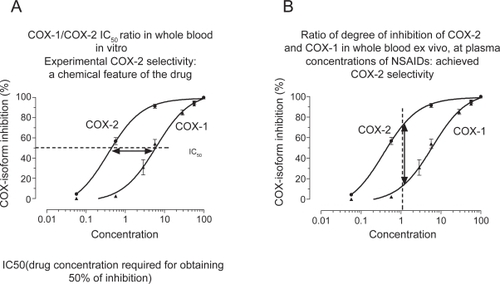
Table 1 Biochemical selectivity, assessed as COX-1/COX-2 IC50 values of several COX inhibitors (derived from CitationPatrignani et al 2008a)
The whole blood assays also enable estimation of achieved COX-isozyme selectivity in humans, which is the ratio of isozyme inhibition at a given plasma concentration (). Importantly, achieved selectivity of NSAIDs varies as a consequence of the dose administered.
There is evidence that the whole blood assays for COX-2 and COX-1 may be candidate surrogate end-points of efficacy and GI toxicity of NSAIDs, respectively. It has been shown that COX-2 inhibition, as determined by PGE2 levels in LPS-stimulated whole blood, can be used as a marker to predict drug efficacy in humans (CitationHuntjens et al 2005). In fact, (concentration required to inhibit the activity of IC80 COX-2 by 80% in vitro) values have been found to correlate directly with the analgesic/anti-inflammatory plasma concentrations of different COX inhibitors (CitationHuntjens et al 2005). Thus, COX-2 inhibition, by assessing PGE2 levels in the whole blood assay in vitro and ex vivo, can be used as a marker to predict drug effects (analgesia, antiinflammatory effects) in humans. Similarly, platelet COX-1 inhibition, by assessing TXB2 levels in the whole blood assay in vitro and ex vivo, can be used as a marker to predict toxicity or efficacy (GI toxicity or antiplatelet effects) in humans by NSAIDs. In fact, CitationCryer and Feldman (1998) reported that the inhibitory effects of tNSAIDs on gastric PGE2 synthesis correlate with COX-1 inhibitory potency in clotting blood.
Now it is critical to establish the CV safety profile of specific individual NSAIDs, be it tNSAIDs or coxibs, and to find valid surrogate biochemical predictors of that risk.
Studies of clinical pharmacology have shown discordant functional implications of inhibition of COX-1 and COX-2. A linear relationship exists between the extent of inhibition of COX-2 and that of prostacyclin in vivo (CitationPatrignani et al 2008a). In contrast, inhibition of TXA2-dependent platelet function in vivo occurs when platelet COX-1-dependent capacity to synthesize TXA2 (as assessed by measuring serum TXB2 levels) is reduced ≥95% (CitationReilly and FitzGerald 1987). In fact, a non-linear relationship of inhibition of platelet TXA2 generation with inhibition of TXA2-mediated platelet aggregation has been found (CitationSciulli et al 2006). This is explained by the fact that even tiny concentrations of TXA2 may activate the platelets. CitationMinuz et al (2006) have shown that low concentrations of TXA2 activate the tyrosine-kinase-based signaling pathway which may translate into a full platelet activation in the presence of weak platelet agonists or subthreshold concentrations of stronger agonists.
These findings lead to the concept of functional COX-2 selectivity by NSAIDs, ie, inhibition of COX-2 in the presence of an insufficient reduction of platelet COX-1 activity to translate into inhibition of platelet function. It has to be pointed out that most NSAIDs are functionally selective for COX-2 at therapeutic doses similarly to coxibs. The only tNSAID that has been shown to be functionally non-selective for the platelet is naproxen at high doses, in some individuals (CitationCapone et al 2004). This depends on the fact that naproxen usually administered at high doses bid is more potent for COX-1 than COX-2, and it is characterized by a pharmacokinetic half-life of approx 14 h (CitationBurke et al 2006). Thus, naproxen is associated with profound inhibition of prostacyclin but the possible CV hazard associated with this effect may be mitigated by a parallel profound and persistent suppression of platelet TXA2 (CitationCapone et al 2004), which translates into a small CV protection or neutral effect (CitationHernández-Díaz et al 2006; CitationKearney et al 2006).
Recent findings suggest that for functional non-selective NSAIDs the extent of inhibition of COX-2-dependent pros-tacyclin may predict the increased incidence of myocardial infarction (MI) (CitationPatrignani et al 2008a). Individual NSAIDs with a degree of COX-2 inhibition <90% at therapeutic concentrations presented a RR of 1.18 (95% CI, 1.02–1.38) while those with a greater COX-2 inhibition had a RR of 1.60 (95% CI, 1.41–1.81).
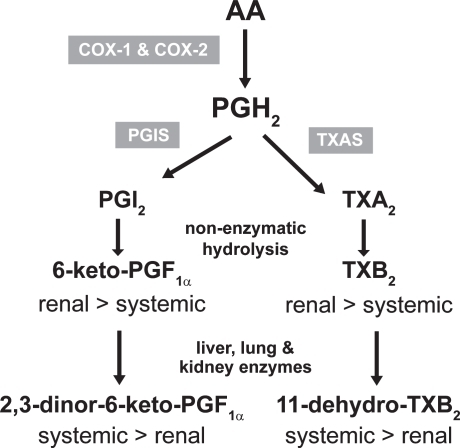
Thus, it has been proposed that the assessment of whole blood COX-2 ex vivo – alone or in combination with the measurement of urinary levels of 2,3-dinor-6- keto-PGF1α [a biomarker of prostacyclin biosynthesis in vivo ()] – in association with genetic biomarkers (such as polymorphisms in the prostacyclin receptor IP) (CitationArehart et al 2008) may be surrogate end-points of CV hazard by pharmacological inhibition of COX-2 (CitationPatrignani et al 2008b).
Figure 4 Biosynthesis and metabolism of prostacyclin (PGI2) and thromboxane (TX) A2.
Etoricoxib
Pharmacology
The NSAID selective for COX-2 etoricoxib, 5-chloro-6′-methyl-3-[4-(methylsulfonyl) phenyl]-2,3′-bipyridine () (CitationRiendeau et al 2001) (Arcoxia®, Tauxib®, Algix®) has been approved in Europe, Latin America, and Asia-Pacific region for the treatment of OA, rheumatoid arthritis (RA), ankylosing spondylitis, acute gouty arthritis, acute pain, and chronic musculoskeletal pain, at doses ranging between 60 and 120 mg daily depending on the different indications.
Figure 5 Chemical structure of etoricoxib: bipyridine with a methyl sulfone side chain 5-chloro-6′-methyl-3-[4-(methylsulfonyl) phenyl]-2,3′-bipyridine.
![Figure 5 Chemical structure of etoricoxib: bipyridine with a methyl sulfone side chain 5-chloro-6′-methyl-3-[4-(methylsulfonyl) phenyl]-2,3′-bipyridine.](/cms/asset/ca8a6c32-92ca-4451-8106-28d279b3c694/dtcr_a_3209_f0005_b.jpg)
As shown in , etoricoxib is 162-fold more potent towards COX-2 than COX-1 in the whole blood assay in vitro. The pharmacokinetics features of etoricoxib are reported in (CitationAgrawal et al 2003). Importantly, it has a long half-life of 22 h and is metabolized via 6’-methyl hydroxylation in human liver microsomes. The reaction is catalyzed by a number of P450 forms, although CYP3A4 accounts for the majority (40%–90%) of the activity. The remainder of the activity is equally divided between a number of other P450s (eg, CYP2D6, CYP2C9, CYP1A2, and possibly CYP2C19). In this regard, the P450 reaction phenotype of etoricoxib is unique and differs from that of other COX inhibitors (CitationKassahun et al 2001).
Figure 6 COX-2 selectivity of etoricoxib in human whole blood. (A) Concentration-response curves assessed in vitro; (B) degree of inhibition of COX-2 and COX-1 by peak plasma concentrations obtained after dosing with etoricoxib 5, 10, 20, 40, and 120 mg; (C) pharmacokinetics features of etoricoxib.
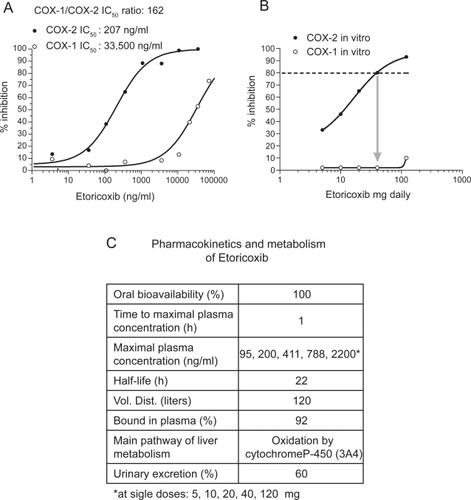
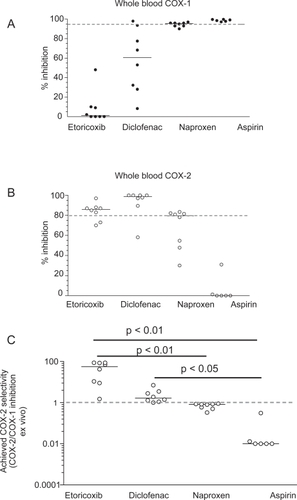
As shown in , etoricoxib administered up to 120 mg is a selective COX-2 inhibitor. This result has been obtained by the extrapolation of COX-1 and COX-2 inhibition in vitro in the human whole blood assays by the maximal plasma concentration achieved after the administration of etoricoxib at single doses of 5, 10, 20, 40, 120 mg. Interestingly, an inhibition of COX-2 by 80% may be reached after the administration of 40 mg, suggesting that the dosage of the drug could be reduced without affecting efficacy. We performed studies of clinical pharmacology to assess achieved COX-2 selectivity ex vivo by the chronic administration of etoricoxib 90 mg daily versus the tNSAIDs diclofenac 75 mg bid, naproxen 500 mg bid, and aspirin 100 mg daily (). As shown in the administration of etoricoxib (at 12 h after dosing), diclofenac (at 4 h after dosing), naproxen (at 12 h after dosing), and low-dose aspirin (at 24 h after dosing) caused a median inhibition of COX-1 of 1%, 60%, 95%, and 98%, respectively. These results show that inhibition of platelet COX-1 ≥95% (which translates into inhibition of platelet function) occurs in all subjected treated with low-dose aspirin and in some of those treated with high-dose naproxen, at the interval between doses, sustaining an anti-platelet effect of high-dose naproxen. In contrast, in all subjects treated with diclofenac and etoricoxib, the degree of inhibition of platelet COX-1 was not adequate to affect significantly TXA2-dependent platelet function. However, our results showed a marked inter-subject variability in the inhibition of platelet COX-1 after the administration of diclofenac (range 8–98%) and to a lesser extent also for etoricoxib (range 0%–48%). As shown in , whole blood COX-2 was profoundly affected by etoricoxib, diclofenac, and naproxen, but not by low-dose aspirin (median inhibition and range: 86% [70–90], 98% [58–100], 79% [30–83], and 0% [0–31], respectively]. These data show that both diclofenac and etoricoxib were administered at supratherapeutic doses, in fact most of individuals had a COX-2 inhibition higher than 80%, considered sufficient to translate into efficacy. From these values we calculated the achieved selectivity (ie the ratio of COX-2 inhibition and COX-1 inhibition ex vivo). As shown in , for etoricoxib and diclofenac the individual values of achieved COX selectivity were placed above 1 (which represents a balanced inhibition of COX-2 and COX-1) showing that circulating concentrations of the two drugs caused a more profound inhibition of COX-2 than COX-1. In contrast, naproxen and aspirin caused a more profound inhibition of COX-1 than COX-2; in fact the ratio of COX-2 /COX-1 inhibition ex vivo was always lower than 1. These data clearly show that diclofenac and naproxen, despite being considered tNSAIDs, are significantly different in selectivity compared with COX-isozymes at therapeutic doses. Interestingly, the achieved selectivity was not different, in a statistically significant fashion, between etoricoxib and diclofenac. These results will be relevant for interpretation of the clinical results, for both efficacy and toxicity.
Figure 7 Whole blood COX-1 (A) and COX-2 (B) inhibition ex vivo after the chronic administration of etoricoxib 90 mg, diclofenac 75 mg bid, naproxen 500 mg bid, and aspirin 100 mg daily to humans; (C) achieved COX-2 selectivity by the drugs.
Prostanoids are autocoids that act in the proximity of the cell where they are generated. In fact, they are subjected to an intense metabolism which leads to inactivation of prostanoids. It proceeds through β-oxidation ω-oxidation, 15-hydroxy dehydrogenation, and double-bond reduction through enzymatic pathways in liver, kidney, and lung (CitationRamwell et al 1980; CitationRoberts 1987) (). The assessment of major enzymatic metabolites of prostanoids represents an index of their systemic biosynthesis. Several lines of evidence support a dominant platelet origin (mostly COX-1-derived) of a major urinary enzymatic metabolite of TXB2, ie 11-dehydro-TXB2, while vasculature (mostly COX-2 derived) represents the dominant origin of a major urinary enzymatic metabolite of prostacyclin, ie, 2,3-dinor-6-keto-PGF1α (CitationCapone et al 2007).
In susceptible patients, inhibition of prostanoid generation by tNSAIDs and coxibs causes sodium retention with resulting edema and hypertension (CitationGrosser et al 2006). Similarly to thrombogenesis, COX-1 inhibition may mitigate the consequence of COX-2 inhibition in the kidney. This is compatible with a COX-2-dependent source of vasodilatory prostacyclin and COX-1-dependent origin of vasoconstrictors, like TXA2 in the kidney (CitationQi et al 2002; CitationGrosser et al 2006). We studied the impact of etoricoxib and naproxen on renal and systemic prostanoid biosynthesis by assessing 6-keto-PGF1α/TXB2 inhibition ratio and 2,3-dinor-6-keto-PGF1α/11-dehydro-TXB2 inhibition ratio, respectively, after the administration of the drugs ().
Figure 8 Effects of the chronic administration of etoricoxib 90 mg daily and naproxen 500 mg bid to humans on prostacyclin/TXA2 inhibition ratio in the kidney (by assessing their urinary non-enzymatic metabolites: 6-keto-PGF1α and TXB2, respectively) (A), versus systemic biosynthesis (by assessing their urinary enzymatic metabolites: PGIM = 2,3-dinor-6-keto-PGF1α [a major urinary enzymatic metabolite of prostacyclin] and TX-M = 11-dehydro-TXB2 [a major enzymatic metabolite of TXA2], respectively) (B).
![Figure 8 Effects of the chronic administration of etoricoxib 90 mg daily and naproxen 500 mg bid to humans on prostacyclin/TXA2 inhibition ratio in the kidney (by assessing their urinary non-enzymatic metabolites: 6-keto-PGF1α and TXB2, respectively) (A), versus systemic biosynthesis (by assessing their urinary enzymatic metabolites: PGIM = 2,3-dinor-6-keto-PGF1α [a major urinary enzymatic metabolite of prostacyclin] and TX-M = 11-dehydro-TXB2 [a major enzymatic metabolite of TXA2], respectively) (B).](/cms/asset/40fd8bbe-c228-4c9d-9b8a-41455ad711fd/dtcr_a_3209_f0008_b.jpg)
As shown in , etoricoxib caused a more profound inhibition of COX-2 than COX-1 both at renal and extrarenal sites. This is predictive of a CV hazard but of reduced GI toxicity by etoricoxib. In contrast, naproxen reduced more profoundly COX-1 both at renal and extrarenal sites (), which may translate into reduced CV but higher GI side effects.
Etoricoxib and GI toxicity
The GI safety of etoricoxib has been assessed in the MEDAL (Multinational Etoricoxib vs Diclofenac Arthritis Long Term) Programme (CitationLaine et al 2007). It was prospectively designed to pool data from 3 randomized, double-blind clinical trials: the MEDAL study, the Etoricoxib vs Diclofenac Sodium Gastrointestinal Tolerability and Effectiveness (EDGE) study, and the EDGE II study. Patients with OA or RA aged 50 years or over were eligible for enrollment if they had a clinical diagnosis of OA of the knee, hip, hand, or spine, or a clinical diagnosis of RA that satisfied at least 4 of 7 of the American Rheumatism Association 1987 revised criteria (CitationArnett et al 1988), and in the judgment of the investigator, would require chronic therapy with an NSAID. Thus, etoricoxib (60 or 90 mg daily) and diclofenac (150 mg daily) were compared in 34,701 patients with OA or RA for upper GI clinical events (bleeding, perforation, obstruction, or ulcer) and the subset of complicated events (perforation, obstruction, witnessed ulcer bleeding, or significant bleeding). As shown in , there were significantly fewer upper GI clinical events with the COX-2 selective inhibitor etoricoxib than with the tNSAID diclofenac due to a decrease in uncomplicated events, but not in the more serious complicated events. Less inhibition of gastric COX-1 by etoricoxib than diclofenac (etoricoxib is a more COX-2 selective drug than diclofenac in vitro and ex vivo [CitationPatrignani et al 1997; CitationTacconelli et al 2002] []) may explain the reduced incidence of uncomplicated GI events. In contrast, the comparable rates of GI bleeding by the two drugs could be due to a similar impact on platelet COX-1, in some individuals, and on healing of gastric lesions. In fact, it has been shown that COX-2 plays an important role in the healing of gastric ulcers and that inhibition of COX-2 delays ulcer healing (CitationKonturek et al 2005). As shown in , etoricoxib and diclofenac have a comparable profound impact on COX-2 (>80%).
Table 2 Risk of GI events with etoricoxib and diclofenac in MEDAL study (derived from CitationLaine et al 2007)
In MEDAL, the use of low-dose aspirin and proton pump inhibitors (PPIs) was allowed. Analyzing the data in aspirin and non-aspirin populations, it has been found that etoricoxib reduced the risk of uncomplicated upper GI events compared with diclofenac in patients taking low-dose aspirin regularly, but the magnitude of the GI benefit decreased with low-dose-aspirin use (aspirin: hazard ratio 0.73, 95% CI 0.64–1.07 versus no-aspirin: 0.52, 0.38–0.71, p = 0.021) (CitationLaine et al 2007). PPIs were used concomitantly for at least 75% of the study period by 40% of patients and treatment effects did not differ significantly in these individuals; however, significantly fewer patients discontinued etoricoxib than diclofenac due to dyspepsia, and this decrease was similar in patients who took PPIs, suggesting that the COX-2-selective inhibitor provided symptomatic benefit even in patients already taking a PPI (CitationLaine et al 2007).
Etoricoxib and CV toxicity
In placebo-controlled RCTs, coxibs (rofecoxib, celecoxib, and valdecoxib) were associated with increased RR of CV events (1.92–3.7) (CitationOtt et al 2003; CitationBresalier et al 2005; CitationNussmeier et al 2005; CitationPfizer 2005; CitationSolomon et al 2005). An overview of data derived from trials with coxibs suggests that MI predominates over stroke (CitationKearney et al 2006). These results led the regulatory authorities such as the Food and Drug Administration (FDA) and the European Agency for the Evaluation of Medicinal Products (EMEA) to restrict the use of these drugs (CitationPatrignani et al 2008c). However, the results of observational studies and a meta-analysis of data derived from trials with coxibs have shown that the CV hazard is also associated with some tNSAIDs, such as diclofenac (CitationHernandez-Diaz et al 2006; CitationKearney et al 2006). A plausible mechanism in increased risk of vascular events in individuals treated with NSAIDs selective for COX-2 and some tNSAIDs is the inhibition of COX-2-dependent prostacyclin unaccompanied by inhibition of COX-1-dependent TXA2 at functional range, ie >95%. In fact, most tNSAIDs are as COX-2 selective as coxibs for platelet function. In addition, coxibs and tNSAIDs inhibit profoundly prostacyclin generation in vivo, as assessed by the measurement of urinary levels of a major enzymatic metabolite 2,3-dinor-6-keto-PGF1α (CitationCatella-Lawson et al 1999; CitationMcAdam et al 1999).
The CV hazard associated with the use of coxibs led to the voluntarily withdrawal from the US and EU market of rofecoxib and valdecoxib, in 2004 and 2005, respectively. Recently, the FDA has formally rejected manufacturer application for approval of etoricoxib as a prescription pain reliever. The FDA decision was influenced by the results of safety analyses from the MEDAL Program (CitationCannon et al 2006). The authors reported that etoricoxib (60 or 90 mg daily, mean duration of therapy was 18 months) and diclofenac (150 mg daily, ie 50 mg tid or 75 mg bid) administered to OA or RA patients have comparable rates, constant over time, of thrombotic CV events (CitationCannon et al 2006). Despite etoricoxib and diclofenac having different COX-2 selectivity in vitro, they are functional COX-2 selective for platelet function. The shared pharmacodynamic translates into comparable incidence of thrombotic events detected in MEDAL.
Aside from the platelet, the relationship between inhibition of prostaglandin formation and prostaglandin dependent function appears to be roughly linear in other cell types. In this context, even an incomplete inhibition of COX-1 might attenuate the functional effect derived from inhibition of COX-2-dependent prostacyclin. This may occur in the kidney where COX-2 is the source of vasodilatory prostacyclin and COX-1 is the source of vasoconstrictor TXA2 (CitationQi et al 2002; CitationGrosser et al 2006). This is sustained by the results of MEDAL where the selective COX-2 inhibitor etoricoxib caused a higher incidence of blood pressure than the less selective tNSAID diclofenac. However, the more pronounced signal from MI rather than stroke showed by the use of NSAIDs (traditional and selective for COX-2) suggests that thrombosis is the dominant component of the CV hazard and thus lesser degrees of selectivity are likely only to mitigate the hazard to a marginal extent.
Both EMEA and FDA and the American Heart Association (CitationAntman et al 2007) issued the recommendations that selective COX-2 inhibitors are contraindicated in patients with ischemic heart disease and/or stroke, that they have to be avoided in patients with risk factors for coronary heart disease, and that all patients have to take the lowest effective dose for the shortest time necessary to control symptoms. Importantly, we have recently shown that the degree of COX-2 inhibition achieved by individual NSAIDs (an index of drug potency/exposure), that do not show functional suppression of platelet COX-1, is a determinant of the CV hazard associated with them (CitationPatrignani et al 2008a, Citationc).
Thus, a possible approach to reduce the CV risk associated with etoricoxib will be to use the lowest effective dose. Studies of efficacy with lower doses of etoricoxib than those used in MEDAL seem to support this possible strategy. Etoricoxib 30 mg/day has been shown efficacious in OA compared with either ibuprofen 800 mg tid (CitationWiesenhutter et al 2005) or diclofenac 50 mg tid (CitationLeung et al 2002). Moreover, it has been recently assessed the efficacy of etoricoxib 30 mg daily versus the recommended therapeutic dose of celecoxib, 200 mg daily, in the treatment of OA patients (CitationBingham et al 2007). The primary hypothesis was that etoricoxib 30 mg would be at least as effective as celecoxib 200 mg for Western Ontario and McMaster (WOMAC) Pain Subscale, WOMAC Physical Function Subscale and Patient Global Assessment of Disease Status. Etoricoxib was non-inferior to celecoxib and both were superior to placebo (p < 0.001) for all three outcomes. The authors suggested that etoricoxib 30 mg was at least as effective as celecoxib 200 mg and had similar safety in the treatment of OA of knee and hip. As shown in , etoricoxib 30 mg seems adequate to reduce whole blood COX-2 at a degree appropriate to translate into clinical efficacy. However, further studies of clinical pharmacology in patients with OA and RA should be performed to verify the impact of etoricoxib 30 mg on biochemical markers of COX inhibition, such as whole blood COX-2 and systemic and renal biosynthesis of prostacyclin and TXA2.
Conclusions
NSAIDs are an important and efficacious class of drugs for the management of musculoskeletal symptoms. The development of NSAIDs selective for COX-2 – to reduce the incidence of serious GI adverse effects compared with tNSAIDs mainly dependent on the inhibition of COX-1 in GI tract and platelets – has unravelled the important protective role of COX-2 for the CV system, mainly through the generation of prostacyclin. In a recent nested-case control study, we found that patients taking NSAIDs (both coxibs and tNSAIDs) had a 35% increase risk of MI. The increased incidence of thrombotic events associated with profound inhibition of COX-2-dependent prostacyclin can be mitigated, even if not removed, by a complete suppression of platelet COX-1 activity. In fact, inhibition of TXA2-dependent platelet function occurs when platelet COX-1 activity, assessed ex vivo, is reduced ≥95% (CitationReilly and FitzGerald 1987). We have introduced the concept of functional COX-2 selectivity (ie, a profound suppression of COX-2 associated with insufficient inhibition of platelet COX-1 to cause inhibition of platelet function) which is a feature shared by most tNSAIDs and coxibs. We have recently shown that inhibition of COX-2 and the functional selectivity with which it is achieved is relevant to CV hazard from NSAIDs and relates to drug potency (exposure) (CitationPatrignani et al 2008a, Citationc).
The development of genetic and biochemical markers will help to identify the responders to NSAIDs or who are uniquely susceptible at developing thrombotic or GI events by COX inhibition. This will lead to the development of individual responder approaches. We propose that the assessment of whole blood COX-2 ex vivo (alone or in combination with urinary 2,3-dinor-6-keto-PGF1α) may represent a valid surrogate end-point to predict CV risk for functionally selective COX-2 inhibitors. The use of these biochemical markers together with genetic biomarkers will help to select patients uniquely susceptible to developing CV risk through inhibition of COX-2-dependent-prostacyclin when exposed to NSAIDs. Their use can lead to a rational selection of doses for efficacy and will help to make decisions in risk management of tNSAIDs and NSAIDs selective for COX-2.
Disclosures
None of the authors has any conflicts of interest to disclose.
References
- AgrawalNGPorrasAGMatthewsCZ2003Single- and multiple-dose pharmacokinetics of etoricoxib, a selective inhibitor of cyclooxygenase-2, in manJ Clin Pharmacol432687612638395
- AlfrancaAIñiguezMAFresnoM2006Prostanoid signal transduction and gene expression in the endothelium: role in cardiovascular diseasesCardiovasc Res704465616458868
- AntmanEMBennettJSDaughertyA2007Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart AssociationCirculation11516344217325246
- ArehartEStithamJAsselbergsFW2008Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation: potential implications for cyclooxygenase-2 inhibitionCirc Res1029869318323528
- ArnettFCEdworthySMBlochDA1988The American rheumatism association 1987 revised criteria for the classification of rheumatiod arthritisArthritis Rheum31315243358796
- AsanoKLillyCMDrazenJM1996Prostaglandin G/H synthase-2 is the constitutive and dominant isoform in cultured human lung epithelial cellsAm J Physiol Lung Cell Mol Physiol271L12631
- BeicheFScheuererSBruneK1996Up-regulation of cyclooxygenase-2 mRNA in the rat spinal cord following peripheral inflammationFEBS Lett39016598706851
- BinghamCO3rdSebbaAIRubinBR2007Efficacy and safety of etoricoxib 30 mg and celecoxib 200 mg in the treatment of osteoarthritis in two identically designed, randomized, placebo-controlled, non-inferiority studiesRheumatology (Oxford)4649650716936327
- BresalierRSSandlerRSQuanH2005Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trialN Engl J Med352109210215713943
- BreyerRMBagdassarianCKMyersSA2001Prostanoid receptors: subtypes and signalingAnnu Rev Pharmacol Toxicol416619011264472
- BurkeASmythEFitzGeraldGA2006Analgesic-antipyretic agents; Pharmacotherapy of goutBruntonLLLazoJSParkerKLGoodman and Gilman‘s The Pharmacological Basis of Therapeutics11th edNew York, USAMcGRAW-HILL, Medical Publishing Division673715
- CannonCPCurtisSPFitzGeraldGA2006Cardiovascular outcomes with etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) programme: a randomised comparisonLancet36817718117113426
- CaponeMLTacconelliSFrancescoLD2008Cardiovascular effects of valdecoxib: transducing human pharmacology results into clinical read-outsExpert Opin Drug Saf7294218171312
- CaponeMLTacconelliSDi FrancescoL2007Pharmacodynamic of cyclooxygenase inhibitors in humansProstaglandins Other Lipid Mediat82859417164136
- CaponeMLTacconelliSSciulliMG2004Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjectsCirculation10914687115037526
- CarlsonMLWilsonETPrescottSM2003Regulation of COX-2 transcription in a colon cancer cell line by Pontin52/TIP49aMol Cancer24214675489
- Catella-LawsonFMcAdamBMorrisonBW1999Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoidsJ Pharmacol Exp Ther2897354110215647
- CryerBFeldmanM1998Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugsAm J Med104413219626023
- EganKMLawsonJAFriesS2004COX-2-derived prostacyclin confers atheroprotection on female miceScience3061954715550624
- FitzgeraldGAPatronoC2001The Coxibs, selective inhibitors of Cyclooxygenase-2N Engl J Med3454334211496855
- FitzGeraldGA2003COX-2 and beyond: Approaches to prostaglandin inhibition in human diseaseNat Rev Drug Discov28799014668809
- FunkCD2001Prostaglandins and leukotrienes: advances in eicosanoid biologyScience2941871511729303
- GrosserTFriesSFitzGeraldGA2006Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunitiesJ Clin Invest11641516395396
- HarrisRCMcKannaJAAkaiY1994Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restrictionJ Clin Invest942504107989609
- Hernández-DíazSVaras-LorenzoCGarcía RodríguezLA2006Non-steroidal antiinflammatory drugs and the risk of acute myocardial infarctionBasic Clin Pharmacol Toxicol982667416611201
- HuntjensDRDanhofMDella PasquaOE2005Pharmacokinetic-pharmacodynamic correlations and biomarkers in the development of COX-2 inhibitorsRheumatology (Oxford)448465915855183
- InoueAIkomaKMoriokaN1999Interleukin-1beta induces substance P release from primary afferent neurons through the cyclooxygenase-2 systemJ Neurochem7322061310537081
- KangYJMbonyeURDeLongCJ2007Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradationProg Lipid Res461082517316818
- KargmanSCharlesonSCartwrightM1996Characterization of prostaglandin G/H synthase 1 and 2 in rat, dog, monkey, and human gastrointestinal tractsGastroenterology111445548690211
- KassahunKMcIntoshISShouM2001Role of human liver cytochrome P4503A in the metabolism of etoricoxib, a novel cyclooxygenase-2 selective inhibitorDrug Metab Dispos298132011353749
- KaufmannWEWorleyPFTaylorCV1997Cyclooxygenase-2 expression during rat neocortical development and in Rett syndromeBrain Dev1925349071487
- KearneyPMBaigentCGodwinJ2006Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trialsBMJ3321302816740558
- KonturekSJKonturekPCBrzozowskiT2005Prostaglandins and ulcer healingJ Physiol Pharmacol56531
- KulmaczRJWangLH1995Comparison of hydroperoxide initiator requirements for the cyclooxygenase activities of prostaglandin H synthase-1 and -2J Biol Chem27024019237592599
- LaineLCurtisSPCryerB2007Assessment of upper gastrointestinal safety of etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) programme: a randomized comparisonLancet3694657317292766
- LeungATMalmstromKGallacherAE2002Efficacy and tolerability profile of etoricoxib in patients with osteoarthritis: a randomized, double-blind, placebo and active-comparator controlled 12-week efficacy trialCurr Med Res Opin18495812017209
- MalkowskiMGGinellSLSmithWL2000The productive conformation of arachidonic acid bound to prostaglandin synthaseScience2891933710988074
- MarnettLJRowlinsonSWGoodwinDC1999Arachidonic acid oxygenation by COX-1 and COX-2: mechanisms of catalysis and inhibitionJ Biol Chem27422903610438452
- McAdamBFCatella-LawsonFMardiniIA1999Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2Proc Natl Acad Sci U S A9627279874808
- MinuzPFumagalliLGainoS2006Rapid stimulation of tyrosine phosphorylation signals downstream of G-protein-coupled receptors for thromboxane A2 in human plateletsBiochem J4001273416859489
- MinuzPGainoSZulianiV2002Functional role of p38 mitogen activated protein kinase in platelet activation induced by a thromboxane A2 analogue and by 8-iso-prostaglandin F2alphaThromb Haemost878889812038794
- MoncadaSHiggsEAVaneJR1977Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potent inhibitor of platelet aggregationLancet1182063657
- MurataTUshikubiFMatsuokaT1997Altered pain perception and inflammatory response in mice lacking prostacyclin receptorNature388678829262402
- NakaoKMuraseAOhshiroH2007CJ-023,423, a novel, potent and selective prostaglandin EP4 receptor antagonist with antihyperalgesic propertiesJ Pharmacol Exp Ther3226869417495127
- NardoneGRoccoAVairaD2004Expression of COX-2, mPGE-synthase1, MDR-1 (P-gp), and Bcl-xL: a molecular pathway of H pylori-related gastric carcinogenesisJ Pathol2023051214991895
- NarumiyaSSugimotoYUshikubiF1999Prostanoid receptors: structures, properties, and functionsPhysiol Rev79119322610508233
- NussmeierNAWheltonAABrownMT2005Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgeryN Engl J Med35210819115713945
- OttENussmeierNADukePC2003Efficacy and safety of the cyclooxygenase 2 inhibitors parecoxib and valdecoxib in patients undergoing coronary artery bypass surgeryJ Thorac Cardiovasc Surg12514819212830070
- PatrignaniPCaponeMLTacconelliS2008cNSAIDs and cardiovascular diseaseHeart94395718347364
- PatrignaniPCampestriniJBransonJ2004Lumiracoxib is a selective inhibitor of cyclooxygenase-2 in patients with osteoarthritisAnn Rheum Dis63368
- PatrignaniPDi FebboCTacconelliS2008bDifferential association between human prostacyclin receptor polymorphisms and the development of venous thrombosis and intimal hyperplasia: a clinical biomarker studyPharmacogenetics and Genomics186112018551041
- PatrignaniPPanaraMRGrecoA1994Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthasesJ Pharmacol Exp Ther2711705127996488
- PatrignaniPPanaraMRSciulliMG1997Differential inhibition of human prostaglandin endoperoxide synthase-1 and -2 by nonsteroidal anti-inflammatory drugsJ Physiol Pharmacol48623319444611
- PatrignaniPSciulliMGManariniS1999COX-2 is not involved in thromboxane biosynthesis by activated human plateletsJ Physiol Pharmacol50661710639016
- PatrignaniPTacconelliSGarcía RodríguezLA2008aRole of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general populationArterioscler Thromb Vasc Biol28e45
- PatronoCCiabattoniGPincaE1980Low dose aspirin and inhibition of thromboxane B2 production in healthy subjectsThromb Res17317277368167
- PatronoCPatrignaniPGarcía RodríguezLA2001Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outsJ Clin Invest10871311435450
- Pfizer. A double-blind randomised placebo-controlled comparative study of celecoxib (SC-58635) for the inhibition of progression of Alzheimer’s disease, protocol IQ5-97-02-001.2000 [online]. Accessed 21 November 2005. URL: http://www.clinicalstudyresults.org/documents/company-study_76_0.pdf
- PrescottSMFitzpatrickFA2000Cyclooxygenase-2 and carcinogenesisBiochim Biophys Acta1470M697810722929
- PulcinelliFMRiondinoSCelestiniA2005Persistent production of platelet thromboxane A2 in patients chronically treated with aspirinJ Thromb Haemost32784916359516
- QiZHaoC-MLangenbachRI2002Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin IIJ Clin Invest11061912093889
- RamwellPWFoeghMLoebR1980Synthesis and metabolism of prostaglandins, prostacyclin, and thromboxanes: the arachidonic acid cascadeSemin Perinatol43136247764
- ReillyIAFitzGeraldGA1987Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugsBlood6918063790723
- RiendeauDPercivalMDBrideauC2001Etoricoxib (MK-0663): preclinical profile and comparison with other agents that selectively inhibit cyclooxygenase-2J Pharmacol Exp Ther2965586611160644
- RistimakiAHonkanenNJankalaH1997Expression of cyclooxygenase-2 in human gastric carcinomaCancer Res571276809102213
- RobertsLJII1987Handbook of eicosanoids: prostaglandins and related lipidsBoca Raton, FLCRC23344
- RobertsonRP1998Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesisDiabetes471379839726224
- RoccaBSpainLMPureE1999Distinct roles of prostaglandin H synthases 1 and 2 in T-cell developmentJ Clin Invest10314697710330429
- RomanoMRicciVMemoliA1998Helicobacter pylori up-regulates cyclooxygenase-2 mRNA expression and prostaglandin E2 synthesis in MKN 28 gastric mucosal cells in vitroJ Biol Chem2732856039786845
- SamadTAMooreKASapirsteinA2001Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivityNature410471511260714
- SciulliMGCaponeMLTacconelliS2005The future of traditional nonsteroidal antiinflammatory drugs and cyclooxygenase-2 inhibitors in the treatment of inflammation and painPharmacol Rep57668516415488
- SciulliMGRendaGCaponeML2006Heterogeneity in the suppression of platelet cyclooxygenase-1 activity by aspirin in coronary heart diseaseClin Pharmacol Ther801152516890573
- ShaoJShengHInoueH2000Regulation of constitutive cyclooxygenase-2 expression in colon carcinoma cellsJ Biol Chem27533951610930401
- ShigetaJTakahashiSOkabeS1998Role of cyclooxygenase-2 in the healing of gastric ulcers in ratsJ Pharmacol Exp Ther2861383909732401
- SimmonsDLBottingRMHlaT2004Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibitionPharmacol Rev5638743715317910
- SmithWDeWittDGaravitoR2000Cyclooxygenases: structural, cellular, and molecular biologyAnnu Rev Biochem691458210966456
- SolomonSDMcMurrayJJPfefferMA2005Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma preventionN Engl J Med35210718015713944
- SolomonSDWittesJFinnPV2008Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials. The Cross Trial Safety AnalysisCirculation11721041318378608
- SwinneyDCMakAYBarnettJ1997Differential allosteric regulation of prostaglandin H synthase 1 and 2 by arachidonic acidJ Biol Chem2721239389139685
- TacconelliSCaponeMLSciulliMG2002The biochemical selectivity of novel COX-2 inhibitors in whole blood assays of COX-isozyme activityCurr Med Res Opin185031112564662
- TakahashiSShigetaJInoueH1998Localization of cyclooxygenase-2 and regulation of its mRNA expression in gastric ulcers in ratsAm J Physiol275G1137459815044
- TonaiTTaketaniYUedaN1999Possible involvement of interleukin-1 in cyclooxygenase-2 induction after spinal cord injury in ratsJ Neurochem7230299886082
- VaneJRBakhleYSBottingRM1998Cyclooxygenases 1 and 2Annu Rev Pharmacol Toxicol38971209597150
- WangDDuboisRN2006Prostaglandins and cancerGut551152216118353
- WiesenhutterCWBoiceJAKoA2005Evaluation of the comparative efficacy of etoricoxib and ibuprofen for treatment of patients with osteoarthritis: a randomized, double-blind, placebo-controlled trialMayo Clin Proc80470915819283
- WilsonDE1991Role of prostaglandins in gastroduodenal mucosal protectionJ Clin Gastroenterol13S65711940199
- YamagataKAndreassonKIKaufmannWE1993Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoidsNeuron11371868352945
- YuYFanJHuiY2007Targeted cyclooxygenase gene (ptgs) exchange reveals discriminant isoform functionalityJ Biol Chem282149850617110378