Abstract
Duloxetine is a balanced selective serotonin norepinephrine reuptake inhibitor (SNRI) which, in 2004, became the first agent to receive regulatory approval for the treatment of painful diabetic neuropathy in the US. This compound has no other significant receptor or channel activities other than the serotonin and norepinephrine reuptake inhibition mechanisms and works to diminish or control the symptoms of diabetic neuropathy. Duloxetine has no known neuroprotective or other effects which prevent the development of neuropathy in patients with diabetes. The purpose of this review article is to discuss the background of painful diabetic neuropathy, the pharmacology of duloxetine, and its safety and efficacy in clinical trials and long-term observations. The authors will also comment on its use in clinical practice. Results from controlled clinical trials reveal that duloxetine administered at 60 mg qd or 60 mg bid is efficacious in treating diabetic neuropathic pain relative to placebo. Positive treatment outcomes are also seen for other measures of pain and quality of life. A minor but statistically significant increase in blood glucose compared with placebo treated patients has been observed in controlled clinical trials. Otherwise, controlled and open-label clinical studies have demonstrated a high degree of safety and tolerability for the compound. These findings provide support for the proposed role of serotonin and norepinephrine as key mediators of the descending pain inhibition pathways of the brain stem and spinal cord.
Introduction
Diabetes mellitus affects over 18 million people in the United States alone. Individuals with long-standing disease are at increased risk for numerous medical complications associated with significant morbidity and mortality (eg, atherosclerotic disease, renal failure, retinopathy, neuropathy). One of the most troubling of these complications is painful diabetic neuropathy, due in large part to its prevalence among diabetics, its effect on the sufferer’s quality of life (QOL), and the limited number of effective, tolerated therapies.
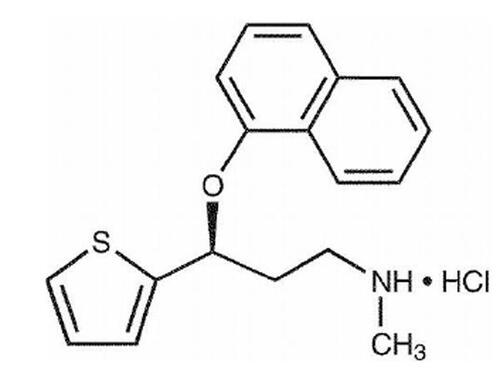
Figure 1 Chemical structure of duloxetine.
Diabetic neuropathy is a symmetrical peripheral polyneuropathy that results from nerve damage after prolonged periods of suboptimal glycemic control and is classified based on the location of the nerves affected. These include:
Peripheral: toes, feet, legs, hands, arms
Autonomic: heart and vasculature, digestive tract, urinary tract, sex organs, sweat glands, and the eyes
Proximal: thighs, hips, buttocks
Focal: eyes, facial muscles, ears, pelvis and lower back, thigh, abdomen.
Approximately 10%–20% develop the painful version of this disorder (CitationHoke and Feasby 2000).
Painful diabetic neuropathy most likely occurs as a result of metabolic changes in the neurons as well as hyperglycemia-associated microvascular damage that lead to abnormal signaling from the peripheral nociceptor that is perceived in the brain as pain (CitationHoke and Feasby 2000; CitationWada and Yagihashi 2005). Over time “central sensitization” occurs, leading to further propagation and continuation of neuropathic pain. Central sensitization refers to neural plasticity and functional changes in the central nervous system (CNS) pain processing pathways that may result in a state of heightened sensitivity to painful stimuli, increased perception of non-painful stimuli as painful, and in some cases, the development of persistent pain in the absence of any stimulus (CitationWoolf and Mannion 1999). In normal neural functioning, descending pain inhibition pathways in the brainstem dampen the strength of incoming nociceptive signaling from the periphery. In chronic diabetic neuropathic pain, the functionality of these descending pain inhibition pathways may be reduced, further amplifying pain perception. Although the exact pathophysiologic mechanism of diabetic neuropathy is not clearly delineated, it is widely recognized that disinhibition and imbalance of 5-hydroxytryptamine (5-HT) and norepinephrine (NE) in endogenous pain inhibitory pathways could contribute to persistent pain mechanisms.
Empirical evidence has suggested that tricyclic antidepressant (TCA) medications are useful as treatment for chronic pain syndromes, including diabetic neuropathy. Those that possess both 5-HT and NE reuptake blockade characteristics (eg, amitriptyline, desipramine) appear to have the best efficacy but their use is limited by side effects (eg, sedation, hypotension, anticholinergic effects, cardiovascular abnormalities; CitationLynch 2001). These side effects are likely due to the affinity of those compounds to cholinergic and adrenergic receptors and possibly due to sodium channel blocking capacities. Duloxetine is a selective inhibitor of reuptake of both 5-HT and NE that is relatively balanced in its activity without significant effects at other sites. Due to its dual effects on 5-HT and NE reuptake, duloxetine has also been studied and approved for use in reducing diabetic neuropathic pain. Because of the prevalence and disabling nature of diabetic neuropathy, this represents a significant addition to the clinician’s armamentarium in providing care for diabetic patients. An understanding of the pharmacology and appropriate medical use of the compound for this painful condition will help ensure that patients receive optimal benefit from its use. This review article pools the data from published RCTs and open-label studies evaluating the efficacy of duloxetine for painful diabetic neuropathy.
Chemistry
Duloxetine hydrochloride, (+)-(S)-N-methyl-gamma-(1-naphthyloxy)-2-thiophenepropylamine hydrochloride, is a selective serotonin and norepinephrine reuptake inhibitor, with molecular weight of 333.88. It is slightly soluble in water and exists as a white to slightly brownish-white solid (Cymbalta®; duloxetine HCl, Eli Lilly, Indianapolis, IN, USA, full prescribing information).
Pharmacodynamic profile
Duloxetine is a selective inhibitor of both serotonin (5-HT) and norepinephrine (NE) reuptake, and is classified as a selective serotonin norepinephrine reuptake inhibitor (SNRI). It possesses central pain inhibitory actions, probably related to its potentiation of serotonergic and noradrenergic activity in the CNS. Both 5-HT and NE have important neurotransmission activities in the descending pain inhibition pathways of the brainstem and spinal cord. Furthermore, these neurotransmitters are felt to act in a synergistic manner to reduce the transmission of pain signals from the periphery to the CNS (CitationYaksh 1985; CitationZhou and Gebhart 1997). Duloxetine has been shown to be effective in animal models of persistent pain, including neuropathic pain. Presumably the analgesic effect of duloxetine is related to augmentation of 5-HT and NE mediated inhibitory pain pathways resulting in the decreased perception of pain.
Importantly, duloxetine has been demonstrated to have no significant activity at muscarinic, histamine-1, α1-adrenergic, dopaminergic, 5-HT1A, 5-HT1B, 5-HT1D, 5-HT2A, 5-HT2C, and opioid receptors. Furthermore, duloxetine has been demonstrated to have no activity on ion channels including Na+ channels (CitationBymaster et al 2001). It has, however, been shown to have balanced activity as an inhibitor of 5-HT and NE reuptake with very low activity on dopamine reuptake (CitationWong et al 1993; CitationWong and Bymaster 2002). Based on what is known about the activity of endogenous inhibitory pathways, a compound with these actions could possibly have good efficacy in treatment of neuropathic pain processes and possess an acceptable side-effect profile.
Duloxetine has been studied extensively in pre-clinical animal models of both persistent neuropathic pain and acute nociceptive pain. Its activity was compared in these experiments to that of a selective serotonin reuptake inhibitor (paroxetine); norepinephrine reuptake inhibitors (thionisoxetine and desipramine), and other SNRIs (venlafaxine, milnacipran, amitriptyline). In the neuropathic pain study paradigms (the formalin model and the L5/L6 nerve ligation model, both in rats), the performance of duloxetine was numerically greater (although not statistically superior) to the comparator drugs at doses which did not lead to neurologic side effects. However, duloxetine did not demonstrate efficacy in the tail-flick model of nociceptive pain, indicating a lack of a primary analgesic or anesthetic effect of the drug. These data demonstrate that duloxetine has potential for the treatment of persistent neuropathic pain owing to its ability to inhibit both 5-HT and NE reuptake (CitationIyengar et al 2004).
Duloxetine has several putative phase I metabolites, including 4-hydroxy-, 5-hydroxy-, 6-hydroxy-, 5-hydroxy-6-methoxy-, 6-methoxy-5-hydroxy-, 5,6-dihydroxy-, and 4,6-dihydroxyduloxetine, as well as phase II glucuronide and sulfate conjugates. In vitro binding studies have shown that none of these circulating metabolites contributes significantly to the pharmacologic activity of duloxetine (CitationKuo et al 2004).
Pharmaceutics
Duloxetine is available in capsules which contain enteric-coated pellets of duloxetine hydrochloride. The drug is degradable in the acidic milieu of the stomach, which necessitates the enteric coated pellets to allow the compound to be principally absorbed in the small intestine. It is extremely important that capsules of duloxetine not be chewed or crushed thereby compromising the integrity of the enteric coating. The capsules are marketed containing 22.4, 33.7, and 67.3 mg of duloxetine hydrochloride to provide equivalent doses of 20, 30, and 60 mg of duloxetine respectively. There are a few inactive ingredients in the capsules, including: FD&C Blue No. 2, gelatin, hypromellose, hydroxypropyl methylcellulose acetate succinate, sodium lauryl sulfate, sucrose, sugar spheres, talc, titanium dioxide, and triethyl citrate. The 20 and 60 mg capsules contain iron oxide yellow as well (CitationAnderson et al 1996).
Pharmacokinetics
Absorption
Following administration of duloxetine hydrochloride there is a 2 hour delay in absorption, owing to the enteric coated pellets previously discussed. It is then well-absorbed, achieving maximal plasma concentrations (Cmax) about 6 hours post dosing (time to maximal concentration, or Tmax). Steady state plasma concentrations are usually accomplished about 3 days after initiation of therapy. The Cmax for duloxetine is not affected by food, but the Tmax is prolonged from 6 hours to 10 hours when given with the food present in the gastrointestinal tract. Furthermore, the overall extent of absorption (area under the curve, or AUC) is reduced by about 10% when administered with food. Studies demonstrate a 3 hour delay in absorption of drug in the evening dose as compared to that of the morning dose.
Distribution
The apparent volume of distribution (VD) of duloxetine is known to be 1640 L. It is highly bound to plasma proteins (>90%), but the interactions between this drug and other highly plasma protein-bound compounds have not been adequately studied. The principal proteins involved in the binding of duloxetine include albumin and α1-acid glycoprotein. The plasma protein binding of duloxetine is not significantly affected by hepatic or renal insufficiency.
Metabolism
The elimination half-life (t1/2) of duloxetine is about 12 hours (range: 8–17 hours). It undergoes extensive hepatic metabolism to inactive compounds. This is mostly carried out by the cytochrome P-450 isoenzymes, 2D6 and 1A2, which catalyze the oxidation of the naphthyl ring. These metabolites are subsequently conjugated and then either eliminated or oxidized further prior to elimination. There are many apparent metabolites as previously discussed, but the two major ones are 4-hydroxy-duloxetine glucuronide and 5-hydroxy-6-methoxy-duloxetine sulfate. All others represent only minor routes of transformation (CitationKuo et al 2004).
Elimination
Less than 1% of the given dose of duloxetine appears in the urine as unchanged parent drug. About 70% of the dose appears in the urine as inactive metabolites. Only about 20% is eliminated in the feces (CitationLantz et al 2003).
Interactions with other drugs
Since the principal metabolism pathways of duloxetine are through the cytochrome P450 isoenzymes 1A2 and 2D6 (CYP1A2 and CYP2D6), it would be anticipated that drugs that interfere with or alter the activity of these hepatic enzymes would potentially alter the blood levels of duloxetine in the patient to which the drugs were co-administered. Since duloxetine is not a substrate, inhibitor, or inducer of the important CYP3A4, there are no concerns regarding co-administration with drugs known to affect that metabolic enzyme, such as macrolide antibiotics or antifungal agents. Duloxetine has no important or measurable effects on and is not a substrate for monoamine oxidase. However, because serious problems have resulted from the co-administration of SSRI with a monoamine oxidase inhibitor (MAOI), duloxetine should not be given in combination with any MAOI or within 2 weeks of discontinuing an MAOI. Further, an MAOI should not be started within 5 days of discontinuing duloxetine. Clinically speaking, there are only a few other concerns of note that involve drugs handled by the CYP1A2 and CYP2D6 systems that require attention by the prescriber.
Inhibitors of CYP1A2
Pharmacokinetic studies looked at the interaction between duloxetine and fluvoxamine, a known potent inhibitor of CYP1A2. The results demonstrated a 5-fold increase in the AUC for duloxetine and a 2.5-fold in the Cmax. The t1/2 of duloxetine was increased approximately 3-fold. Other drugs known to inhibit CYP1A2 include cimetidine and quinolone antibiotics such as ciprofloxacin.
Inhibitors of CYP2D6
Paroxetine is a moderate inhibitor of CYP2D6. A pharmacokinetic study was done looking at the serum concentration curves of duloxetine when concomitantly administered with low to moderate doses of paroxetine (20 mg), showing increases in both the AUC and Cmax of 60% for duloxetine. Other medications with similar actions on CYP2D6, such as fluoxetine and quinidine, would be expected to have similar effects on the concentration of duloxetine and should be used with caution together with this drug (CitationSkinner et al 2003).
Drugs metabolized by CYP1A2
Duloxetine has no ability to induce CYP1A2, but is known to have mild inhibitory effects on the enzyme in vitro. In clinical pharmacokinetic studies, there were no significant effects seen in the concentration of CYP1A2 substrates such as theophylline when co administered with duloxetine given 60 mg bid. Therefore, duloxetine is not felt to have significant effects on drugs metabolized by the CYP1A2 system (CitationDiVirgilio 2002).
Drugs metabolized by CYP2D6
Duloxetine is a moderate inhibitor of the actions of CYP2D6 and requires caution when co-administering agents metabolized by that system. Duloxetine has been shown to increase the AUC of desipramine 2.9-fold and its Cmax by 70% when coadministered (CitationSkinner et al 2003). Because of this effect, certain other drugs including other TCAs (nortriptyline, amitriptyline, and imipramine), phenothiazines, and type 1C anti-arrhythmics (flecainide, propafenone) should be administered at a lower dose than usual and monitored carefully. Because of the risk of fatal arrhythmia, duloxetine should not be co-administered with thioridazine under any circumstance.
Other drug–drug interaction concerns
There are theoretical concerns that the co-administration of duloxetine with medications that raise the gastrointestinal pH could result in a hastened dissolution of the enteric coating of the pellets of duloxetine. The concern would be that the drug might be more readily and rapidly absorbed under these conditions. However, co-administration studies done with aluminum and magnesium hydroxide suspension and with famotidine showed no change in the pharmacokinetics of a 40 mg dose of duloxetine. Studies have not been conducted with proton pump inhibitors (CitationSathirakul 2002).
Any CNS-acting medication should be used with caution when administered to patients already on other CNS-acting medications. Sedation or other impairment of CNS functioning could occur sporadically and without warning in individual patients. Therefore cautious use is warranted. Specific pharmacokinetic interactions with benzodiazepines have not been demonstrated in studies. There appears to be no significant interaction with alcohol on initial evaluation (CitationSkinner and Weerakkody 2002).
Clinical efficacy
The clinical efficacy of duloxetine for pain associated with diabetic peripheral neuropathy has been demonstrated in three double-blind, placebo-controlled, randomized studies. These studies (n = 1139) were 12-week fixed-dose trials (CitationGoldstein et al 2005; CitationRaskin et al 2005; CitationWernicke et al 2006a) that enrolled Type I or Type II diabetics with painful diabetic neuropathy for greater than 6 months duration with at least moderate 24-hour pain severity. All three trials randomized participants to a duloxetine 60 mg qd, duloxetine 60 mg bid, or placebo treatment arm. CitationGoldstein et al (2005) also included a 20 mg qd treatment arm. In all three trials, investigators excluded patients with major depressive disorder (MDD). The primary efficacy end point for all studies was mean change on average daily pain severity (measured using an 11-point Likert-type pain scale from baseline to the end of the 12-week dosing period. Secondary endpoints included other pain diary outcomes (24-hour worst pain, night pain), clinician/patient global impressions (CGI, PGI), scales from the Brief Pain Inventory (BPI), a QOL measure (Short Form-McGill Pain Questionniare; SF-MPQ), dynamic allodynia, and the Hamilton Depression Rating Scale (HAM-D17).
Participant profile and weekly outcomes
shows the baseline characteristics of patients for all studies. A total of 77.8% of subjects completed the trials. shows that in all 3 trials, subjects in the duloxetine 60 mg qd or 60 mg bid treatment arm reported a greater decrease in 24-hour average pain severity relative to placebo after the first week and this effect was maintained throughout the 12-week dosing period. In the CitationGoldstein et al (2005) trial, subjects in the 20 mg arm did not show significant pain reduction relative to placebo. also shows that in all 3 studies, the reduction in 24-hour average pain severity was consistent with a dose dependent response.
Figure 2 Primary efficacy measure (24 hour average pain severity score) in duloxetine-treated patients with pain associated with diabetic neuropathy. Reproduced with permission from CitationGoldstein D, Lu Y, Detke MJ et al 2005. Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain, 116:109–18. Copyright © IASP®.
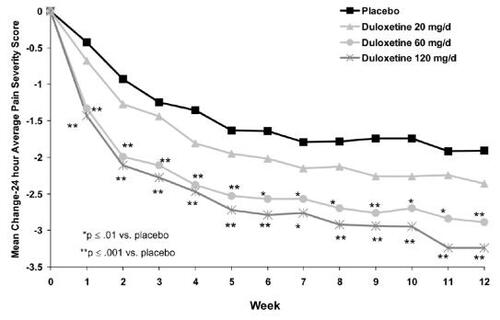
Table 1 Baseline characteristics of patients
Pooled outcomes and analysis of effect sizes
In order to gain a better sense of treatment effects across all studies and to assess more accurately the magnitude of differences among treatment arms and placebo (termed “effect size”), outcome data were combined across studies. shows the change in the primary outcome; the 24-hour pain diary score from baseline to week 12 and change in two other pain diary measures (24-hour worst pain and night pain). shows the change in the BPI scales (severity, interference, general activity) from baseline to week 12. Both the 60 mg qd and the 60 mg bid arms showed significant reductions (all p < 0.001) across all pain diary measures and BPI scales relative to changes seen in the placebo arm. shows the mean change from baseline to week 12 on the CGI, PGI, SF-MPQ, dynamic allodynia, and HAM-D17 for each treatment arm. Duloxetine 60 mg qd showed significant improvement on CGI, SF-MPQ and decreased dynamic allodynia relative to placebo. Duloxetine 60 mg bid showed significant improvement on CGI and SF-MPQ relative to placebo.
Figure 3 Pooled mean change (baseline-week 12) for pain diary scores in duloxetine-treated patients. *Both treatment arms showed significant improvement (p < 0.001) from baseline to end of treatment period on all measures relative to change seen in placebo.
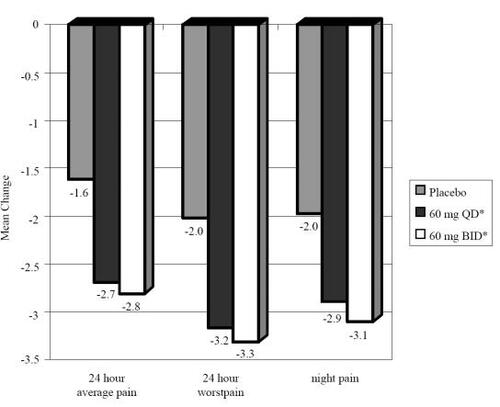
Figure 4 Pooled mean change (baseline-week 12) for Brief Pain Inventory (BPI) subscales. *Both treatment arms showed significant improvement (p < 0.001) from baseline to end of treatment period on all measures relative to change seen in placebo.
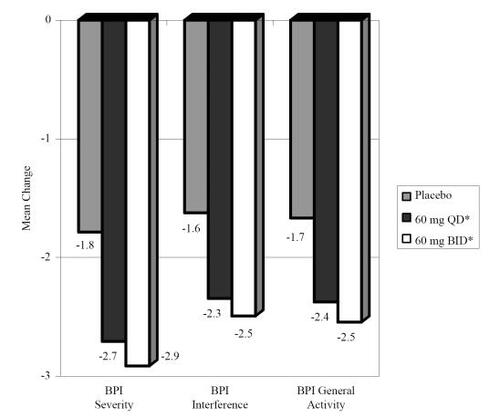
Table 2 Mean change and 95% CI for mean difference between treatment arm and placebo from baseline on secondary outcome measures
shows the mean difference in effect size (ES; with 95% CI for the effect) for each outcome. ES was calculated using Cohen’s d. Looking at ES allows the reader to better ascertain the magnitude of group differences as well as provide a hint of whether this difference is considered “clinically relevant” (where higher ES is reflective of a greater clinical relevance). By convention, Cohen’s d <0.2 = negligible difference; 0.2–0.49 = small; 0.5–0.79 = medium; ≥0.8 = large. The table shows that there was a medium effect for improvement in average 24-hour pain for both treatment arms relative to placebo. There also was a medium ES seen when comparing change in the duloxetine 60 mg bid and placebo arms for the following: 24-hour worst pain, BPI severity, CGI, and SF-MPQ. There was a small ES seen when comparing change in the duloxetine 60 mg bid and placebo arms for the following: night pain, BPI general activity and BPI interference. There was a small ES seen when comparing change in the duloxetine 60 mg qd arms and placebo for the following: 24-hour worst pain, night pain, BPI severity, BPI general activity, BPI interference, CGI, and SF-MPQ. There was also a statistically significant difference between change in dynamic allodynia between the duloxetine 60 mg qd and placebo arms; however, the ES was considered negligible and is thus of little or no clinical relevance.
Table 3 Effect sizes for mean differences comparing duloxetine 60 mg qd and 60 mg bid with placebo
The investigators in all three studies concluded that duloxetine 60 mg qd and 60 mg bid were effective for treating pain associated with diabetic neuropathy. All three studies argued that the data confirmed the proposed role of 5-HT and NE as key mediators of descending pain pathways. They also suggested that 5-HT and NE reuptake inhibition by duloxetine may offer an effective and safe alternative for the treatment of persistent pain states. Moreover, since differences in pain did not correlate with changes in mood (as measured by change in the HAM-D17) and patients with MDD were excluded, it was apparent that the pain relief was not due to improvement in depression.
Safety and tolerability
Safety and tolerability of controlled trials
Safety and tolerability was evaluated for all three controlled trials as well as three long term (52 week) open label studies (CitationRaskin et al 2006; CitationWernicke et al 2006a, Citationb, Citation2007). We will first report the safety and tolerability findings of the blinded trials. Overall, 67/339 (19.7%) discontinued during the study period. shows the pooled percentage that discontinued from each arm among the controlled trials (CitationGoldstein et al 2005; CitationRaskin et al 2005; CitationWernicke et al 2006a, Citationb). In regards to serious adverse events (SAEs), a total of 41/1139 (3.6%) patients reported at least one SAE; however, SAEs did not differ among groups. CitationGoldstein et al (2005) and CitationWernicke et al (2006a, Citationb) included the treatment emergent adverse events (TEAEs) for each group as well as lipid profiles. shows the percentages of individuals who experienced the most common TEAEs. shows the pooled changes in lipid profiles and glycosylated hemoglobin (HbA1c). The only group difference was for high density lipoprotein (HDL) between the duloxetine 60 mg bid and placebo treatment arms; however, the difference (0.027) is not clinically significant.
Figure 5 Percentage of patients discontinuing treatment per arm.
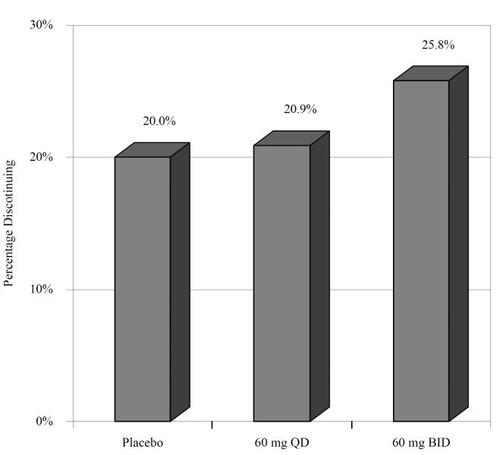
Figure 6 Treatment-emergent adverse events in controlled trials (CitationGoldstein et al 2005; CitationWernicke et al 2006a, Citationb).
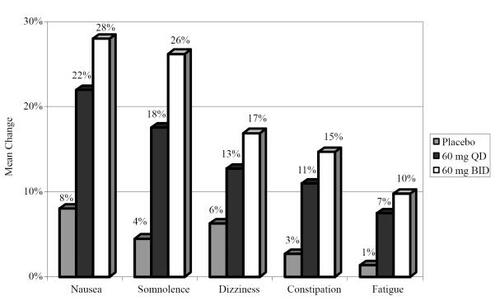
Table 4 Mean change in HbA1c and lipid profile from baseline to end point for controlled trials
Safety and tolerability of long-term open label trials
Discontinuation and adverse events
Within the long-term open label trials (CitationRaskin et al 2006; CitationWernicke et al 2006a, Citationb, 2007) participants were assigned to either a duloxetine 60 mg bid or routine care arm (at a 2:1 ratio, respectively). The completion rate was 77.6% (450/580) for the duloxetine 60 mg bid arm and 83.6% (240/287) for the routine care arm. A total of 150/867 (17.3%) patients reported at least one SAE; however, SAEs did not differ between arms. A total of 6.3% (18/287) in the routine care arm discontinued due to a SAE compared with 10.2% (77/689) in the duloxetine 60 mg bid arm (this difference was not significant). CitationRaskin et al (2006) and Wernicke et al (2007) reported no significant group differences for TEAEs. Wernicke et al (2006) reported 8 TEAEs that occurred more frequently in the routine care group. The only TEAE that was reported by >5% in the duloxetine 60 mg BID arm for all three studies was nausea. No TEAE was reported by >5% in the routine care arm for all three studies.
Lipid profiles and HbA1c
Within the open label trials, 52-week lipid profile change analysis showed no consistent pattern of group differences. CitationRaskin et al (2006) reported a greater decrease in HDL in the routine care group (p < 0.001), CitationWernicke et al (2006a, Citationb) reported a grater decrease in low density lipoprotein (LDL) in the routine care group (p < 0.001), and Wernicke et al (2007) reported a greater increase in HbA1c in the duloxetine group.
Analysis of chemistry/urinalysis
summarizes the changes in chemistry analytes and urinalysis from baseline to endpoint. Significant differences were seen between groups on six laboratory values. Albumin g/L, alanine transaminase/serum glutamate pyruvate transaminase (ALT/SGPT) U/L, aspartate transaminase/serum glutamate oxaloacetic transaminase (AST/SGOT) U/L, total cholesterol mmol/L, gamma-glutamyl transferase (GGT) U/L, and fasting glucose all showed a greater increase among those in the duloxetine 60 mg bid arm relative to the increase among those in the routine care arm. Further examination of the ES indicated that all of the differences were negligible except for glucose and cholesterol (where the ES was small).
Table 5 Mean change in chemistry/urinalysis profiles from baseline to end point for long-term open-label studies
QOL measures
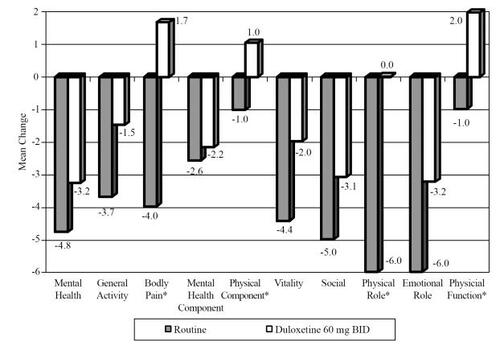
shows the changes in SF-36 measures from baseline to endpoint. A decrease on the SF-36 indicates a poorer QOL. Those in the routine care group showed greater decreases on all SF-36 subscales relative to change in the duloxetine 60 mg bid arm. These differences were significant for the bodily pain, physical component, and physical role subscales. In addition, those in the routine care arm had a significantly greater decrease from baseline to endpoint as measured by the European QOL (indicating a poorer QOL) scale relative to change among those in the duloxetine 60 mg bid arm. A decrease on the SF-36 scales and European QOL indicate a poorer QOL.
Figure 7 SF-36 scales change from baseline in long-term open label studies. *p < 0.05.
Conclusions and expert opinion
Research and clinical experience in the last decade has contributed to new and clearer understanding of the physiology surrounding descending pain inhibition pathways of the brainstem and spinal cord. The advantages of balanced 5-HT and NE reuptake inhibition as a pharmacotherapeutic modality are essentially without challenge. Duloxetine has the unique profile of dual 5-HT and NE reuptake inhibition in a balanced fashion with little or no other receptor or channel effects. This relatively “clean” mechanism of action places this compound in position to provide relief of chronic pain syndromes, such as painful diabetic neuropathy, without the significant side effect issues associated with routine current and past therapies.
Data from both short and long term trials shows that duloxetine consistently provides significantly more diabetic neuropathic pain relief than either placebo or routine care. This is seen in both pain measures and QOL measures assessing the impact pain has on functioning. In long-term trials, there are few side effects from using duloxetine that would decrease patient adherence. There is a small (but clinically insignificant) increase in HDL among those taking duloxetine relative to placebo in short-term controlled trials. In the long-term open label studies, small differences are seen in fasting glucose, albumin, GGT, AST/SGOT, ALT/SGPT, and total cholesterol among those taking duloxetine relative to routine care. When considering the current findings, it is important to note that the USPI for duloxetine notes small increases in fasting glucose in short-term controlled trials and small increases in HbA1c in long-term trials relative to placebo or routine care (CitationEli Lilly and Company 2007). However, the magnitude of these differences is not clinically significant and thus should not be a major concern for most patients.
The pain of diabetic peripheral neuropathy is one of the more severe and debilitating complications of diabetes mellitus. Diminished physical functioning, poor sleep, and psychologic comorbidities contribute to poorer QOL among those with this condition. Accordingly, one can easily understand how patients with painful diabetic neuropathy begin to slip into a downward spiral of decreasing physical activity, depression, poor self-care, worsening hyperglycemia, and progressive neuropathic pain. The regulatory approval of duloxetine for use in diabetic neuropathy is a milestone in pain management as it represents the first approved agent for this condition. Other agents have since followed, further contributing to a better understanding of the mechanisms of central sensitization and the neuroplastic changes associated with chronic and neuropathic pain.
References
- AndersonNOrenPOguraTDuloxetine enteric pelletsOfficial Gazette of the United States Patent and Trademark Office Patents199611851954
- BymasterFDreshfield-AhmadLThrelkeldPComparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes and other neuronal receptorsNeuropsychopharmacology2001258718011750180
- DiVirgilioSGonzalesCKnadlerMEffect of duloxetine (DU) on CYP1A2-mediated drug metabolism and the pharmacokinetics (PK) of theophylline (THEO)Clin Pharmacol Ther20027163
- GoldsteinDLuYDetkeMJDuloxetine vs. placebo in patients with painful diabetic neuropathyPain20051161091815927394
- HokeAFeasbyTHumesHDisorders of the peripheral nervous systemKelley’s Textbook of Internal Medicine2000PhiladelphiaLippincott Williams and Wilkins29801
- IyengarSWebsterAHemrick-LueckeSEfficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models in ratsJPET200431157684
- KuoFGillespieTKulanthaivelPSynthesis and biological activity of some known and putative duloxetine metabolitesBioorg Med Chem Lett2004143481615177457
- LantzRGillespieTRashTMetabolism, excretion, and pharmacokinetics of duloxetine in healthy human subjectsDrug Metab Dispos20033111425012920170
- LynchMAntidepressants as analgesics: a review of randomized controlled trialsJ Psychiatry Neurosci20012630611212591
- RaskinJPritchettYLWangFA double-blind, randomized mutlicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic painPain Med200563465616266355
- RaskinJSmithTRWongKDuloxetine versus routine care in the long-term management of diabetic peripheral neuropathic painJ Palliat Med20069294016430342
- SathirakulKTengLYeoKImpact of gastric pH and the presence of activated charcoal on the absorption of duloxetineInt J Clin Pharmacol20027118
- SkinnerMKuanHPanADuloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteersClin Pharmacol Ther200373170712621382
- SkinnerMWeerakkodyGDuloxetine does not exacerbate the effects of alcohol on psychometric testsClin Pharmacol Ther20027153
- Eli Lilly and CompanyCymbalta (duloxetine) package insert [online]2007 Accessed March 1, 2007. URL: http://pi.lilly.com/us/cymbalta-pi.pdf
- WadaRYagihashiSRole of advanced glycation end products and their receptors in development of diabetic neuropathyAnn N Y Acad Sci2005104359860416037282
- WernickeJFPritchettYLD’SousaDNA randomized controlled trial of duloxetine in diabetic peripheral neuropathic painNeurology2006a6714112017060567
- WernickeJFRaskinJRosenADuloxetine in long-term management of diabetic peripheral neuropathic pain: an open-label, 52 week extension of a randomized controlled clinical trialCurr Ther Res Clin Exp2006b67283304
- WernickeJFWangFPritchettYLAn open-label 52 week clinical extension comparing duloxetine with routine care in patients with diabetic peripheral neuropathic painPain MedfIn press
- WongDBymasterFDual serotonin and noradrenalin reuptake inhibitor class of anti-depressants: potential for greater efficacy or just hype?Prog Drug Res20025816922212079200
- WongDBymasterFMayleDLY248686, a new inhibitor of serotonin and norepinephrine uptakeNeuropsychopharmacology1993823338424846
- WoolfCMannionRNeuropathic pain: aetiology, symptoms, mechanisms and managementLancet199935319596410371588
- YakshTPharmacology of spinal adrenergic systems which modulate spinal nociceptive processingPharmacol Biochem Behav198522845582861606
- ZhouMGebhartGBiphasic modulation of spinal nociceptive transmission from the medullary raphe nuclei in the ratJ Neurophysiol199778746589307109