Abstract
High plasma homocysteine levels are a known risk factor in heart failure and sudden cardiac death. The G proteins, Gs (stimulatory) and Gi (inhibitory), are involved in calcium regulation; overexpression has pathological consequences. The aims of this study were to examine the differential expression of Gs G protein and Gi in the hearts of hyperhomocysteinemic (Hhcy) mice, and to determine if homocysteine (Hcy) acts as an agonist in cell culture to mediate the change in G protein isoforms. To create Hhcy, heterozygous cystathionine-β-synthase (CBS) knockout (KO) mice were used. Mice were sacrificed, hearts were excised, cardiac tissue homogenates were prepared, and Western blots were performed. The results suggested that Gs G protein was downregulated in cardiac tissue of heterozygous CBS KO mice to 46% that of control hearts. However, the intracellular Gi G protein content remained the same in heterozygous CBS KO mice. Transformed cardiomyocyte HL-1 cells were treated with varying concentrations of homocysteine. The results suggested no detectable differential Gs and Gi expression. This suggested that Hcy did not act as an agonist in vitro to alter G protein content, but that Hcy produced some other in vivo effects to incur these results.
Introduction
Homocysteine (Hcy) is a nonprotein amino acid. It differs from cysteine only in its side chain that contains an additional methylene group before the thiol group.Citation1,Citation2 Moreover, Hcy is the result of methionine metabolism and is converted to cystathionine by cystathionine-β-synthase (CBS); an established model for creating hyperhomocysteinemia (Hhcy) is with heterozygous CBS knockout mice (CBS KO mice) since these mice are unable to convert Hcy to cystathionine, resulting in excess levels of plasma Hcy.Citation3
A high plasma Hcy level has recently been suggested to be a major risk factor surrounding chronic heart failure (CHF).Citation4–Citation9 Recently, Hcy has been shown to increase ERK phosphorylation in cultured microvascular endothelial cells (MVEC).Citation10 Hcy-induced ERK phosphorylation was suppressed by pertussis toxin (PTX).Citation10 Since PTX is known to block Gi and Gq-mediated pathways, this suggested the involvement of G protein-coupled receptors (GPCRs) in initiating signal transduction by Hcy that leads to ERK activation.Citation10 GPCRs are integral membrane-spanning proteins that have G proteins attached intracellularly; upon activation of a GPCR by an agonist, the G-proteins detach from the receptor and proceed to activate other effectors.Citation11 There is a large class of GPCRs that utilize a great variety of different G proteins. Cardiac-adrenergic receptors are an example of GPCRs that are important regulators of cardiac function.Citation12
Stimulation of GPCRs can occur with the following: sympathetic or parasympathetic neuronal activation, by circulating catecholamines, by adrenergic agonists that increase heart rate (chronotropism), force of cardiac contraction (inotropism), rate of cardiac relaxation (lusitropism), and automaticity.Citation12 The prominent β1 subtype is mainly responsible for positive chronotropic and inotropic effects of catecholamines.Citation13 Activation of a GPCR coupled to Gs increases the activity of adenylyl cyclase and increases intracellular calcium concentration.Citation12 Calcium is a known modulator of cardiac function.
Net G protein content available for signaling is imperative for maintaining homeostasis. Many studies indicate cardiomyopathy from overexpression of G proteins. Cultured neonatal cardiac myocytes from transgenic mice overexpressing cardiac G showed a five-fold higher Icalcium compared with wild type controls.Citation14 In TG mice overexpressing the Gs sub-unit, heart rate and left ventricular ejection fraction (LVEF) was elevated compared with littermate wild-type mice.Citation15
In summary, there is an association of high plasma Hcy levels and heart failure.Citation4–Citation9
Moreover, calcium regulation is crucial to cardiac function. Evidence from our lab suggested the involvement of GPCRs in modulating calcium-induced ERK activation in MVEC.Citation10 Moreover, Gs and Gi are fundamental effectors that regulate calcium homeostasis in cardiomyocytes; overexpression can result in pathology.Citation12,Citation14,Citation15 We, therefore, hypothesized that high plasma Hcy levels could modulate net G protein content available for signaling in cardiomyocytes in vitro and in vivo, thereby modulating calcium homeostasis. Moreover, we were unsure whether content would increase or decrease since our results could indicate a contribution or mitigation to cardiomyopathy. Cultured HL-1 transformed cardiomyocytes were used as models for cardiac response to determine if homocysteine acts as an agonist in isolation to alter G protein content and if these results could be replicated in a more complex living system.
Materials and methods
Animals
All animal procedures were in accordance with the National Institute of Health Guidelines for animal research and were approved by the Institutional Animal Care and Use Committee of the University Of Louisville School Of Medicine. There were 4 mice used in each of 2 groups: control, CBS KO. The CBS KO mice used were also used in a previous study: 3-Deazaadenosine mitigates arterial remodeling and hypertension in hyperhomocysteinemic mice.Citation16 Wild-type (WT; B6.129PF2/J) and breeding pairs of CBS heterozygote (–/+) knockout (CBSKO; B6.129P2-Cbstm1Unc) mice aged 8–12 wks were obtained from Jackson Laboratories (Bar Harbor, ME). Mice were bred at the breeding facility at University of Louisville School of Medicine. Homozygous mutants, completely lacking CBS, have 40- to 50-fold higher plasma Hcy levels and a very short life span; hence, they were not used for the study. In this study, we used male heterozygous animals with plasma Hcy levels between 25 and 30 μM, a well established model of HHcy. Each group received a standard lab chow for that duration. After 12 weeks of treatment, mice were sacrificed, hearts were excised and stored at −70 °C until further experimentation. Homogenates were prepared from ventricle of the hearts.
Plasma Hcy measurements
The level of Hcy in plasma was quantitatively analyzed with the Bio-Rad microplate enzyme immunoassay homocysteine assay (Bio-Rad Laboratories, Hercules, CA) and a Spectra-Max M2 Analyzer (Molecular Devices, Sunnyvale, CA) according to the manufacturers’ instructions to ensure Hcy levels of CBS KO were between 25 and 30 μM.
Chemicals
The antibodies against Gs and Gi were obtained from Santa Cruz (Santa Cruz, CA). The antibody against-actin was obtained from Sigma (St. Louis, MO). Plain DMEM/F-12 50/50 medium was obtained from Mediatech, Inc (Herndon, VA). Fetal Bovine Serum was acquired from Gemini Bio-Products, (West Sacramenta, CA). Claycomb medium with penicillin, streptomycin, trypsin-EDTA, and Hanks’ balanced salt solution (HBSS) were purchased from GIBCO-BRL (Grand Island, NY); DL-Hcy, NaCl, sodium orthovanadate, Tris, EDTA, EGTA, dithiothreitol, NP-40, protease inhibitor cocktail, fibronectin, agarose, from Sigma (St. Louis, MO); D,l-homocysteine was obtained from Sigma (St. Louis, MO).
Cell culture and treatments
Cultured HL-1 cells were received as a gift from LSU, (New Orleans, LA) The complete Claycomb medium was supplemented with sterile 100 μM norepinephrine bitartrate (10 mL/L of a 10 mM norepinephrine stock solution), 4 mM L-glutamine (Catalog No. 59202C) and 10% FBS prior to use. A T-25 flask was coated with fibronectin overnight before plating cells. Cells were grown to near confluence, and serum starved overnight when treated with the indicated reagents for Western blot analysis. Claycomb Medium is designed for use in a 5% CO2 humid environment. HL-1 cells require higher than normal cell densities for optimal growth and behavior.
Preparation of samples, Western blot analysis, and immunodetection
After treatment, medium was aspirated from six-well culture dishes and HL-1 were washed twice with ice-cold 1 × PBS. Ice-cold lysis buffer (in mM: 50 Tris · HCl, pH 7.4, 150 NaCl, 1% Triton X-100, and 1 EGTA) and freshly prepared inhibitors (1 mM PMSF, 1 μg/ml leupeptin, 200 μM sodium orthovandate, and 1 μg/ml aprotinin) were added to each well. Plates were scraped on ice, and the supernatant containing cytosolic protein was collected and centrifuged at 5500 g for 10 min at 4 °C, and resolved by SDS-PAGE on 10% polyacrylamide gels and blotted onto a polyvinylidene difluoride membrane. After being transferred, blots were washed with Tris-buffered saline (TBS) for 5 min at room temperature and incubated in blocking buffer for 1 h at room temperature. The blots were then incubated with the indicated primary antibodies [appropriate dilutions in 3% Milk solution of TBST, (0.1% Tween 20 + TBS: TBST)] overnight at 4 °C using gentle agitation. The blots were washed four times (5-min wash each time) with TBST and incubated with HRP-conjugated secondary antibody (1:3,000 dilution in 3% Milk-TBST). After being washed with TBST 4 times (10 minutes wash each time), the proteins of interest were detected using an ECL plus kit (Amersham Biosciences, Piscataway, NJ). The membranes were then stripped using 0.2 M NaOH solution for 30 min at room temperature and reprobed with a standard, -actin.
Statistical analysis
Statistical analysis was performed using an unpaired Student’s t-test for comparison between control, Hcy, and other pharmacological treatment groups. P < 0.05 was considered statistically significant. All results are averages ± SE of at least three different experiments.
Results
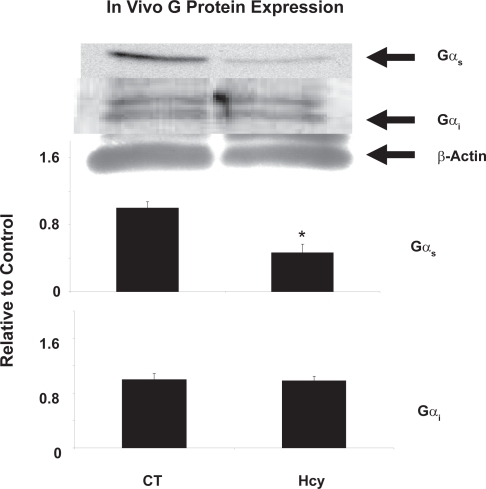
We report that Gs expression in left ventricular heart tissue decreased to 46% of the control value of expression (P < 0.01, n = 4) (). Gi expression remained unchanged in heart homogenates of both CBS KO mice and wild type mice. Equal protein concentration was determined using Bradford dye experiment, while equal loading was confirmed using -actin.
Figure 1 In vivo G protein content of Gs and Gi in wild type and CBS KO mice. β-actin was used as a standard for both protein samples. Gs content in CBS KO mice was reduced to 46% (P < 0.01, n = 4) that of control values based on 3 different tissue homogenates. Pixel intensity was digitized using UnScanIt Software.
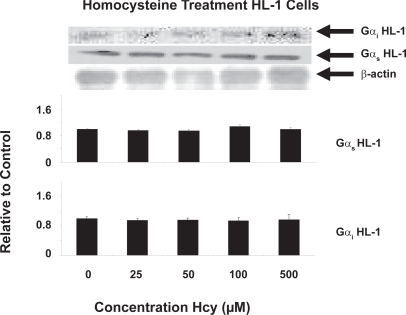
The level of Gs expression and Gi expression remained the same in HL-1 transformed cardiomyocytes cultured cells treated with the following concentrations of Hcy for 24 hours: 0 μM, 25 μM, 50 μM, 100 μM, 500 μM. There were no significant changes in expression (P < 0.05, n = 3) ().
Figure 2 In vitro G protein content of Gs and Gi in serum-starved HL-1 cells using the following concentrations for 24 hours: 0 μM, 25 μM, 50 μM, 100 μM, 500 μM. No differential expression was detected (P < 0.05, n = 3). Pixel intensity was digitized using UnScanIt Software.
Discussion
There is a consistent association of high plasma Hcy with clinical and echocardiographic measures of CHF.Citation9 In fact, data suggested that Hhcy is not only associated with an increased incidence of CHF, but also with severity of disease.Citation4 Moreover, Hhcy could evoke cardiac remodeling that features interstitial and perivascular fibrosis.Citation4 The presence of elevated plasma Hcy is also an independent predictor of progression of coronary plaque load that results in atherosclerosis.Citation7 In heart failure, the sympathetic system is activated, cardiac receptor number and function are decreased, and downstream mechanisms are altered.Citation13 Our CBS KO mice demonstrated indicators of heart failure such as hypertension, hypertrophy, and arterial remodeling.Citation16 It is important to note that metabolic, structural and functional effects on the heart may differ fundamentally if Hhcy is induced by methionine, homocystine or B-vitamin deficiency.
Downregulation of GPCRs can clearly result in differences of G protein content since G proteins are attached to their respective receptors via cytoplasmic C-terminal and cytoloops.Citation17–Citation19 Several studies show differential β-adrenergic downregulation in conjunction with the differential G protein expression that we observed in vivo. The decrease resulted from receptor internalization, sequestration, and consumption.Citation20 In a study using a pacing tachycardia HF model in canines, HF resulted in a uniform reduction in β1-AR density in surface and T-tubule membrane.Citation21 In a pulmonary in vivo model of β2-adrenoceptor desensitization and internalization, it was found that albuterol or salmeterol induced a downregulation of Gs·Citation22,Citation23 Another fluorescent study of Gs showed that internalization of β2 adrenergic receptors and Gs subunit occurs simultaneously, providing another link for receptor downregulation occurring in conjunction with differential G protein content.Citation24
It is important to note that there are several GPCRs that utilize the Gs and Gi G proteins other than the β-adrenergic receptors.Citation25 It is likely that the decrease in Gs can be attributed to downregulation of several different GPCRs that utilize this G protein. Western blot analysis showed Gs content is decreased to 46% that of control levels in CBS KO mice ().
Varying levels of G proteins and GPCRs can influence cardiomyopathy. The result of a disrupted balance of β-ARs can be seen in a study of TG mice overexpressing β1-adrenoceptors that encounter mortality sooner.Citation26 β-AR blockage by propranolol treatment was salutatory in these TG mice, and can prevent the development of cardiomyopathy.Citation27 The model of volume overload cardiac hypertrophy produced our same results for CBS KO mice: decreased content of Gs, but not of Gi in heart.Citation28 These results have a clear link to Hcy-induced CHF and hypertrophy; it is already known Hhcy leads to pathological ventricular hypertrophy in normotensive rats.new1 Clinical and experimental studies have indicated a role of homocysteine in ventricular hypertrophy via angiotensin II mechanism.new2 Moreover, a human study showed an association between plasma homocysteine concentrations and cardiac hypertrophy in end-stage renal disease.Citation7,Citation31
G protein content is important for proper cardiac function. It was found that activation of Gs via β-adrenergic receptors enhanced the activity of cardiac voltage-dependent Ca2+ channels of the l-type.Citation33 One other study showed TG mice with cardiac Gs overexpression exhibited enhanced inotropic and chronotropic responses to sympathetic stimulation, and TG mice developed cardiomyopathy with age. Other TG models with selective induction of Gi subunit expression showed novel downstream events in hypertrophic signaling that could be critical factors leading to cellular electrophysiological remodeling and cardiac arrhythmias in hypertrophic cardiomyopathy.Citation32
Hcy treatment of HL-1 transformed cardiomyocytes at varying doses for 24 hours produced no detectable changes in intracellular Gs or Gi protein content, which suggested that downregulation of any GPCRs utilizing these G proteins did not occur in isolated treatment of Hcy agonist (). It is known that treatment of cells with receptor agonists for many minutes or hours results in a loss of cellular receptors (downregulation).Citation6 Hence, this treatment would have been sufficient to evoke receptor downregulation and variable G protein content if Hcy is, indeed, an agonist to GPCRs utilizing these G proteins. Since this is not the case, this suggested that Hcy does not act as an agonist to evoke GPCR downregulation and modulate Gs content. Therefore, this suggested that Hcy acts via some other mechanism in vivo to evoke differential Gs content.
Conclusion
Results indicated that Gs is the only G protein of interest decreased in vivo, this limits available Gs for calcium signaling. Moreover, our results suggested a compensatory decrease of calcium signaling via G protein pathway in spite of greater chronotropy and ionotropy characteristic of heart failure. Moreover, Hcy did not act as an agonist in vitro to incur differential G protein content, which suggested that Hcy incurs some other in vivo effects that lead to a decrease in Gs in the living system.
Limitations
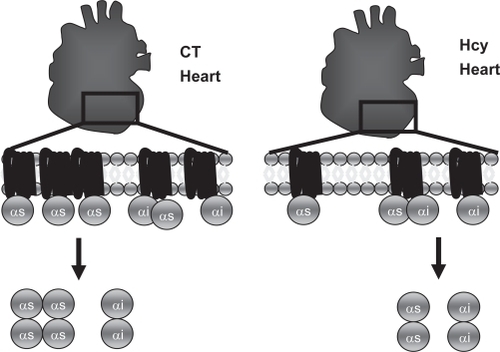
We did not test whether the levels of expression of G protein isoforms of interest are the same in CBS KO mice at the early stage (normal cardiac function) and at the late stage (heart failure); this could have served as another control to ensure the CBS gene did not affect G protein isoforms. Our interest in this study laid in determining net G protein content available for signaling before discovering which GPCRs that contain the respective G proteins are modulated. We speculate that β1-AR downregulation, which contains Gs G protein, is responsible for our measured decrease in Gs G protein in in vivo data since β1-AR is consistently downregulated in cases of heart failure (). Since HL-1 cells are known to demonstrate the same phenotype as primary culture cardiomyocytes, we did not view this as a limitation. The duration of time for treatment in vitro (24 hours) was chosen since it is known that treatment of cells with receptor agonists for many minutes or hours resulted in a loss of cellular receptors (downregulation).Citation6 If downregulation is going to occur, it is likely to be captured within this period of time.
Figure 3 Proposed in vivo model G protein content based on decrease in Gs content. High homocysteine levels created by CBS KO model decreased Gs content available for calcium signaling. A decrease in Gs content decreased chronotropic and ionotropic response to circulating GPCR agonists that utilize this G protein.
Acknowledgements
This research was supported in part by NIH Grants HL-71010, HL-74185, HL-88012 and NS-51568.
References
- RefsumHUelandPMNygardOVollsetSEHomocysteine and cardiovascular diseaseAnnu Rev Med19984931629509248
- SeshadriSPlasma homocysteine as a risk factor for dementia and Alzheimer’s diseaseN Eng J Med200234647683
- OvechkinAVTyagiNSenULominadzeD3-Deazaadenosine mitigates arterial remodeling and hypertension in hyperhomocysteinemic miceAm J Physiol Lung Cell Mol Physiol2006291L9051116815886
- HerrmannMTaban-ShomalOHübnerUBöhmMHerrmannWA review of homocysteine and heart failureEur J Heart Fail20068571616504575
- NiMZhangXHJiangSLZhangYHomocystinemia as an independent risk factor in the Chinese population at a high risk of coronary artery diseaseAm J Cardiology20071004558
- GibelinPSerreSCanditoMPrognostic value of homocysteinemia in patients with congestive heart failureClin Chem Lab Med2006448131616776625
- RasouliMLNasirKBlumenthalRSParkRAzizDCBudoffMJPlasma homocysteine predicts progression of atherosclerosisAtherosclerosis20051811596515939068
- JosephJJosephLShekhawatNSHyperhomocysteinemia leads to pathological ventricular hypertrophy in normotensive ratsAm J Phsyiol Heart Circ Physiol2003285H67986
- HerrmannMKindermannIMüllerSRelationship of plasma homocysteine with the severity of chronic heart failureClin Chem20055115121516040845
- MoshalKSSenUTyagiNRegulation of homocysteine-induced MMP-9 by ERK ½ pathwayAm J Physiol Cell Physiol2006290C8839116251475
- GoughNSTKE focus issue on GPCRs: The evolution of G protein-coupled receptor signalingSci STKE200110412
- PostSHammondKInselPadrenergic receptors and receptor signaling in heart failureAnnu Rev Pharmacol Toxicol1999393436010331088
- LohseMEngelhardtSEschenhagenTWhat is the role of B-adrenergic signaling in heart failureCirc Res20039389690614615493
- LaderASXiaoYFIshikawaYCardiac Gsalpha overexpression enhances L-type calcium channels through an adenylyl cyclase independent pathwayProc Natl Acad Sci U S A1998959669749689139
- HardtSGengY-JMontagneOAccelerated cardiomyopathy in mice with overexpression of cardiac Gsalpha and a missense mutation in the alpha-myosin heavy chainCirculation20021056142011827928
- OvechkinAVTyagiNSenULominadzeDSteedMMMoshalKS3-Deazaadenosine mitigates arterial remodeling and hypertension in hyperhomocysteinemic miceAm J Physiol Lung Cell Mol Phsyiol2006291L90511
- FilizolaMWeinsteinHThe structure and dynamics of GPCR oligomers: a new focus in models of cell-signaling mechanisms and drug designCurr Opin Drug Discov Devel2005857784
- LuttrellLMTransmembrane signaling by G protein-coupled receptorsMethods Mol Biol200633234916878684
- NatarajanKBerkBCCrosstalk coregulation mechanisms of G protein-coupled receptors and receptor tyrosine kinasesMethods Mol Biol2006332517716878685
- Barros RdeAOkoshiMPCicognaACBeta-adrenergic pathway in healthy and hypertrophied heartsArq Bras Cardiol19997264956
- HeJ-GBalijepalliRHaworthRCrosstalk of B-adrenergic receptor subtypes through Gi blunts B-adrenergic stimulation of L-type Ca2+ channels in canine heart failureCirc Res2005975667316100050
- FinneyPABelvisiMGDonnellyLEAlbuterol-induced down-regulation of Gsalpha accounts for pulmonary beta(2)-adrenoceptor desensitization in vivoJ Clin Invest20001061253510880056
- FinneyPDonnellyLBelvisiMChronic systemic administration of salmeterol to rats promotes pulmonary B2-adrenoceptor desensitization and down-regulation of GsalphaBr J Pharmacol200113212617011250877
- AllenJAYuJZDonatiRJRasenickMMBeta-adrenergic receptor stimulation promotes G alpha s internalization through lipid rafts: a study in living cells: a study in living cellsMol Pharmacol200567149350415703379
- HammondHKMechanisms for myocardial B-adrenergic receptor desensitization in heart failureCirculation19938765248381061
- FoersterKGronerFMatthesJKochWJBirnbaumerLHerzigSCardioprotection specific for the G protein Gi2 in chronic adrenergic singaling through B2-adrenoceptorsProc Natl Acad Sci U S A2003100144758014612574
- AsaiKYangG-PGengY-JBeta-adrenergic receptor blockade arrests myocyte damage and preserves cardiac function in the transgenic Gs mouseJ Clin Invest1999104551810487769
- Di FuscoFHashimSAnand-SrivastavaMVolume overload cardiac hypertrophy exhibits decreased expression of Gsalpha and not of Gialpha in heartAm J Physiol Cell Physiol2000279C990811003579
- JosephJJosephLShekhawatNSHyperhomocysteinemia leads to pathological ventricular hypertrophy in normotensive ratsAm J Physiol Heart Circ Physiol2003285H6798612730062
- KassabSGaradahTAbu-HijlehMThe angiotensin type 1 receptor antagonist valsartan attenuates pathological ventricular hypertrophy induced by hyperhomocysteinemia in ratsJ Renin Angiotensin Aldosterone Syst200672061117318789
- PoyrazoğluHMDüşünselRNarinFHomocysteine and left ventricular hypertrophy in children with chronic renal failurePediatr Nephrol200419193814658062
- RuanHMitchellSVainorieneMGi alpha 1-mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathyCirculation200711659660517646583
- BlumensteinYIvaninaTShistikERegulation of cardiac L-type Ca2+ channel by coexpression of Galphas in xenopus oocytesFEBS Lett1999444788410037152