Abstract
Statin-induced inhibition of HMG-CoA reductase reduces cholesterol production and prevents the formation of many non-steroidal isoprenoid compounds, such as farnesylpyrophosphate and geranylgeranylpyrophosphate, that act as lipid attachments for the post-translational modification of various proteins, including the G-proteins and transcription factors involved in a number of cell processes. However, the blockade of isoprenylation elicited by statin treatment also has biological effects on cell function that go beyond the decrease in cholesterol synthesis: these are the so-called “pleiotropic” effects that mainly relate to vascular function. Endothelial dysfunction is an independent predictor of cardiovascular events that correlates with inflammation markers/mediators and robust predictors of cardiovascular diseases such as increased high-sensitivity C-reactive protein levels. The results of in vivo and in vitro studies indicate that the statins have beneficial effects unrelated to cholesterol lowering, such as improving endothelial function, increasing myocardial perfusion, and enhancing the availability of nitric oxide. This review describes the pleiotropic effects of statins that may be involved in modulating/preventing endothelial dysfunction and inflammatory processes, as well as the cellular and molecular mechanisms through which they improve endothelial function.
Introduction
The endothelium is a monocellular layer lining the inside of vessels that normally provides a non-adhesive, non-thrombogenic surface for blood constituents, and acts as a dynamic interface regulating blood vessel functions (CitationBehrendt and Ganz 2002). It influences responses to environmental and endogenous factors by generating paracrine and autocrine mediators that control the biology of the entire vessel wall. The endothelium plays a pivotal role in regulating vascular tone, but also controls other physiological process such as inflammation, coagulation and thrombosis.
Persistent hemodynamic or inflammatory factors activate the endothelium and lead to it becoming dysfunctional. In particular, endothelial cell activation by cytokines or other inflammatory mediators increases the expression of a variety of cell surface adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), procoagulants and anticoagulants, and substances regulating vasomotor tone. One hallmark of endothelium dysfunction is an altered response to endothelium-dependent and endothelium-independent stimuli, such as acetylcholine and bradykinin (CitationHarrison 1997). These effects are due to a reduced bioavailability of nitric oxide (NO), which may be caused by a decrease in the synthesis, release and/or activity of endothelial-derived NO.
It is now known that abnormal endothelium function can be detected before the establishment of obvious intimal lesions in patients with risk factors for atherosclerosis (CitationCelermajer et al 1992), and endothelial dysfunction of the coronary or peripheral arteries is an independent predictor of cardiovascular events, even after adjusting for traditional factors (CitationGanz and Vita 2003). Endothelial dysfunction correlates with inflammation markers/mediators and robust predictors of cardiovascular diseases, such as increased high-sensitivity C-reactive protein (hs-CRP) levels in subjects with coronary artery disease (CitationFichtlscherer et al 2000). This observation is of particular interest because CRP stimulates the expression of VCAM-1, thus highlighting once again the link between inflammation and endothelial dysfunction (CitationPasceri et al 2000). Furthermore, it has been suggested that enhanced endothelial function may contribute to improved clinical status (CitationAnderson et al 1995; CitationTreasure et al 1995).
Experimental and clinical studies have shown that hypercholesterolemia, a major risk factor for vascular diseases, impairs endothelium function (CitationCreager et al 1990; CitationEgashira et al 1993), and LDL apheresis alone rapidly exerts beneficial effects on endothelial vasodilator function within a few hours (CitationTamai et al 1997).
It has been shown that, in addition to reducing atherosclerosis and cardiovascular events, lipid-lowering therapies and particularly 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors (commonly known as statins) improve endothelium function, and numerous clinical studies have demonstrated that this is not necessarily related to a detectable decrease in serum cholesterol levels.
The first evidence that statins may inhibit cardiovascular events regardless of their effects on blood cholesterol levels came from the WOSCOPS study, which found that the incidence of cardiovascular events in a subgroup of patients treated with placebo or statin with the same LDL-cholesterol level was markedly lower in the statin group (CitationWOSCOPS Study Group 1998). The beneficial effects of statins may occur relatively soon after the start of therapy and are different from those observed after the reduction of plasma cholesterol levels (CitationBuchwald et al 1995). The cholesterol-independent, anti-thrombotic, anti-oxidative and anti-inflammatory vascular effects of statins are known as pleiotropic effects (CitationDavignon 2004; CitationHalcox and Deanfield 2004). The most important underlying mechanism is mediated by a reduction in the synthesis of mevalonate, which is not only a precursor of cholesterol, but also of a variety of non-steroidal isoprenoid compounds that are essential for normal cell activity (CitationCorsini et al 1999; CitationWolfrum et al 2003). Isoprenoids, such as farnesylpyrophosphate and geranylpyrophosphate, are essential for the cell membrane attachment of important regulatory proteins, particularly small GTPase: by inhibiting its synthesis, statins deplete cells of these lipids and thus elicit the retention of small GTPase in the cytosol, where they cannot exert their biological actions.
Statins and endothelial NO synthase (eNOS) expression
The initial studies of endothelial function concentrated on vasomotion, which became the major parameter of endothelial health. Endothelium-dependent vasodilatation primarily occurs via the release of a humoral mediator (identified as NO), rather than prostacyclin and an endothelium-derived hyperpolarisation factor (CitationKansui et al 2004).
Well documented data from experimental and clinical studies show that statins increase eNOS expression and activation, which may be the principal mechanism by which statins improve endothelial dysfunction in addition to reduce cholesterol levels. Interestingly, statins can modulate NO bioavailability by increasing mRNA expression or increasing eNOS activity: the first is a late effect and involves inhibiting the isoprenylation of Rho, small GTPase proteins, whereas the second is much more rapid and requires a lower statin concentration. It must also be remembered that statins maintain NO availability by preventing its degradation by free radical molecules (CitationKoh 2000).
NO bioavailability is mediated by Rho inhibition
Rho proteins are small GTPases that regulate cytoskeleton organization and cell adhesion, thus contributing to cell migration and endothelial permeability (CitationNobes and Hall 1999; CitationRidley 1995). Their function is strictly regulated by their membrane localization, which is favored by prenylation, a post-translational modification that helps anchor them to membranes.
Statins modulate the stability and activity of Rho GTPases by acting on their sub-cellular localization and inhibiting the synthesis of farnesyl- and geranylgeranyl pyrophosphate, the isoprenoids required for the prenylation of Rho proteins. They also alter Rho expression at transcriptional level, although the underlying mechanism is still unclear.
However, the inhibition of Rho (and particularly RhoA) is a determinant factor in stabilizing the mRNA of endothelial NO synthase (eNOS) and improving endothelium-dependent relaxation ().
Figure 1 Statins inhibit the activation of Rho signaling, which negatively influences eNOS mRNA stability and activity, leading to an increased NO bioavailability.
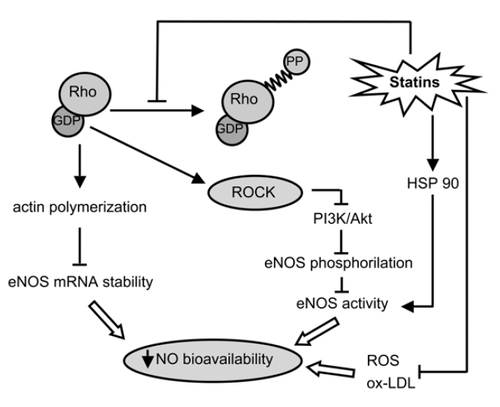
It has been demonstrated that RhoA affects vasomotion by activating Rho-kinases (ROCK), which inactivates myosin light chain phosphatase (MLCP) and reduces the expression of eNOS (CitationSomlyo 2002). Direct inhibition of ROCK by specific inhibitors such as hydroxyfasudil and Y27632 increases the half-life and expression of eNOS mRNA (CitationRikitake et al 2005).
Furthermore, ROCK inhibits the serin/threonine kinase Akt phosphorylation and activity, and thus has negative effects on eNOS activity. Akt can phosphorylate eNOS on Ser 1179, and that phosphorylation enhances the enzyme’s ability to generate NO (CitationFulton et al 1999). Interestingly, the membrane compartmentalization of both proteins (inside the Golgi region and plasma membrane of endothelial cells) is required for Akt’s functional interaction with eNOS. It is not fully understood how the phosphorylation of eNOS enhances NO release, but it seems to be mediated by the introduction of a negative charge that ‘opens’ the structure and thus permits activated calmodulin binding at lower calcium concentrations (CitationSalerno et al 1997). In vitro data suggest that enzyme activity is enhanced in a Ca2+-independent manner, but is due to greater Ca2+-sensitization (CitationDimmeler et al 1999).
The bioavailability of NO may also be influenced by RhoA in a ROCK-independent manner. Non-filamentous actin (G-actin) interacts with eNOS mRNA, and changes in actin polymerization affect eNOS mRNA stability and down-regulate eNOS expression (CitationSearles et al 2004). Recent studies have demonstrated that cytoplasmatic filaments and microtubules are necessary to transport mRNA within the cytoplasm and anchor them at specific sub-cellular locations (CitationBassell and Singer 1997; CitationNasmyth and Jansen 1997). The cytoskeleton anchoring of mRNAs, and their co-localisation with ribosomes and RNA-binding protein complexes are necessary for their translational expression and stability. The Rho-controlled reorganisation of the actin cytoskeletron may therefore play a key role in the movement and compartmentalisation of specific mRNAs.
Endothelial dysfunction and vasoactive agonists
The partial reversion of endothelial dysfunction induced by statin treatment is not totally due to improved NO bioavailability, but also to the better regulated expression of vasoactive factors. The regulation of vascular tone is a complex process that involves the concerted action of many factors; in particular, endothelin-1 (ET-1) and angiotensin II (Ang II) elicit contractile and proliferative activities in the vascular smooth muscle layer. An imbalance between Ang II and NO is often caused by a loss of NO due to endothelial dysfunction and oxidative stress and/or the enhancement of Ang II local tissue activity (CitationDzau 2001).
Ang II, which is the primary effector of the renin-angiotensin system (RAS), is a multifunctional hormone that plays important autocrine and paracrine roles in vascular function (CitationCampbell and Habener 1986). Albeit indirectly, Ang II and NO interact with each other in the vasculature to influence vascular tone, cell growth, apoptosis and inflammation (). Via AT1 receptor-coupled G protein, Ang II activates phospholipase C (PLC) which in turn produces inositol 1,4,5-triphosphate (IP3), and stimulates Ca2+ mobilisation. The Ca2+-mediated activation of myosin light-chain kinase (MLCK) leads to the phosphorylation of MLCK and smooth muscle contraction. Ang II also induces Ca2+ sensitization of the contractile apparatus by activating RhoA/Rho kinase, which in turn inactivates myosin light-chain phosphatases (MLCP). A recent in vivo study has shown that Ang II infusion decreases NO production and uncouples eNOS in rat aortas, thus causing superoxide rather than NO production (CitationMollnau et al 2002). Interestingly, the long-term infusion of Ang II causes endothelial dysfunction associated with decreases in guanylyl cyclase (GC) expression and cGMP-dependent protein kinase (PKG) activity in rat aorta (CitationMollnau et al 2002).
Figure 2 Ang II and NO functions interplay to influence vascular tone, through different effects on RhoA pathway.
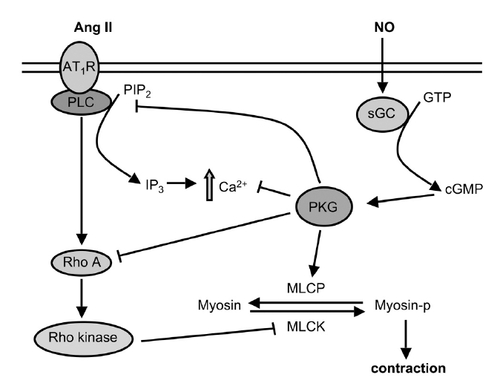
On the contrary, by activating soluble guanylyl cyclase (sGC), NO stimulates PKG and reduces Ca2+ levels by downregulating IP3 production and decreasing Ca2+ mobilization. PKG inactivates the RhoA/Rho kinase signaling pathway to inhibit RhoA-induced Ca2+ sensitization. The results of a number of experimental studies suggest that NO may directly inhibit ACE activity reducing plasma Ang II concentrations and decrease AT1 receptor mRNA expression at transcriptional level (CitationKumar and Das 1997; CitationAckermann et al 1998; CitationWu et al 2000).
Recent studies have demonstrated that statins may prevent harmful Ang II-induced events, such as the production of reactive oxygen species in vascular smooth muscle cells (VSMCs), cardiac hypertrophy and end-organ damage (CitationWassmann et al 2001; CitationPark et al 2000). In particular, it has been found in an in vivo model of arterial neointimal thickening that fluvastatin inhibits Ang II-mediated ERK phosphorylation, and the tyrosine- and serine-induced phosphorylations of STAT1 and STAT3 that are known to be activated by many extracellular signaling proteins, including cytokines, growth factors, and Ang II via the AT1 receptor. Furthermore, in vitro and in vivo studies have shown that the effects of statins on AT1 receptor-mediated actions affecting VSMCs may also be mediated by decreasing AT1 receptor expression (CitationIchiki et al 2001; CitationWassmann et al 2001). Various findings indicate that patients with high levels of LDL-cholesterol may also increase vascular responses to Ang II, and it is known that hypercholesterolemia is closely associated with AT1 receptor upregulation (CitationNickenig et al 1997, Citation1999). Statin treatment improves vascular responsiveness to Ang II (although not in a dose-dependent manner), and seems to be closely related to serum cholesterol levels.
In vascular endothelial cells, statins also affect the expression of pre-pro-endothelin-1, a precursor of endothelin-1 (ET-1), which elicits potent contractile and proliferative action in VSMCs (CitationHirata 1996). Statins-mediated inhibition of the activity of Rho proteins downregulates pre-pro-endothelin-1 gene expression, an effect that is independent of their action on NO.
Statins and caveolae
The endothelial cell plasma membrane consists of liquid-ordered microdomains (lipid rafts), that are assembled from lipid constituents and have distinct biophysical characteristics and limited random movement (CitationBrown and London 2000; CitationSimons and Ehehalt 2002). These regions are involved in the local sequestration of proteins that mediate signal transduction in a variety of cell types, including endothelial and vascular smooth muscle cells. In certain pathological situations, such as hyperlipidemia, the composition of some membrane microdomains are altered and thus contribute to the mechanisms of atherogenesis in vascular cells.
Caveolae are the most widely studied lipid rafts. Their principal component is the protein caveolin, a scaffolding element that efficiently binds cholesterol and interacts with various signalling macromolecules, including G proteins (CitationSmart et al 1999; CitationGargalovic and Dory 2003). Caveolin also inhibits eNOS by blocking its access to cofactors, and regulates the production of NO in the endothelium (CitationJu et al 1997; CitationFeron et al 1998).
The high caveolae levels under condition of hypercholesterolemia are associated with reduced endothelial NO synthesis and increased superoxide levels, SMC proliferation and leukocyte adhesion (CitationVergnani et al 2000). The therapeutic benefit of statins is mainly due to their restoration of normal endothelial NO levels by means of various mechanisms, including the upregulation of eNOS expression (CitationLaufs et al 1998). They also stimulate endothelial NO production by greatly decreasing plasma membrane caveolin levels: a recent study on endothelial cells (ECs) found that atorvastatin reduced the abundance of caveolin-1 in the absence or presence of LDL-cholesterol, and promoted NO production regardless of the level of extracellular LDL-cholesterol (CitationFeron et al 2001). These results highlight the central role of inhibiting the mevalonate pathway in peripheral cells by reducing the synthesis of isoprenoid intermediates regardless of cholesterol synthesis. Moreover, the beneficial effect of atorvastatin on eNOS activity was greater in the cells expressing high levels of caveolin. Finally, the statin promoted the agonist-induced association of eNOS and chaperone Hsp90, thus leading to increased activity (CitationFeron et al 2001).
In addition to modulating the physical and chemical properties of membrane lipids, hypercholesterolemia has also been associated with the disruption of active L-arginine transport, which affects the capacity of endothelial cells to generate NO: ie, L-arginine deficit leads eNOS to overproduce superoxide from oxygen instead of NO. By improving L-arginine uptake through amino acid transport, statins may also enhance NO production and interfere with superoxide formation.
Effects of statins on endothelial dysfunction
Many studies have demonstrated the beneficial clinical effects of statins on endothelial dysfunction, but the underlying mechanisms remain largely unknown. Nevertheless, all researchers believe that, in addition to the reduction in cholesterol, a NO-dependent process is also involved. In vitro and in vivo studies have confirmed that statins enhance the expression of eNOS by means of post-transcriptional/translational mechanisms (CitationLaufs et al 1998, Citation2000). In particular, the use of different animal models has been useful in improving our understanding of the role of individual risk factors, such as hypertension, hypertriglyceridemia, hyperinsulinemia, hyperglycemia and insulin resistance in endothelial dysfunction, and correlating the improvement in endothelial function due to statins treatment with changes in these factors.
Animal studies
Chronic treatment with simvastatin improves endothelium-dependent acetylcholine relaxations of aorta from hypertensive rats (SHR) by means of a mechanism that is independent of the cyclo-oxygenase pathway (Citationde Sotomayor et al 1999). The improved endothelial function in the treated animals can be attributed to the normalization of deranged NOS activity, partly mediated by the promotion of superoxide dismutase (SOD) (CitationCarneado et al 2002).
The same authors have also shown that simvastatin improves endothelium-dependent acetylcholine relaxations in vessels from aged Wistar rats. The mechanisms involved enhanced endothelial NO vasodilatation due to increased eNOS expression, decreased participation of TXA2 associated with the decreased expression of the COX-2 isoform, and enhanced vessel antioxidant properties (Citationde Sotomayor et al 2005).
Statins improve endothelial health in many situations, but have failed in the well-known model of cardiovascular disease offered by DOCA-salt rats that develop hypertension, cardiovascular hypertrophy, inflammation and endothelial dysfunction. At a dose that decreased plasma cholesterol levels, rosuvastatin attenuated aortic media thickness and vascular hypertrophy, but did not affect the developing hypertension. It surprisingly increased aortic responses to acetylcholine in male Wistar rats, but had no effect on the reduced responses to noradrenaline, sodium nitroprusside and acetylcholine of DOCA-salt rats. These results may be attributed to species-related differences and variations in the capacity of statins to penetrate vascular cell membranes, but it is also conceivable that lowering blood pressure is necessary to improve endothelial dysfunction in DOCA-salt rats (CitationLoch et al 2006).
A recent study investigated in vivo a possible molecular mechanism of vascular dysfunction and the effects of fluvastatin in obese Zucker rats, a model of diabetes mellitus (CitationNishimatsu et al 2005). Fluvastatin partially (but signicantly) reduced Ang II-induced vasoconstriction and improved endothelium-dependent vasorelaxation via the phosphatidylinositol 3-kinase/protein kinase Akt (PI3K/Akt)-dependent and NO/cGMP-dependent pathways in rat aorta. This had previously been observed in an in vitro study of endothelial cells in which statins seemed to stimulate the membrane translocation of Akt and its activating phosphorylation by PI3-kinase (CitationSkaletz-Rorowski et al 2003): statin stimulation promoted the association of tyrosine phosphorylated protein with the p85 subunit of PI3-kinase, and Akt translocation was inhibited by mevalonate and wortmannin, a PI3-kinase inhibitor, thus leading to the inactivation of the enzyme. It has been reported that the Akt-dependent phosphorylation of eNOS is necessary for the full activation of eNOS and endothelium-dependent vasorelaxation, and so impaired PI3K/Akt activation may have been involved in the reduced endothelium-dependent vasorelaxation (CitationDimmeler et al 1999; CitationFulton et al 1999).
Furthermore, Akt signaling is subject to regulation by a rapidly exchanging pool of cholesterol within cells. Some authors suggest that this endothelial cholesterol pool is more sensitive to the statin-mediated inhibition of endogenous cholesterol synthesis than it is to changes in exogenous cholesterol delivery from the serum by LDLs. In this regard, it may be relevant that PI3-kinase activity in fibroblasts is negatively regulated by the recruitment of caveolin-1, an intracellular cholesterol transport protein, to PI3-kinase-associated receptor complexes within lipids rafts (CitationZundel et al 2000). It has also been shown that the statin-induced inhibition of cholesterol synthesis in endothelial cells can improve the inhibitory action of caveolin-1 on eNOS (CitationFeron et al 2001), the activity of which is controlled by Akt-mediated phosphorylation (CitationFulton et al 1999; CitationDimmeler et al 1999).
Cerivastatin treatment improves endothelial dysfunction in Otsuka Long-Evans Tokushima Fatty (OLETF) rats, a useful model of obese type 2 diabetes. The restoration of endothelial function was related to an increased in the aortic expression of CD36, a gene encoding a fatty acid transporter, and PPAR-γ. This study is an interesting example of an inter-relationship between cholesterol and fatty acid metabolism which may lead to marked beneficial effects on endothelial function in patients with diabetic hyperlipidemia and insulin-resistance syndromes (CitationNakamura et al 2004).
Human studies
Strong evidence that lowering LDL alone is not enough to improve endothelial dysfunction has been provided by the inability of ezetimibe, in comparison with atorvastatin alone or in combination with ezetimibe, to improve endothelial vasodilator function in the forearm circulation of patients with coronary artery disease (CAD), despite its LDL-cholesterol lowering effect (CitationFichtlscherer et al 2006). These findings suggest that the lipid-lowering capacity of atorvastatin is not the primary mechanism underlying the beneficial effects of short-term atorvastatin therapy in patients with CAD.
In subjects with moderately high total serum cholesterol levels, the vasodilatator response to acetylcholine and baseline blood flow significantly increased after four weeks’ treatment with simvastatin, which simultaneously increased the vasoconstrictor response to L-NMMA, whereas the response to the endothelium-independent vasodilator sodium nitroprusside remained unchanged. None of these effects were related to the decrease in cholesterol levels (CitationO’Driscoll et al 1997). Statin therapy also improves endothelial function in normocholesteremic patients with chronic heart failure (CHF) and, if the treatment is long, stabilizes neurohormonal imbalances and provides measurable clinical benefits. Furthermore, these beneficial effects on the endothelium are dose dependent (Citationvan der Harst et al 2005).
Some new aspects have been highlighted by a recent study of atorvastatin in hyperlipidemic patients. The improvement in endothelial function preferentially occurred in patients with pre-existing endothelial dysfunction and completely disappeared within the 36 hours following the withdrawal of the statin (CitationTaneva et al 2006). This last observation corresponds to results from cell cultures and animal experiments in which the lowering of eNOS and/or NO levels occurred after the discontinuation of statins (CitationLaufs et al 2000; CitationGertz et al 2003; CitationXing et al 2005). One possible molecular mechanism of this may be related to the increase in membrane Rho expression after statin withdrawal that has been found both in vitro and in vivo (CitationLaufs et al 2000).
However, as the findings conflict with the results obtained in non-diabetic subjects, it is intriguing to note that statins do not seem to improve endothelial function consistently in patients with type 2 diabetes. Only a few published clinical studies have assessed the effect of statins on the microcirculation in subjects with type 2 diabetes, and their results were varied. Some found that statin therapy failed to improve endothelial dysfunction, and the authors suggested that lowering LDL alone may not be sufficient to improve endothelial function in the absence of glycemic control (CitationMansourati et al 2001; Citationvan Etten et al 2002; Citationvan Venrooij et al 2002; CitationFegan et al 2005). Another recently suggested explanation for the failure of statins to improve endothelial function in such cases is the diabetes-specific accumulation of advanced glycosylation end-product (AGE) products (CitationSowers 2002) that leads to vascular thickening, loss of elasticity, and the cross-linking of subendothelial structural proteins.
However, two studies have demonstrated improved endothelial function with statins in diabetes and, although CRP levels did not decrease significantly, the change correlated with the change in endothelium-dependent vasodilatation (CitationTsunekawa et al 2001; CitationTan et al 2002).
Anti-inflammatory properties of statins
Inflammation plays a pivotal role in all stages of atherosclerosis, from the nascent lesion to acute coronary syndromes (CitationLibby et al 2002).
A number of in vitro studies have described the beneficial effects of statins in decreasing the levels of CD 11b adhesion molecules (CitationWeber et al 1997), leukocyte function antigen-1 (LFA-1) (CitationWeitz-Schmidt et al 2001), and ICAM-1 and VCAM-1 (CitationBernot et al 2003; CitationZapolska-Downar et al 2004; Landsberger et al 2006), and other studies have shown that they reduce the secretion of pro-inflammatory cytokines (such as interleukin IL-6, IL-1β and TNF-α) and chemokines, such as IL-8 and MCP-1 (CitationRomano et al 2000; CitationWang et al 2005). Interestingly, statins inhibit the production of TNF-α in endothelial cells as well as the CRP-stimulated activation of NF-kB, a strong biomarker of systemic inflammation in cardiovascular diseases. It has been shown that CPR induces plasminogen activator inhibitor (PAI-1) expression and complement activation, and decreases eNOS expression, thus leading to a propensity for thrombosis, inflammation and endothelial dysfunction. Recent clinical studies have found that various statins reduce CRP levels, and that this is at least partially independent of their lipid-lowering properties (CitationRidker 2003; CitationNawawi et al 2003; CitationSugiyama et al 2005).
Numerous in vitro and in vivo experiments have shown that the anti-inflammatory effects of statins are partially mediated by vascular endothelial NO (CitationScalia et al 2001; CitationStalker et al 2001). This cholesterol-independent effect of statins is absent in eNOS-deficent mice, thus suggesting that eNOS mediates the protective vascular effects of statins (CitationStalker et al 2001).
NO mediates the beneficial effects of statins on vascular health
Recent studies have shown that endothelial dysfunction plays an important role as an independent risk factor (CitationWidlansky et al 2003) and, together with inflammation, triggers cardiovascular diseases (CitationDrexler et al 1992; CitationTousoulis et al 2005). Furthermore, it has been recognized that endothelium-derived NO is an anti-inflammatory and anti-arteriosclerotic molecule as it protects nuclear transcription factor (NF-kB) from activation by oxidized LDL or cytokines, and thus prevents or attenuates the transcription and expression of adhesion molecules (CitationMarui et al 1993). It has also been shown that the inhibition of NO synthesis in cultured endothelial cells increases the expression of the gene coding for MCP-1, and that MCP-1 expression is associated with the activation of NF-kB (CitationZeiher et al 1995). Therapies that increase NO bioactivity may reduce the synthesis of pro-inflammatory proteins on the endothelial cell surface, which may reduce inflammation.
The capacity of statins to improve endothelial dysfunction and reduce inflammation has been demonstrated in numerous experimental studies. Long-term treatment with simvastatin normalizes acetylcholine-induced relaxation in rats treated with L-NAME without affecting response to the nitrovasodilator itself (CitationPerez-Guerrero et al 2003). The inhibition of NO synthesis by N-nitro-arginine methyl ester induces early inflammation characterized by increased monocyte infiltration coronary vessels and increased MCP-1 expression (CitationTakemoto et al 1997; CitationTomita et al 1998). In the same animal model, pravastatin and cerivastatin inhibited vascular inflammation by increasing eNOS expression and restoring NO-generating capacity by inhibiting Rho activity (CitationNi et al 2001). A recent study has suggested a novel molecular mechanism by which statins regulate vascular inflammation by finding that simvastatin increased NO production in human aortic endothelial cells (HAECs), and this covalently modified N-ethylmaleimide sensitive factor (NSF), a key regulator of endothelial exocytosis. The nitrosylation of NSF blocked the externalization of P-selectin to the endothelial surface, which otherwise activates leukocyte rolling, the first step in leukocyte inflammation (CitationYamakuchi et al 2005). The statin also modified the second step in leukocyte trafficking by blocking the interaction of LFA-1 with intercellular adhesion molecule-1 (VCAM-1) but, interestingly, not in eNOS knockout mice.
In patients with heart failure, atorvastatin treatment significantly improves forearm vasodilatory response to reactive hyperemia and reduced serum levels of IL-6, TNF-α and soluble VCAM-1, but has no effects on MCP-1 (CitationTousoulis et al 2005). Statins also improve arterial stiffness and decrease the plasma levels of hsCRP, a sensitive marker of the chronic inflammation of arteriosclerotic lesions in patients with hypercholesterolemia (CitationMatsuo et al 2005).
In hypercholesterolemic patients with angiographically-documented coronary artery disease, simvastatin significantly improved the percent flow-mediated dilator response to hyperemia, whereas the response to nitroglycerin was not significantly modified (CitationKoh et al 2003). In the same patients, it significantly lowered the plasma levels of TNF-α, CRP, fibrinogen and ICAM-1, but had no effect on E-selectin and VCAM-1; furthermore, the greatest reduction in plasma TNF-α and CRP levels occurred in the patients with the highest baseline levels. It is interesting to note that there was a significant inverse correlation between the percentage of flow-mediated dilatation and plasma TNF-α levels, and a positive correlation between the latter and changes in plasma nitrate levels.
High-dose atorvastatin acutely increased endothelium-dependent forearm blood flow (FBF) in subjects with normal vascular function, and rapidly decreased the levels of the inflammation marker hs-CPR (CitationLaufs et al 2001). Furthermore, its withdrawal has been found to induce a rapid deterioration in endothelial function, a rebound-like decrease in NO bioavailability, and increased inflammation in clinical and experimental studies (CitationThomas and Mann 1998; CitationLaufs and Liao 2000; CitationLaufs et al 2000). These data are in line with the recent finding that the discontinuation of statin treatment induces vascular complications in patients with acute coronary syndromes (CitationHeeschen et al 2002; CitationLi et al 2006).
Statin modulates thrombosis and coagulation
Nitric oxide is the major mediator synthesized by the endothelium. It regulates vascular homeostasis and blood flow, and a decrease in its bioavailability is related to vasoconstriction, vascular smooth muscle proliferation, platelet aggregation and endothelial-leukocyte adhesion (CitationPalmer et al 1987; CitationRadomski et al 1992; CitationGauthier et al 1995). These pathological conditions (together known as endothelial dysfunction) is considered to be an early marker of the atherothrombosis that underlies cardiovascular, and particularly coronary heart disease. Numerous experimental and clinical studies have shown that statins have beneficial effects on atherothrombosis by reducing the progression of the atheroma and the incidence of acute thrombosis-related vascular events. The mechanisms by which statins inhibit thrombosis have not been totally clarified, although several pathways seem to be involved. In particular, they increase the stability of the plaque whose rupture leads to thrombosis by exposing blood to the highly thrombogenic contents of its lipid core (CitationLiao 2002). Rather than reducing lipid levels (which reduces plaque size and modifies the physiochemical compositions of the lipid core) (CitationKoh 2000; CitationTakemoto et al 2001), statins exert their beneficial effects by decreasing the infiltration and activity of macrophages and T-lymphocytes within the plaque, and inhibiting proteolytic enzymes such as matrix metalloproteinases (MMP), which are thought to be responsible for the plaque rupture induced by the thinning, ulceration and fissuring of the fibrous cap (CitationBellosta et al 1998; CitationAikawa et al 2001).
Platelet hyperactivity is an important factor contributing to the enhanced risk of thrombotic complications in hypercholesterolemic patients (CitationOpper et al 1995). It is linked to increases in the biosynthesis of thromboxane A2 (TXA2), platelet α2-adrenergic density, and cytosolic calcium (CitationBaldassarre et al 1997; CitationNotarbartolo et al 1995). Studies of platelets taken from statin-treated hypercholesterolemic patients have led to contrasting results: platelet aggregation has been found to be reduced, unchanged or increased by lovastatin treatment (CitationColli et al 2004); some studies have found that fluvastatin reduces platelet aggregation as well as soluble P-selectin (a marker of α-granule platelet secretion or endothelial cell dysfunction) and ICAM-1 levels (CitationOsamah et al 1997; CitationRomano et al 2000); and it has also been found that simvastatin inhibits the production of TXA2, and urinary excretion of its metabolite 11-dehydrothromboxane B2 (CitationNotarbartolo et al 1995). These discrepant results may be explained by the different experimental conditions used to assess platelet function.
Statins also inhibit the expression of tissue factor (TF), a transmembrane glycoprotein whose binding with coagulation factor VII initiates blood coagulation by activating proteolytically factor IX and X (CitationColli et al 1997). Various studies have demonstrated that statins affect this pathway by inhibiting Rho/Rho-kinase and the activation of Akt (CitationEto et al 2002). The (ATROCAP) study provides definite in vivo evidence that statins affect TF expression and activity, and macrophage infiltration in human vessels (CitationCortellaro et al 2002). These data strongly indicate that statins attenuate atherosclerotic plaque thrombogenicity by reducing cell-mediated thrombin generation. Studies of the effects of statins on plasma fibrinogen and factor VII levels have led to very contrasting results (CitationColli et al 2004).
Additional features of endothelial cell dysfunction include atheroma fibrinolytic imbalance. In advanced lesions, a state of hypofibrinolysis prevails because of the high levels of plasminogen activator inhibitor-1 (PAI-1) released by activated cells within the atheroma and the platelets incorporated in mural thrombi (CitationRobbie et al 1996). Although, in vitro and ex vitro studies have shown that different statins induce tissue-type plasminogen activators (t-PA) and reduce PAI-1, the results of in vivo studies are conflicting (CitationColli et al 2004). These results may be explained by differences in metabolic profiles and genetic backgrounds, which are known to have a considerable effect on PAI-1 levels.
Conclusion
Many of the beneficial pleiotropic effects of statins occur as a result of modulated endothelial function and reduced inflammatory processes. Attempting to understand these properties of statins is an exciting field of research that will also improve our understanding of vascular biology in health and disease, and thus enable the better use of this drug class in clinical practice.
References
- AckermannAFernandez-AlfonsoMSSanchezdRModulation of angiotensin-converting enzyme by nitric oxideBr J Pharmacol199812429189641545
- AikawaMRabkinESugiyamaSAn HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitroCirculation20011032768311208689
- AndersonTJMeredithITYeungACThe effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotionN Engl J Med1995332488937830729
- BaldassarreDMoresNColliSPlatelet alpha 2-adrenergic receptors in hypercholesterolemia: relationship between binding studies and epinephrine-induced platelet aggregationClin Pharmacol Ther199761684919209252
- BassellGSingerRHmRNA and cytoskeletal filamentsCurr Opin Cell Biol19979109159013679
- BehrendtDGanzPEndothelial function. From vascular biology to clinical applicationsAm J Cardiol20029040L48L
- BellostaSViaDCanavesiMHMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophagesArterioscler Thromb Vasc Biol199818167189812903
- BernotDBenolielAMPeirettiFEffect of atorvastatin on adhesive phenotype of human endothelial cells activated by tumor necrosis factor alphaJ Cardiovasc Pharmacol2003413162412548094
- BrownDALondonEStructure and function of sphingolipid- and cholesterol-rich membrane raftsJ Biol Chem200027517221410770957
- BuchwaldHCamposCTBoenJRDisease-free intervals after partial ileal bypass in patients with coronary heart disease and hypercholesterolemia: report from the Program on the Surgical Control of the Hyperlipidemias (POSCH)J Am Coll Cardiol19952635177608434
- CampbellDJHabenerJFAngiotensinogen gene is expressed and differentially regulated in multiple tissues of the ratJ Clin Invest1986783193013940
- CarneadoJAlvarezdSPerez-GuerreroCSimvastatin improves endothelial function in spontaneously hypertensive rats through a superoxide dismutase mediated antioxidant effectJ Hypertens2002204293711875310
- CelermajerDSSorensenKEGoochVMNon-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosisLancet19923401111151359209
- ColliSEliginiSLalliMVastatins inhibit tissue factor in cultured human macrophages. A novel mechanism of protection against atherothrombosisArterioscler Thromb Vasc Biol199717265729081680
- ColliSWerbaJPTremoliEStatins in atherothrombosisSemin Vasc Med200444071515861322
- CorsiniABellostaSBaettaRNew insights into the pharmacodynamic and pharmacokinetic properties of statinsPharmacol Ther1999844132810665838
- CortellaroMCofrancescoEArbustiEAtorvastatin and thrombogenicity of the carotid atherosclerotic plaque: the ATROCAP studyThromb Haemost20028841712152675
- CreagerMACookeJPMendelsohnMEImpaired vasodilation of forearm resistance vessels in hypercholesterolemic humansJ Clin Invest199086228342195060
- DavignonJBeneficial cardiovascular pleiotropic effects of statinsCirculation2004109III394315198965
- de SotomayorMAPerez-GuerreroCHerreraMDEffects of chronic treatment with simvastatin on endothelial dysfunction in spontaneously hypertensive ratsJ Hypertens1999177697610459874
- de SotomayorMAPerez-GuerreroCHerrreraMDImprovement of age-related endothelial dysfunction by simvastatin: effect on NO and COX pathwaysBr J Pharmacol200514611303816231003
- DimmelerSFlemingIFisslthalerBActivation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylationNature1999399601510376603
- DrexlerHHayozDMunzelTEndothelial function in chronic congestive heart failureAm J Cardiol19926915966011598876
- DzauVJTheodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesisHypertension20013710475211304501
- EgashiraKInouTHirookaYImpaired coronary blood flow response to acetylcholine in patients with coronary risk factors and proximal atherosclerotic lesionsJ Clin Invest19939129378423226
- EtoMKozaiTCasentinoFStatin prevents tissue factor expression in human endothelial cells: role of Rho/Rho-kinase and Akt pathwaysCirculation20021051756911956113
- FeganPGShoreACMawsonDMicrovascular endothelial function in subjects with Type 2 diabetes and the effect of lipid-lowering therapyDiabetes Care20052216706
- FeronODessyCDesagerJPHydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundanceCirculation20011031131811136695
- FeronOSaldanaFMichelJBThe endothelial nitric-oxide synthase-caveolin regulatory cycleJ Biol Chem1998273312589452418
- FichtlschererSRosenbergerGWalterDHElevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery diseaseCirculation20001021000610961964
- FichtlschererSSchmidt-LuckeCBojungaSDifferential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for ‘pleiotropic’ functions of statin therapyEur Heart J20062711829016621868
- FultonDGrattonJPMcCabeTJRegulation of endothelium-derived nitric oxide production by the protein kinase AktNature199939959760110376602
- GanzPVitaJATesting endothelial vasomotor function: nitric oxide, a multipotent moleculeCirculation200310820495314581383
- GargalovicPDoryLCaveolins and macrophage lipid metabolismJ Lipid Res200344112112518018
- GauthierTWScaliaRMuroharaTNitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemiaArterioscler Thromb Vasc Biol199515165297583540
- GertzKLaufsULindauerUWithdrawal of statin treatment abrogates stroke protection in miceStroke200334551712574574
- HalcoxJPDeanfieldJEBeyond the laboratory: clinical implications for statin pleiotropyCirculation2004109II42815173062
- HarrisonDGCellular and molecular mechanisms of endothelial cell dysfunctionJ Clin Invest1997100215379410891
- HeeschenCHammCWLaufsUWithdrawal of statins increases event rates in patients with acute coronary syndromesCirculation200210514465211914253
- HirataYEndothelin peptidesCurr Opin Nephrol Hypertens199651258834156
- IchikiTTakedaKTokunouTDownregulation of angiotensin II type 1 receptor by hydrophobic 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors in vascular smooth muscle cellsArterioscler Thromb Vasc Biol200121189690111742861
- JuHZouRVenemaVJDirect interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activityJ Biol Chem19972721852259228013
- KansuiYFujiiKGotoKEffects of fluvastatin on endothelium-derived hyperpolarizing factor- and nitric oxide- mediated relaxations in arteries of hypertensive ratsClin Exp Pharmacol Physiol200431354915191411
- KohKKEffects of statins on vascular wall: vasomotor function, inflammation, and plaque stabilityCardiovasc Res2000476485710974215
- KohKKAhnJYChoiYMVascular effects of step I diet in hypercholesterolemic patients with coronary artery diseaseAm J Cardiol2003927081012972113
- KumarKVDasUNEffect of cis-unsaturated fatty acids, prostaglandins, and free radicals on angiotensin-converting enzyme activity in vitroProc Soc Exp Biol Med199721437499111529
- LandsbergerMWolffBJantzenFCerivastatin reduces cytokine-induced surface expression of ICAM-1 via increased shedding in human endothelial cellsAtherosclerosis2007190435216529752
- LaufsUEndresMCustodisFSuppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcriptionCirculation200010231041011120702
- LaufsULa FataVPlutzkyJUpregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitorsCirculation1998971129359537338
- LaufsULiaoJKPost-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPaseJ Biol Chem199827324266719727051
- LaufsULiaoJKDirect vascular effects of HMG-CoA reductase inhibitorsTrends Cardiovasc Med200010143811239793
- LaufsUWassmannSHilgersSRapid effects on vascular function after initiation and withdrawal of atorvastatin in healthy, normocholesterolemic menAm J Cardiol2001881306711728362
- LiaoJKBeyond lipid lowering: the role of statins in vascular protectionInt J Cardiol20028651812243846
- LiJJLiYSChenJRebound phenomenon of inflammatory response may be a major mechanism responsible for increased cardiovascular events after abrupt cessation of statin therapyMed Hypotheses200666119920416413682
- LibbyPRidkerPMMaseriAInflammation and atherosclerosisCirculation200210511354311877368
- LochDLevickSHoeyARosuvastatin attenuates hypertension-induced cardiovascular remodeling without affecting blood pressure in DOCA-salt hypertensive ratsJ Cardiovasc Pharmacol20064739640416633082
- MansouratiJNewmanLGRomanSHLipid lowering does not improve endothelial function in subjects with poorly controlled diabetesDiabetes Care2001242152311723099
- MaruiNOffermannMKSwerlickRVascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cellsJ Clin Invest1993921866747691889
- MatsuoTIwadeKHirataNImprovement of arterial stiffness by the antioxidant and anti-inflammatory effects of short-term statin therapy in patients with hypercholesterolemiaHeart Vessels20052081215700196
- MollnauHWendtMSzocsKEffects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signalingCirc Res200290E586511884382
- NakamuraTSaitoYOhyamaYEffect of cerivastatin on endothelial dysfunction and aortic CD36 expression in diabetic hyperlipidemic ratsHypertens Res2004275899815492479
- NasmythKJansenRPThe cytoskeleton in mRNA localization and cell differentiationCurr Opin Cell Biol199793964009159073
- NawawiHOsmanNSYusoffKReduction in serum levels of adhesion molecules, interleukin-6 and C-reactive protein following short-term low-dose atorvastatin treatment in patients with non-familial hypercholesterolemiaHorm Metab Res2003354798512953165
- NiWEgashiraKKataokaCAntiinflammatory and antiarteriosclerotic actions of HMG-CoA reductase inhibitors in a rat model of chronic inhibition of nitric oxide synthesisCirc Res2001894152111532902
- NickenigGBaumerATTemurYStatin-sensitive dysregulated AT1 receptor function and density in hypercholesterolemic menCirculation19991002131410571970
- NickenigGSachinidisAMichaelsenFUpregulation of vascular angiotensin II receptor gene expression by low-density lipoprotein in vascular smooth muscle cellsCirculation19979547389008466
- NishimatsuHSuzukiESatonakaHEndothelial dysfunction and hypercontractility of vascular myocytes are ameliorated by fluvastatin in obese Zucker ratsAm J Physiol Renal Physiol2005288H17706
- NobesCDHallARho GTPases control polarity, protrusion, and adhesion during cell movementJ Cell Biol199914412354410087266
- NotarbartoloADaviGAvernaMInhibition of thromboxane biosynthesis and platelet function by simvastatin in type IIa hypercholesterolemiaArterioscler Thromb Vasc Biol199515247517749833
- O’DriscollGGreenDTaylorRRSimvastatin, an HMG-Coenzyme A reductase inhibitor, improves endothelial function within 1 monthCirculation1997951126319054840
- OpperCClementCSchwarzHIncreased number of high sensitive platelets in hypercholesterolemia, cardiovascular diseases, and after incubation with cholesterolAtherosclerosis1995113211177605360
- OsamahHMiraRSorinaSReduced platelet aggregation after fluvastatin therapy is associated with altered platelet lipid composition and drug binding to the plateletsBr J Clin Pharmacol19974477839241100
- PalmerRMFerrigeAGMoncadaSNitric oxide release accounts for the biological activity of endothelium-derived relaxing factorNature198732752463495737
- ParkJKMullerDNMervaalaEMCerivastatin prevents angiotensin II-induced renal injury independent of blood pressure- and cholesterol-lowering effectsKidney Int20005814203011012877
- PasceriVWillersonJTYehETDirect proinflammatory effect of C-reactive protein on human endothelial cellsCirculation20001022165811056086
- Perez-GuerreroCAlvarezdSJimenezLEffects of simvastatin on endothelial function after chronic inhibition of nitric oxide synthase by L-NAMEJ Cardiovasc Pharmacol2003422041012883323
- RadomskiMWReesDDDutraAS-nitroso-glutathione inhibits platelet activation in vitro and in vivoBr J Pharmacol199210774591335336
- RidkerPMHigh-sensitivity C-reactive protein and cardiovascular risk: rationale for screening and primary preventionAm J Cardiol20039217K22K
- RidleyAJRho-related proteins: actin cytoskeleton and cell cycleCurr Opin Genet Dev1995524307749321
- RikitakeYKimHHHuangZInhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protectionStroke2005362251716141422
- RobbieLABoothNABrownAJInhibitors of fibrinolysis are elevated in atherosclerotic plaqueArterioscler Thromb Vasc Biol199616539458624776
- RomanoMDiomedeLSironiMInhibition of monocyte chemotactic protein-1 synthesis by statinsLab Invest2000801095110010908155
- RomanoMMezzettiAMarulliCFluvastatin reduces soluble P-selectin and ICAM-1 levels in hypercholesterolemic patients: role of nitric oxideJ Investig Med2000481839
- SalernoJCHarrisDEIrizarryKAn autoinhibitory control element defines calcium-regulated isoforms of nitric oxide synthaseJ Biol Chem199727229769779368047
- ScaliaRGooszenMEJonesSPSimvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient miceCirculation2001103259860311382730
- SearlesCDIdeLDavisMEActin cytoskeleton organization and posttranscriptional regulation of endothelial nitric oxide synthase during cell growthCirc Res2004954889515256481
- SimonsKEhehaltRCholesterol, lipid rafts, and diseaseJ Clin Invest200211059760312208858
- Skaletz-RorowskiALutchmanMKureishiYHMG-CoA reductase inhibitors promote cholesterol-dependent Akt/PKB translocation to membrane domains in endothelial cellsCardiovasc Res2003572536412504836
- SmartEJGrafGAMcNivenMACaveolins, liquid-ordered domains, and signal transductionMol Cell Biol199919728930410523618
- SomlyoAVNew roads leading to Ca2+ sensitizationCirc Res20029183412142337
- SowersJREndothelial vasodilation effects of statins in type 2 diabetic patients: response to van Venrooij et alDiabetes Care2002251242312087027
- StalkerTJLeferAMScaliaRA new HMG-CoA reductase inhibitor, rosuvastatin, exerts anti-inflammatory effects on the microvascular endothelium: the role of mevalonic acidBr J Pharmacol20011334061211375257
- SugiyamaMOhashiMTakaseHEffects of atorvastatin on inflammation and oxidative stressHeart Vessels200520133616025360
- TakemotoMEgashiraKUsuiMImportant role of tissue angiotensin-converting enzyme activity in the pathogenesis of coronary vascular and myocardial structural changes induced by long-term blockade of nitric oxide synthesis in ratsJ Clin Invest199799278879005996
- TakemotoMLiaoJKPleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitorsArterioscler Thromb Vasc Biol20012117121911701455
- TamaiOMatsuokaHItabeHSingle LDL apheresis improves endothelium-dependent vasodilatation in hypercholesterolemic humansCirculation19979576828994420
- TanKCChowWSTamSCAtorvastatin lowers C-reactive protein and improves endothelium-dependent vasodilation in type 2 diabetes mellitusJ Clin Endocrinol Metab200287563811836286
- TanevaEBoruckiKWiensLEarly effects on endothelial function of atorvastatin 40 mg twice daily and its withdrawalAm J Cardiol2006971002616563905
- ThomasMMannJIncreased thrombotic vascular events after change of statinLancet1998352183019851392
- TomitaHEgashiraKKubo-InoueMInhibition of NO synthesis induces inflammatory changes and monocyte chemoattractant protein-1 expression in rat hearts and vesselsArterioscler Thromb Vasc Biol1998181456649743235
- TousoulisDAntoniadesCBosinakouEEffects of atorvastatin on reactive hyperemia and inflammatory process in patients with congestive heart failureAtherosclerosis20051783596315694946
- TreasureCBKleinJLWeintraubWSBeneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery diseaseN Engl J Med199533248177830728
- TsunekawaTHayashiTKanoHCerivastatin, a hydroxymethylglutaryl coenzyme a reductase inhibitor, improves endothelial function in elderly diabetic patients within 3 daysCirculation2001104376911468195
- van der HarstPWagenaarLJBuikemaHEffect of intensive versus moderate lipid lowering on endothelial function and vascular responsiveness to angiotensin II in stable coronary artery diseaseAm J Cardiol2005961361416275178
- van EttenRWde KoningEJHoningMLIntensive lipid lowering by statin therapy does not improve vasoreactivity in patients with type 2 diabetesArterioscler Thromb Vasc Biol20022279980412006393
- van VenrooijFVvan de ReeMABotsMLAggressive lipid lowering does not improve endothelial function in type 2 diabetes: the Diabetes Atorvastatin Lipid Intervention (DALI) Study: a randomized, double-blind, placebo-controlled trialDiabetes Care20022512111612087021
- VergnaniLHatrikSRicciFEffect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production : key role of L-arginine availabilityCirculation20001011261610725285
- WangHRLiJJHuangCXFluvastatin inhibits the expression of tumor necrosis factor-alpha and activation of nuclear factor-kappaB in human endothelial cells stimulated by C-reactive proteinClin Chim Acta2005353536015698590
- WassmannSLaufsUBaumerATInhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPaseMol Pharmacol2001596465411179461
- WeberCErlWWeberKSHMG-CoA reductase inhibitors decrease CD11b expression and CD11b-dependent adhesion of monocytes to endothelium and reduce increased adhesiveness of monocytes isolated from patients with hypercholesterolemiaJ Am Coll Cardiol1997301212179350917
- Weitz-SchmidtGWelzenbachKBrinkmannVStatins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin siteNat Med200176879211385505
- WidlanskyMEGokceNKeaneyJFJrThe clinical implications of endothelial dysfunctionJ Am Coll Cardiol20034211496014522472
- WolfrumSJensenKSLiaoJKEndothelium-dependent effects of statinsArterioscler Thromb Vasc Biol2003237293612615672
- WOSCOPS Study GroupInfluence of pravastatin and plasma lipids on clinical events in the West of Scotland Coronary Prevention Study (WOSCOPS)Circulation199897144059576423
- WuLGirouardHde ChamplainJInvolvement of the cyclic GMP pathway in the superoxide-induced IP3 formation in vascular smooth muscle cellsJ Hypertens20001810576410953997
- XingYChenHHuDYEffects of withdrawal of statins on nitric oxide production in vascular endothelial cellsZhonghua Nei Ke Za Zhi20054422415769392
- YamakuchiMGreerJJCameronSJHMG-CoA reductase inhibitors inhibit endothelial exocytosis and decrease myocardial infarct sizeCirc Res20059611859215905463
- Zapolska-DownarDSiennickaAKaczmarczykMSimvastatin modulates TNFalpha-induced adhesion molecules expression in human endothelial cellsLife Sci200475128730215234187
- ZeiherAMFisslthalerBSchray-UtzBNitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cellsCirc Res19957698067758169
- ZundelWSwierszLMGiacciaACaveolin 1-mediated regulation of receptor tyrosine kinase-associated phosphatidylinositol 3-kinase activity by ceramideMol Cell Biol20002015071410669728