Abstract
Low ethanol intake is known to have a beneficial effect on cardiovascular disease. In cardiovascular disease, insulin resistance leads to altered glucose and lipid metabolism resulting in an increased production of aldehydes, including methylglyoxal. Aldehydes react non-enzymatically with sulfhydryl and amino groups of proteins forming advanced glycation end products (AGEs), altering protein structure and function. These alterations cause endothelial dysfunction with increased cytosolic free calcium, peripheral vascular resistance, and blood pressure. AGEs produce atherogenic effects including oxidative stress, platelet adhesion, inflammation, smooth muscle cell proliferation and modification of lipoproteins. Low ethanol intake attenuates hypertension and atherosclerosis but the mechanism of this effect is not clear. Ethanol at low concentrations is metabolized by low Km alcohol dehydrogenase and aldehyde dehydrogenase, both reactions resulting in the production of reduced nicotinamide adenine dinucleotide (NADH). This creates a reductive environment, decreasing oxidative stress and secondary production of aldehydes through lipid peroxidation. NADH may also increase the tissue levels of the antioxidants cysteine and glutathione, which bind aldehydes and stimulate methylglyoxal catabolism. Low ethanol improves insulin resistance, increases high-density lipoprotein and stimulates activity of the antioxidant enzyme, paraoxonase. In conclusion, we suggest that chronic low ethanol intake confers its beneficial effect mainly through its ability to increase antioxidant capacity and lower AGEs.
Introduction
Ethanol intake is a common lifestyle factor found across many cultures and geographic regions. Since cardiovascular disease is also found ubiquitously and accounts for more than 16 million deaths per year worldwide, it is essential to understand how they relate to each other (CitationWHO 2003a). Various epidemiological and controlled clinical studies have investigated the effects of varying levels and patterns of ethanol intake on cardiovascular health (CitationReynolds et al 2003; CitationCorrao et al 2004). In contrast to high ethanol intake which is detrimental to cardiovascular health, chronic low ethanol has been shown to have a beneficial effect (CitationCamargo et al 1997; CitationOkubo et al 2001; CitationCorrao et al 2004; CitationPiano 2005). Understanding the biological mechanism of this beneficial effect will aid in identifying potential preventative or therapeutic agents. In this review, we discuss the factors involved in the development and progression of cardiovascular disease and the possible biochemical mechanisms by which low ethanol counters these factors to prevent or attenuate this disease.
Cardiovascular disease
Cardiovascular disease includes atherosclerosis and hypertension. Hypertension affects more than 600 million people worldwide and results in 13% of the total deaths globally (CitationWHO 2003b). Approximately 90% of all hypertension is classified as “essential” meaning that the cause is not known. Essential hypertension involves endothelial dysfunction with alterations in nitric oxide (NO) bioavailability and calcium handling, smooth muscle cell proliferation, thickening of the vessel walls, and increased peripheral vascular resistance and blood pressure (CitationResnick 1993; CitationOshima and Young 1995; CitationTaddei et al 2003; CitationPortaluppi et al 2004). Risk factors for hypertension include family history, diabetes, obesity, smoking, excessive alcohol intake, and a diet high in salt and/or low in antioxidant nutrients. Most of these risk factors are modifiable through lifestyle changes such as participating in moderate physical activity and eating a well-balanced diet. Healthy lifestyle choices also include not smoking and limiting alcohol intake (CitationWHO 2003b). Individuals with hypertension are at increased risk for atherosclerotic diseases such as stroke, and heart and kidney disease.
Atherosclerosis is a leading cause of death in the world. Heart disease and stroke are the two leading causes of death in adults in developed countries and are responsible for a third of all deaths in developing countries (CitationWHO 2003a). Atherosclerosis is an inflammatory condition of the blood vessels (CitationRoss 1999). Damage to, or activation of, the endothelium promotes entry of modified low-density lipoprotein (LDL) into the intima, a process enhanced by an elevation in circulating levels of LDL. Alteration in endothelium also increases the expression of adhesion molecules on the cell surface resulting in recruitment of monocytes and platelet adhesion. The monocytes transmigrate to the sub-endothelial space where they differentiate into macrophages. Modified LDL is scavenged by macrophages in the interstitial space transforming over time into foam cells. Accumulation of foam cells and other cellular debris evolve into atherosclerotic plaques (CitationChakarvarti et al 1991; CitationWitztum and Steinberg 1991; CitationPalinski et al 1995; CitationRoss 1999; CitationSima and Stancu 2002; CitationHansson 2005). Although atherosclerotic lesions generally occur at junctions of large- and medium-size vessels, they may also arise throughout the vasculature (CitationTegos et al 2001; CitationHansson 2005). Through stenosis or embolytic occlusion, lesions within the coronary, cerebral, peripheral or renal vessels result in the clinical manifestations of angina, myocardial infarction, stroke, peripheral arterial disease or renal failure (CitationTegos et al 2001). Atherosclerosis also involves increased smooth muscle cell migration and proliferation. Stiffening of the vessel walls may hinder elasticity exacerbating hypertension. Hypertension is considered a risk factor for atherosclerosis, as increased blood pressure itself can contribute to vascular injury, making vessels more susceptible to inflammation (CitationChobanian and Alexander 1996). However, studies show that lowering blood pressure alone does not completely eliminate the risk of cardiovascular disease (CitationNeutel 2000). Other risk factors include smoking, diabetes mellitus, obesity, dyslipidemias, and a diet high in saturated fat (CitationWHO 2003a, Citation2003b). Hypertension and atherosclerosis share similar risk factors and both are characterized by modified vascular structure and function (CitationPepine and Handberg 2001). These cardiovascular conditions are also linked by another common feature, insulin resistance.
Etiological factors of cardiovascular disease
Insulin resistance
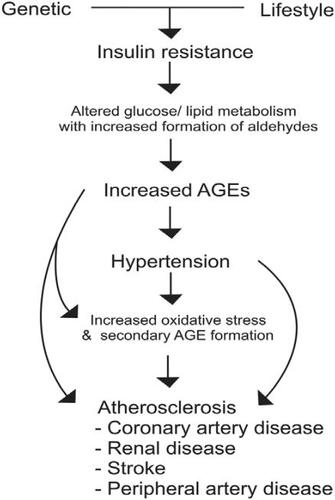
We suggest that the key etiological factor in cardiovascular disease is insulin resistance (). Insulin resistance is characterized by an inadequate glucose uptake in peripheral tissues at a given concentration of plasma insulin. It involves an impairment of the nonoxidative (glycolytic) pathways of intracellular glucose metabolism (CitationFerrannini et al 1987). Insulin resistance has been well documented in hypertension and atherosclerosis. Humans with essential hypertension and normotensive offspring of essential hypertensives have insulin resistance (CitationFerrannini et al 1987; CitationSaad et al 2004; CitationVlasakova et al 2004). Abnormalities in glucose metabolism exist in up to 80% of subjects with essential hypertension (CitationFerrannini et al 1987; CitationFlack and Sowers 1991). Insulin resistance has also been documented in humans with atherosclerosis (CitationDeFronzo and Ferrannini 1991; CitationHoward et al 1996; CitationShinozaki et al 1996; CitationReaven 2003). It has been suggested that hypertensives who are insulin resistant are at increased risk for atherosclerotic disease (CitationReaven 2003; CitationLiao et al 2004). In metabolic syndrome, also known as syndrome X or insulin resistance syndrome, primary insulin resistance is linked to a group of co-existing conditions including hypertension, dyslipidemias, diabetes, and atherosclerotic cardiovascular disease (CitationDeFronzo and Ferrannini 1991).
Figure 1 Mechanism of cardiovascular disease. In insulin resistant state, excess aldehydes formed due to altered glucose/lipid metabolism react with proteins to form advanced glycation end products (AGEs). AGEs alter the functions of cellular proteins including vascular ion channels, and metabolic and antioxidant enzymes, with oxidative stress leading to hypertension and atherosclerosis.
Aldehydes
Under normal physiological conditions, glucose is metabolized via the glycolytic pathway to glyceraldehyde-3-phosphate (G3P) which is converted to 1,3-diphosphoglycerate by the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), with further metabolism to pyruvate. Any factor which affects GAPDH, whether through inhibition or upregulation, has an impact on the rate of glucose metabolism. It has been shown that GAPDH is upregulated by insulin (CitationAlexander et al 1988). In insulin resistant states, altered insulin function may down regulate GAPDH, slowing glucose metabolism through the glycolytic pathway, thus increasing metabolism via the polyol pathway. This may result in a build-up of G3P leading to an increase in the highly reactive aldehyde, methylglyoxal (CitationMayes 1983; CitationThornalley 1993; CitationBeisswenger et al 2003). Methylglyoxal itself has been shown to inhibit GAPDH, resulting in further abnormalities in glucose metabolism (CitationMorgan et al 2002). Methylglyoxal also induces aldose reductase which is known to stimulate flux of glucose through the polyol pathway with further formation of methylglyoxal (CitationChang et al 2002). An excess of dietary sugar or fat, or both, typical of western diets, may overload these pathways and exacerbate this altered metabolism. Insulin resistance is also associated with dyslipidemia (CitationFuh et al 1987; CitationLiao et al 2004); elevated LDL without the mitigating antioxidant effect of high high-density lipoprotein (HDL), may contribute to an increase in reactive aldehydes (CitationDargel 1992).
Advanced glycation end products
Advanced glycation end products (AGEs) are formed when aldehydes (eg, methylglyoxal, glyoxal, glucose) react nonenzymatically with free sulfhydryl (SH) and amino (NH2) groups of amino acids including cysteine, arginine or lysine, of proteins (CitationSchauenstein et al 1977; CitationThorpe and Baynes 2003; CitationThornalley et al 2003). This direct modification of protein structure results in functional changes (CitationMorgan et al 2002; CitationNagaraj et al 2002; CitationKarachalias et al 2003). AGE-modified protein has also been shown to stimulate receptors of AGEs (RAGEs) and various scavenger receptors to influence protein function and expression (CitationSchmidt et al 2001; CitationWautier et al 2001; CitationHoriuchi et al 2003). Normally, methylglyoxal is formed but kept at a low level through catabolism via the glutathione-dependent glyoxalase enzyme system or by binding to cysteine and being excreted in bile and urine (CitationSchauenstein et al 1977). It has been suggested that AGEs formed in low concentrations contribute to the regulation of normal tissue remodeling (CitationKirstein et al 1992) but when found in excess are pathogenic (CitationVasdev et al 2000a, Citation2000b; CitationMizutani et al 2002; CitationAlderson et al 2003; CitationBidasee et al 2003; CitationKarachalias et al 2003; CitationStitt et al 2004; CitationVasdev et al 2005). It has also been proposed that certain AGEs have normal biological functions whereas others referred to as toxic AGEs, play a pathophysiological role (CitationTakeuchi and Yamagishi 2004). Whatever the case, several specific AGEs including carboxymethyl-lysine, carboxyethyl-lysine, argpyrimidine, and glycoaldehyde-pyridine, have been identified, and have been implicated in the pathology of hypertension and atherosclerosis (CitationAnderson et al 1999; CitationOya et al 1999; CitationBaynes and Thorpe 2000; CitationNagai et al 2002; CitationAlderson et al 2003; CitationWang et al 2004).
Causative role of advanced glycation end products in cardiovascular disease
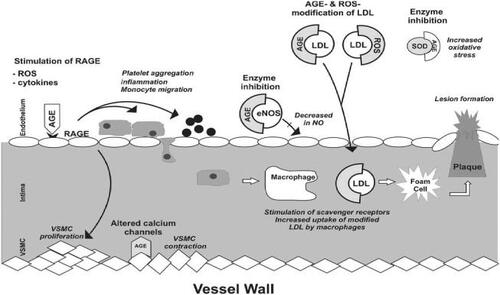
It is known that AGEs act directly or via receptors to alter the function of cellular proteins including calcium channels, metabolic and antioxidant enzymes, receptors and structural proteins leading to endothelial dysfunction, inflammatory responses, and increased oxidative stress (). Thus, it is becoming increasingly clear that AGEs play a major causative role in hypertension and atherosclerosis. We have shown that methylglyoxal given in the diet to Wistar-Kyoto (WKY) rats increased tissue AGEs and caused hypertension (CitationVasdev et al 1998). Levels of tissue methylglyoxal and AGEs are higher in spontaneously hypertensive rats (SHR) and sugar-induced hypertensive rats (CitationVasdev et al 2000a, Citation2000b; CitationWu and Juurlink 2002; CitationMidaoui et al 2003; CitationWang et al 2004). Although research on AGEs in human essential hypertension is scant, in preeclampsia, a hypertensive condition of pregnancy, RAGE expression was increased in vascular tissue (CitationCooke et al 2003). AGE-mediated crosslinks in collagen and elastin, also contribute to arterial stiffening, hindering vessel elasticity, and exacerbating hypertension (CitationAronson 2003). There is strong evidence in diabetes, another insulin resistant state, that AGEs are responsible for cellular protein modifications which contribute to diabetic atherosclerotic complications (CitationKislinger et al 2001; CitationDegenhardt et al 2002; CitationNagaraj et al 2002; CitationBabaei-Jadidi et al 2003; CitationKarachalias et al 2003; CitationAlt et al 2004; CitationStitt et al 2004). Evidence supporting the concept that AGEs are involved in the etiology of cardiovascular disease shows that compounds which lower AGEs also lower blood pressure and attenuate atherosclerotic conditions (CitationVasdev et al 2000a, Citation2000b, Citation2002; CitationKislinger et al 2001; CitationDegenhardt et al 2002; CitationMizutani et al 2002; CitationAlderson et al 2003; CitationBabaei-Jadidi et al 2003; CitationMidaoui et al 2003; CitationSakaguchi et al 2003; CitationSmit and Lutgers 2004; CitationStitt et al 2004).
Figure 2 Atherosclerotic and hypertensive effects of advanced glycation end products (AGEs) on blood vessels. AGEs act directly and via receptors of AGEs (RAGES) to alter the function of cellular proteins including calcium channels, endothelial nitric oxide synthase (eNOS), antioxidant enzyme superoxide dismutase (SOD) resulting in a decrease in NO and an increase of reactive oxygen species (ROS), cytokines, imflammation, platelet aggregation and vascular smooth muscle cell (VSMC) proliferation. AGEs and ROS also modify low density lipoprotein (LDL) increasing uptake by macrophages contributing to the formation of plaque. These alterations lead to hypertension and atherosclerosis.
Vascular dysfunction
Maintaining normal endothelial function is essential to blood pressure homeostasis and vessel integrity. One of the major factors involved in regulation of endothelial function is NO. Endothelium derived NO is not only a potent vasodilator but it inhibits platelet aggregation, vascular smooth muscle cell (VSMC) proliferation and intimal migration, and monocyte adhesion, thus regulating blood pressure and protecting vascular function (CitationTaddei et al 2003). Abnormalities in NO have been demonstrated in both hypertension and atherosclerosis (CitationTaddei et al 2003; CitationShinozaki et al 2004).
The ability of vascular tissue to form adequate amounts of NO depends on the availability of its substrate, arginine, and the endothelial enzyme, nitric oxide synthase (eNOS) (CitationTaddei et al 2003). Methylglyoxal reacts with arginine residues to form several AGEs including argpyrimidine (CitationShamsi et al 1998; CitationOya et al 1999; CitationThornalley et al 2003), which may limit substrate availability (CitationDegenhardt et al 2002). eNOS is an SH-dependent enzyme with a cysteine residue identified at its active site (CitationChen et al 1994). AGEs have been shown to inhibit eNOS activity and expression (CitationVerbeke et al 2000; CitationRojas et al 2000). We have shown that methylglyoxal given in the diet to WKY rats increased tissue AGEs and decreased plasma NO (CitationVasdev et al 1998). NO production is also regulated by insulin acting on specific receptors on the cell surface (CitationTrovati et al 1997; CitationKahn et al 2000). In insulin resistant states, aldehydes may alter receptor function decreasing NO formation (CitationSechi et al 1996; CitationVasdev et al 1998, Citation2005). NO has a very short half life. Under physiological conditions, it has been shown to readily react with SH groups of plasma proteins forming biologically stable adducts (S-nitrosothiols) which represent a pool of available NO. Low molecular weight thiols such as cysteine and glutathione act as transfer agents for NO from these pools to NO's site of action (CitationScharfstein et al 1994; CitationAlencar et al 2003). Since aldehydes also readily react with SH groups, this suggests a competitive role limiting both protein binding sites and transfer agent availability resulting in an increase in NO degradation and possible endothelial dysfunction (CitationFarkas and Menzel 1995; CitationThorpe and Baynes 2003).
Vascular calcium channels are dependent on SH groups for normal function (CitationSchauenstein et al 1977; CitationZaidi et al 1989) and their alteration can lead to increased cytosolic free calcium, abnormal contractile activity and increased peripheral vascular resistance. AGEs impair type 2 ryanodine receptor calcium release channels (calcium receptors which regulate cardiac contractility) during chronic diabetes (CitationBidasee et al 2003) and AGE-modified protein increases intracellular calcium (CitationScivattaro et al 2000). We have shown increased vascular AGEs and platelet cytosolic free calcium in SHRs, a genetic model of hypertension and in methylglyoxal- and fructose-treated WKY rats, dietary models of hypertension (CitationVasdev et al 1998, Citation2000a, Citation2000b).
Since calcium plays a key role in platelet activation, elevated intracellular free calcium may contribute to the enhanced platelet aggregation seen in hypertension and atherosclerosis (CitationDing 1996). AGEs may also contribute to platelet aggregation and thrombus formation. AGEs increased superoxide production and aggregation in human platelets in vitro (CitationHangaishi et al 1998). It has been suggested that AGEs stimulate externalization of phosphatidylserine which activates clotting factors leading to platelet adhesion (CitationWang et al 2005).
Oxidative stress
Under normal physiological conditions, reactive oxygen species (ROS) are produced in low concentrations and act as signaling molecules to regulate VSMC contraction-relaxation, and VSMC growth (CitationTouyz and Schiffrin 2004). Oxidative stress occurs when ROS outweigh the body's antioxidant capacity. Oxidative stress is controlled in part by antioxidant enzymes, including glutathione peroxidase and glutathione reductase (CitationKaul et al 1993). These enzymes have SH and NH2 groups at their active sites and AGEs inhibit their activity increasing oxidative stress (CitationMorgan et al 2002; CitationWu and Juurlink 2002; CitationPark et al 2003). Excess ROS can lead to the secondary production of aldehydes such as malondialdehyde and hydroxynonenal, through lipid peroxidation (CitationBrooks and Klamerth 1968; CitationSchauenstein et al 1977; CitationBenedetti et al 1980; CitationThornalley et al 1984; CitationDargel 1992; CitationTouyz and Schiffrin 2004). These lipid-derived aldehydes have been shown to react with cysteine or lysine residues of proteins to form a type of AGE known as advanced lipoxidation end products (ALEs) (CitationBaynes and Thorpe 2000; CitationThorpe and Baynes 2003; CitationStitt et al 2004).
There is strong evidence that oxidative stress contributes to the progression of essential hypertension and the development of atherosclerosis (CitationChakravarti et al 1991; CitationKumar and Das 1993; CitationChobanian and Alexander 1996; CitationStocker and Keaney 2004; CitationTouyz and Schiffrin 2004). Oxidative modification of LDL increases uptake by macrophages via scavenger receptors (further discussed in dislipidemia section). This results in an increased production and accumulation of foam cells in the vascular intima leading to the formation of atherosclerotic lesions. Oxidized LDL also stimulates atherogenic processes including inflammation, platelet aggregation, and VSMC proliferation (CitationRoss 1999; CitationStocker and Keaney 2004; CitationHansson 2005).
Oxidative stress impairs endothelial function. ROS promote uncoupling of eNOS resulting in a decreased production of NO (CitationAlp and Channon 2004; CitationShinozaki et al 2004). ROS also react directly with NO, forming peroxynitrite, also known as reactive nitrogen species. This not only limits NO bioavailability, but peroxynitrite and peroxynitrous acid are powerful and cyto-toxic oxidants which may cause damage to vascular tissue (CitationBeckman and Koppenol 1996). Methylglyoxal and AGEs have been shown to increase oxidative stress (CitationAnderson et al 1999; CitationScivittaro et al 2000; CitationWautier et al 2001; CitationMizutani et al 2002; CitationWu and Jurrlink 2002; CitationMidaoui et al 2003; CitationWong et al 2003) and in VSMC in vitro induced significant generation of superoxide radical and peroxynitrite (CitationChang et al 2005).
Dyslipidemia
Dyslipidemia has long been associated with atherosclerosis, and controlling elevated cholesterol and abnormal lipoprotein ratios is a recognized preventative cardiovascular health strategy (CitationFodor et al 2000). It is well known that high levels of HDL offer protective cardiovascular effect, and that elevated LDL is associated with higher risk of cardiovascular disease.
However, the roles of these lipoproteins are complex and have not yet been fully elucidated. It has been proposed that in its native state LDL is not atherogenic but becomes so under conditions where its structure is modified (). Alterations to lysine residues of apolipoprotein B of LDL result in a decreased binding by LDL receptors and an increased stimulation of macrophage scavenger receptors (CitationWitztum and Steinberg 1991; CitationHaberland et al 1992; CitationHoriuchi et al 2003). Oxidatively modified LDL initiates atherogenic processes including inflammation, platelet aggregation and smooth muscle cell proliferation (CitationStocker and Keaney 2004). Oxidized LDL has been identified as a main component in atherosclerotic lesions, and hypertensive subjects exhibit an enhanced susceptibility to LDL oxidation (CitationWitztum and Steinberg 1991; CitationMaggi et al 1993).
AGEs can form on lipoproteins themselves and AGE-LDL has been shown to have similar atherogenic properties as oxidized LDL (). AGE-LDL and AGE-modified protein are ligands for class AI/AII scavenger receptors (CitationHoriuchi et al 2003). Binding to these receptors leads to endocytic uptake of LDL and accumulation in human monocytes-macrophages (CitationHaberland et al 1992; CitationHoriuchi et al 2003). AGE-LDL has been identified in the cytoplasm of foam cells and extracellularly in the core of atherosclerotic lesions in humans and animals (CitationPalinski et al 1995; CitationNagai et al 2002; CitationSima and Stancu 2002).
HDL's function in reverse cholesterol transport plays a key role in its cardioprotective effect. HDL removes cholesterol from vascular tissue and either transfers it to very low density lipoprotein (VLDL) or LDL for transport to the liver, or it carries cholesterol directly to the liver where it may be recycled or excreted. In dyslipidemia, where HDL is low, cholesterol may be left to accumulate in vascular tissue. It has been suggested that AGEs interfere with reverse cholesterol transport by suppressing scavenger receptor B1 (SR-B1)-mediated uptake of cholesterol ester from HDL by liver and SR-B1-mediated cholesterol efflux from peripheral cells (CitationHoriuchi et al 2003). AGEs have been shown to cause cholesterol and cholesterol ester accumulation in macrophages in vitro (CitationBrown et al 2005).
HDL also has antioxidant ability. It is closely associated with the antioxidant enzyme paraoxonase. Low HDL and paraoxonase levels are associated with oxidative stress, hypertension and coronary heart disease (CitationSuh et al 1992; CitationMackness et al 2001; CitationUzun et al 2004; CitationMancia et al 2005). Paraoxonase protects both HDL and LDL from oxidation and lipid peroxidation (CitationAviram, Rosenblat, et al 1998; CitationAviram, Billecke, et al 1998; CitationRao et al 2003). In vitro studies show that paraoxonase lowers levels of oxidized LDL by converting them to biologically inactive products (CitationRao et al 2003). Blockage of free SH groups (cysteine residues) of paraoxonase reduced its ability to protect LDL from oxidation (CitationAviram, Billecke, et al 1998) suggesting a possible inhibitory role for AGEs (CitationHedrick et al 2000; CitationFerretti et al 2006).
HDL has also been shown to have antiinflammatory properties. It inhibited cytokine-induced expression of adhesion proteins and reduced neutrophil infiltration into the wall of injured arteries (CitationBarter et al 2004). AGE-modification of HDL resulted in a loss of these anti-inflammatory properties (CitationHedrick et al 2000; CitationFerretti et al 2006).
Low ethanol intake
Effect on hypertension and atherosclerosis
It has been well documented that excessive alcohol intake can be detrimental to cardiovascular health (CitationThun et al 1997; CitationReynolds et al 2003; CitationCorrao et al 2004). Although some studies of ethanol intake of greater than 30 g/day have shown improvement in cardiovascular risk, this level of intake increases risk of other diseases and death by other means (CitationCamargo et al 1997; CitationFagrell et al 1999). Therefore, we have focused our attention in this review on the effects of low ethanol intake (). Since there is no standard definition of “low” ethanol intake, we have chosen a level of up to, and including, 24 g per day (equivalent to 2 drinks), for the purposes of this discussion. A drink has been defined as a 12 oz bottle or can of beer (4%–5% ethanol), a 4 oz glass of table wine (10%–12% ethanol), or a 1–1.5 oz shot of liquor or spirits (40% ethanol) (CitationEllison et al 2004).
Table 1 Effect of low ethanol on hypertension and atherosclerosis
We have shown in rat models of hypertension that chronic low ethanol is effective in decreasing blood pressure (CitationVasdev et al 1999). Two studies of low ethanol intake in humans, one using up to 9 g/day and the other up to 18 g/day, showed antihypertensive effects (CitationJackson et al 1985; CitationOkubo et al 2001). Most animal research that looked specifically at atherosclerotic vascular changes, used moderate to high levels of ethanol. However, one study which fed low ethanol to an atherosclerotic model of rabbit showed that it attenuated atherosclerosis with a decrease in the extent of neointimal proliferation, lipid oxidation, and number of foam cells (CitationMerritt et al 1997). In humans, low ethanol consumption reduced risk for both cardiovascular and non-cardiovascular mortality (CitationKlatsky et al 1990; CitationCamargo et al 1997; CitationThun et al 1997) and was associated with a significantly reduced risk of stroke (CitationReynolds et al 2003) and ischemic heart disease (CitationGenchev et al 2001). In diabetic patients, low ethanol intake was associated with a 47% reduction in the prevalence of acute coronary syndrome (CitationPitsavos et al 2005) and in individuals known to be at risk for cardiovascular disease, a 20% reduction in risk of coronary artery disease (CitationCorrao et al 2004).
Mechanism of beneficial effect
Low ethanol has been shown to improve the clinical manifestations of cardiovascular disease (), but what is the biochemical mechanism of this beneficial effect? We suggest that low ethanol intake has the ability to increase antioxidant capacity, improve insulin resistance and decrease AGEs, thus preventing subsequent hypertensive and atherosclerotic consequences. To begin, we need to understand the metabolism of ethanol (). Ethanol is metabolized differently at high and low concentrations. With chronic high ethanol intake, the microsomal ethanol oxidizing system (MEOS) is induced. MEOS has a relatively high Km for ethanol, and at high concentrations ethanol is metabolized to acetaldehyde without producing reduced nicotinamide adenine dinucleotide (NADH). Instead, this pathway utilizes reduced nicotinamide adenine dinucleotide phosphate (NADPH), another reducing equivalent, thus producing an oxidative environment (CitationLieber 1990). Additionally, chronic high ethanol consumption inhibits aldehyde dehydrogenase resulting in a significant decrease in the ability of rat mitochondria to oxidize acetaldehyde. This high ethanol intake thus associated with an enhanced rate of metabolism by the MEOS pathway resulting in a decrease in reducing equivalents, elevated acetaldehyde, and increased oxidative stress. These factors may account for the detrimental effects of high ethanol intake. However, at low ethanol blood levels, ethanol is metabolized very efficiently by low Km alcohol dehydrogenase to acetaldehyde and then by aldehyde dehydrogenase to acetate, producing NADH in both reactions (CitationLieber 1990; CitationBello et al 1994). NADH has a major influence on total antioxidant capacity of the body. NADH is a key component of the electron transport chain and, due to its high reducing potential functions to promote regeneration of endogenous vitamin radicals back to their reduced form (). Although ethanol is primarily metabolized in the liver, it is also metabolized in other tissues, including vascular tissue. This ability of ethanol at low concentrations to create a strong reducing environment, possibly at the site of atherosclerotic activity, may enhance its cardiovascular protective effect.
Figure 3 Metabolism of high versus low concentrations of ethanol. In high concentrations, ethanol is metabolized by the microsomal ethanol oxidizing system (MEOS) system. In this reaction, reduced nicotinamide adenine dinucleotide phosphate (NADPH) is converted to oxidized nicotinamide adenine dinucleotide phosphate (NADP+) creating an oxidative environment. In low concentrations, ethanol is metabolized by the enzymes alcohol and aldehyde dehydrogenase producing reduced nicotinamide adenine dinucleotide (NADH) from oxidized nicotinamide adenine dinucleotide (NAD+) by both reactions, increasing antioxidant capacity. At the low levels produced, acetate, which is a normal metabolite of glucose and fatty acid metabolism, is further metabolized in the citric acid cycle.
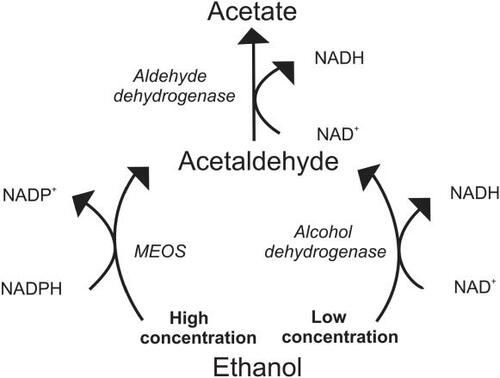
Figure 4 Antioxidant activity of low ethanol. Free radicals (ROO−) are reduced (ROOH) by vitamins which become oxidized in the process. These vitamin radicals are reduced by nicotinamide adenine dinucleotide (NADH) which forms oxidized nicotinamide adenine dinucleotide (NAD+). Ethanol in low concentrations converts NAD+ back into NADH, via its metabolism to acetate. At this low level, acetate, which is a normal metabolite of glucose and fatty acid metabolism, is further metabolized in the citric acid cycle.
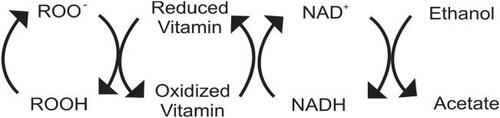
An increase in antioxidant capacity would offer protection against oxidative stress ( and ) and secondary production of aldehydes through lipid peroxidation (CitationBello et al 1994). In vitro, ethanol has been shown to act as an antioxidant by decreasing the rate of consumption of LDL antioxidants and reducing the formation of lipid peroxides (CitationBonnefont-Rousselot et al 2001). In humans, it decreased urinary 8-hydroxydeoxyguanosine, a measure of oxidative stress (CitationYoshida et al 2001). NADH treatment has been shown to reduce blood pressure and lipid peroxidation in SHRs (CitationBushehri et al 1998). NADH may also increase overall antioxidant capacity, increasing tissue levels of cysteine by converting cystine to cysteine, via the NADH dependent enzyme, cystine reductase (CitationRodwell 1983). Cysteine is a precursor of glutathione, a major endogenous antioxidant. Additionally, glutathione is a cofactor in methylglyoxal catabolism and cysteine has the ability to bind aldehydes to foster excretion and reduce AGE formation (CitationSchauenstein et al 1977; CitationVasdev et al 1998).
Figure 5 Mechanism of action of low ethanol.
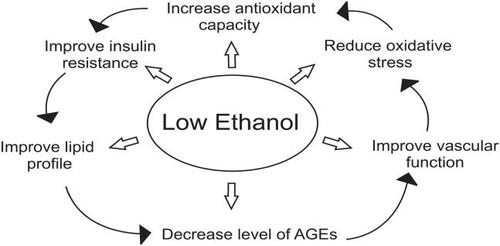
An increase in the low molecular weight thiols, cysteine and glutathione, may also enhance the transfer of NO from protein-bound reserves improving endothelial dysfunction (CitationScharfstein et al 1994). Decreasing levels of AGEs may also preserve eNOS and prevent breakdown of NO. Low ethanol increased the expression of eNOS (CitationVenkov et al 1999) and stimulated calcium-activated potassium channels increasing production of NO in vascular endothelial cells in culture (CitationKuhlmann et al 2004). Humans consuming low amounts of ethanol showed significant dilatation of brachial artery at rest and at reactive hyperaemic conditions (CitationVlachopoulos et al 2003). Low ethanol decreases cytosolic free calcium, an initiator of smooth muscle cell contraction. We have shown in rat models of essential hypertension that low ethanol reduced AGEs and platelet cytosolic free calcium (CitationVasdev et al 1999). Low ethanol activated sarco/endoplasmic reticulum uptake of calcium in platelets (CitationMitidieri and de Meis 1995) which would lower cytosolic free calcium concentration. This effect of ethanol on vascular dilation/contraction regulators may contribute to its beneficial cardiovascular effect ().
Low alcohol intake increased insulin sensitivity in humans (CitationFacchini et al 1994; CitationFlanagan et al 2000) and in rats (CitationFuruya et al 2003). Improving insulin resistance, the source of excess aldehydes, would limit formation of AGES and their subsequent hypertensive and atherosclerotic complications (). Since aldehydes and AGEs have also been shown to inhibit GAPDH (CitationMorgan et al 2002; CitationBeisswenger et al 2003), likely further exacerbating insulin resistance, reducing AGE formation may limit the effect of this cycle. Low ethanol may also work to improve insulin resistance by lowering plasma free fatty acids and improving glucose uptake and metabolism (CitationAvogaro et al 2002).
Low ethanol intake has a beneficial effect on lipoprotein profiles (). Low ethanol has been shown to increase HDL (CitationFacchini et al 1994; CitationDe Oliviera e Silva et al 2000; CitationEllison et al 2004). This increase may be a result of mediating the AGE-induced inhibitory effect on reverse cholesterol transport. In hypertensive patients low ethanol decreased lipoprotein a (Lp[a]), an independent predictor for atherosclerosis (CitationCatena et al 2003). It has been suggested that this may be due to an increase in the transportation rate of apolipoprotein AI and AII (CitationDe Oliviera e Silva et al 2000) or to an ethanol-induced redistribution of cholesteryl ester transport proteins between HDL and VLDL (CitationHannuksela et al 1996). Chronic low ethanol intake stimulated paraoxonase activity by upregulating liver paraoxonase mRNA in rats and humans (CitationRao et al 2003). Ethanol may also prevent inhibition of paraoxonase by lowering AGEs.
Implication for treatment
We have suggested that low ethanol intake affords beneficial cardiovascular effects due to its ability to improve insulin resistance and its subsequent detrimental effects. However, due to the addictive nature of alcohol, recommending ethanol intake at any level may not be prudent. It may be more appropriate to suggest other agents which act in a similar fashion and may provide the same beneficial effects without the complication of addiction. Supplementation with antioxidants including vitamin C, E, or B6, N-acetylcysteine, lipoic acid and coenzyme Q10 has been shown to lower blood pressure in animal models and humans with essential hypertension (CitationDuffy et al 1999; CitationSingh et al 1999; CitationVasdev et al 2000a, Citation2000b, Citation2002; CitationMorcos et al 2001; CitationBarrios et al 2002; CitationBoshtam et al 2002). In humans, antioxidants have also shown a beneficial effect on atherosclerotic endpoints in several studies (CitationStampfer et al 1993; CitationLosonczy et al 1996; CitationStephens et al 1996). As well, various anti-AGE therapies, which either prevent or reverse AGE formation, such as pyridoxamine, thiamine, metformin, alagebrium, or soluble RAGE have been shown to attenuate hypertension and atherosclerotic disease (CitationKislinger et al 2001; CitationDegenhardt et al 2002; CitationMizutani et al 2002; CitationAlderson et al 2003; CitationBabaei-Jadidi et al 2003; CitationMidaoui et al 2003; CitationSakaguchi et al 2003; CitationSmit and Lutgers 2004; CitationStitt et al 2004).
Conclusion
In summary, insulin resistance leads to altered glucose and lipid metabolism. A subsequent increase in AGEs, oxidative stress, and endothelial dysfunction leads to the development of hypertension and atherosclerosis. Low ethanol intake provides cardiovascular beneficial effect primarily through its ability to increase antioxidant capacity, improving insulin resistance and decreasing AGEs. Considering the potential for addiction with ethanol consumption, agents such as antioxidants which have similar antihypertensive and anti-atherosclerotic mechanisms, may provide an appropriate alternative.
Acknowledgements
We would like to thank the Canadian Institutes of Health Research for their financial support.
References
- AldersonNLChachichMEYoussefNNThe AGE inhibitor pyridoxamine inhibits lipemia and development of renal and vascular disease in Zucker obese ratsKidney Int20036321233312753299
- AlencarJLLobyshevaIGeffardMRole of s-nitosation of cysteine residues in long-lasting inhibitory effect of nitric oxide on arterial toneMol Pharmac200363114858
- AlexanderMCLomantoMNasrinNInsulin stimulates glyceraldehyde-3-phosphate dehydrogenase gene expression through CIS-acting DNA sequencesProc Natl Acad Sci U S A198885509262839830
- AlpNJChannonKMRegulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular diseaseArterioscler Thromb Vasc Biol2004244132014656731
- AltNCarsonJAAldersonNLChemical modification of muscle protein in diabetesArch Biochem Biophys2004425200615111128
- AndersonMMRequenaJRCrowleyJRThe myeloperoxidase system in human phagocytes generates Nɛ-(carboxymethyl)lysine on proteins: a mechanism for producing advanced glycation end products at sites of inflammationJ Clin Invest19991041031310393704
- AronsonDCross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetesJ Hypertens20032131212544424
- AviramMRosenblatMBisgaierCLParaoxonase inhibits high-density lipoprotein oxidation and preserves its functionJ Clin Invest19981011581909541487
- AviramMBilleckeSSorensonRParaoxonase active site required for protection against LDL oxidation involves its free sulfhydryl group and is different from that required for its arylesterase/paraoxonase activities. Selective action of human paraoxonase allozymes Q nd RArterioscler Thromb Vasc Biol1998181617249763535
- AvogaroAWatanabeRMGottardoLGlucose tolerance during moderate alcohol intake: Insights on insulin action from glucose/lactate dynamicsJ Clin Endocrinol Metab2002871233811889193
- Babaei-JadidiRKarachaliasNAhmedNPrevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamineDiabetes20035221102012882930
- BarriosVCalderonANavarro-CidJN-Acetylcysteine potentiates the antihypertensive effect of ACE inhibitors in hypertensive patientsBlood Pressure2002112353912361192
- BarterPJNichollsSRyeKAAntiinflammatory properties of HDLCirc Res2004957647215486323
- BaynesJWThorpeSRGlycoxidation and lipoxidation in atherogenesisFree Rad Biol Med20002817081610946212
- BeckmanJSKoppenolWHNitric oxide, superoxide and peroxynitrite: the good, the bad and the uglyAm J Physiol1996271C1424378944624
- BeisswengerPJHowellSKSmithKGlyceraldehyde-3-phosphate dehydrogenase activity as an independent modifier of methylglyoxal levels in diabetesBiochim Biophys Acta200316379810612527413
- BelloATBoraNSLangeLGCardioprotective effects of alcohol: mediation by human vascular alcohol dehydrogenaseBiochem Biophys Res Commun19942031858647945338
- BenedettiAComportiMEsterbauerHIdentification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipidsBiochim Biophys Acta1980620281966254573
- BidaseeKRNallaniKYuYChronic diabetes increases advanced glycation end products on cardiac ryanodine receptors/calcium-release channelsDiabetes20035218253612829653
- Bonnefont-RousselotDRouscillesABizardCAntioxidant effect of ethanol toward in vitro peroxidation of human low-density lipoproteins initiated by oxygen free radicalsRadiat Res20011552798711175662
- BoshtamMRafieiMSadeghiKVitamin E can reduce blood pressure in mild hypertensivesInt J Vitam Nutr Res2002723091412463106
- BrooksBRKlamerthOJInteraction of DNA with bifunctional aldehydesEuro J. Biochem1968517882
- BrownBEDeanRTDaviesMJGlycation of low-density lipoproteins by methylglyoxal and glycoaldehyde gives rise to the in vitro formation of lipid-laden cellsDiabetologia200548361915660260
- BushehriNJarrellSTLiebermanSOral reduced B-nicotinamide adenine dinucleotide (NADH) affects blood pressure, lipid peroxidation, and lipid profile in hypertensive rats (SHR)Geriat Nephrol Urol1998895100
- CamargoCAHennekensCHGazianoJMProspective study of moderate alcohol consumption and mortality in US male physiciansArch Intern Med199715779858996044
- CatenaCNovelloMDottoLSerum lipoprotein(a) concentrations and alcohol consumption in hypertension: possible relevance for cardiovascular damageJ Hypertens200321281812569257
- ChakravartiRNKirshenbaumLASingalPKAtherosclerosis: Its pathophysiology with special reference to lipid peroxidationJ Appl Cardiol1991691112
- ChangKCPaekKSKimHJSubstrate-induced up-regulation of aldose reductase by methylglyoxal, a reactive oxoaldehyde elevated in diabetesMol Pharmacol20026111849111961137
- ChangTWangRWuLMethylglyoxal-induced nitric oxide and peroxynitrite production in vascular smooth muscle cellsFree Radic Biol Med2005382869315607912
- ChenPTsaiAWuKKCysteine 184 of endothelial nitric oxide synthase is involved in heme coordination and catalytic activityJ Biol Chem19942692506267523378
- ChobanianAVAlexanderRWExacerbation of atherosclerosis by hypertensionArch Intern Med1996156195268823148
- CookeCLMBrockelsbyJCBakerPNThe receptor for advanced glycation end products (RAGE) is elevated in women with preeclampsiaHypertens Preg20032217384
- CorraoGBagnardiVZambonAA meta-analysis of alcohol consumption and the risk of 15 diseasesPrevent Med20043861319
- DargelRLipid peroxidation - a common pathogenetic mechanism?Exp Toxic Pathol19924416981
- DeFronzoRAFerranniniEInsulin resistance: A multifaceted syndrome responsible for NIDDM, obesity, hypertension and atherosclerotic cardiovascular diseaseDiabetes Care199114173942044434
- DegenhardtTPAldersonNLArringtonDDPyridoxamine inhibits early renal disease and dyslipidemia in streptozotocin-diabetic ratKidney Int2002619395011849448
- De Oliveira e SilvaERFosterDMcGee HarperMAlcohol consumption raises HDL cholesterol levels by increasing the transport rate of apolipoproteins A-I and A-IICirculation200010223475211067787
- DingYAThrombogenic and lipid risk factors in hypertension and coronary artery diseaseJpn Circ J19966075848683858
- DuffySJGokceNHolbrookMTreatment of hypertension with ascorbic acidLancet19993542048910636373
- EllisonRCZhangYQureshiMMLifestyle determinants of high-density lipoprotein cholesterol: The National Heart, Lung, and Blood Institute Family Heart StudyAm Heart J20041475293514999205
- FacchiniFChenYDIReavenGMLight-to-moderate alcohol intake is associated with enhanced insulin sensitivityDiabetes Care199417115197907975
- FagrellBDe FaireUBondySThe effects of light to moderate drinking on cardiovascular diseasesJ Intern Med19992463314010583704
- FarkasJMenzelEJProteins lose their nitric oxide stabilizing function after advanced glycosylationBiochim Biophys Acta19951245305108541305
- FerrettiGBacchettiTNegre-SalvayreAStructural modifications of HDL and functional consequencesAtheroscler200618417
- FerranniniEBuzzigoliGBonadonnaRInsulin resistance in essential hypertensionN Engl J Med198731735073299096
- FlackJMSowersJREpidemiologic and clinical aspects of insulin resistance and hyperinsulinemiaAm J Med19919111S21S1867224
- FlanaganDEHMooreVMGodslandIFAlcohol consumption and insulin resistance in young adultsEuro J Clin Invest200030297301
- FodorJGFrohlichJJGenestJJGRecommendations for the management and treatment of dyslipidemia. Report of the Working Group on Hypercholesterolemia and Other DyslipidemiasCMAJ20001621441710834048
- FuhMMSheihSMWuDAAbnormalities of carbohydrate and lipid metabolism in patients with hypertensionArch Intern Med1987147103583296980
- FuruyaDTBinsackRMachadoUFLow ethanol consumption increases insulin sensitivity in Wistar ratsBraz J Med Biol Res2003361253012532236
- GenchevGDGeorgievaLMWeijenbergMPDoes alcohol protect against ischemic heart disease in Bulgaria? A case-control study of non-fatal myocardial infarction in SofiaCentr Eur J Public Health20019836
- HaberlandMEFlessGMScanuAMMalondialdehyde modification of lipoprotein(a) produces avid uptake by human monocyte-macrophagesJ Biol Chem19922674143511531481
- HangaishiMTaguchiJMiyataTIncreased aggregation of human platelets produced by advanced glycation end products in vitroBiochem Biophys Res Comm1998248285929675128
- HannukselaMLRantalaMKesaniemiYAEthanol-induced redistribution of cholesteryl ester transfer protein (CEPT) between lipoproteinsAtheroscler Thromb Vasc Biol19961621321
- HanssonGKInflammation, atherosclerosis, and coronary artery diseaseN Engl J Med200535216859515843671
- HedrickCCThorpeSRFuMXGlycation impairs high-density lipoprotein functionDiabetalogia20004331220
- HoriuchiSSakamotoYSakaiMScavenger receptors for oxidized and glycated proteinsAmino Acids2003252839214661091
- HowardGO'LearyDHZaccaroDInsulin sensitivity and atherosclerosisCirculation1996931809178635260
- JacksonRStewartABeagleholeRAlcohol consumption and blood pressureAm J Epidemiol19851221037444061438
- KahnNNAcharyaKBhattacharyaSNitric oxide: The “second messenger” of insulinIUBMB Life2000494415010902577
- KarachaliasNBabaei-JadidiRAhmedNAccumulation of fructosyl-lysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic ratsBiochem Soc Trans2003311423514641079
- KaulMSiveski-IiskovicMHillMFree radicals and the heartJ Pharmacol Toxicol Methods19933055678298182
- KirsteinMAstonCHintzRReceptor-specific induction of insulin-like growth factor I in human monocytes by advanced glycosylation end product–modified proteinsJ Clin Invest199290439461322940
- KislingerTTanjiNWendtTReceptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null miceArterioscler Thromb Vasc Biol2001219051011397695
- KlatskyALArmstrongMAFriedmanGDRisk of cardiovascular mortality in alcohol drinkers, ex-drinkers and nondrinkersAm J Cardiol1990661237422239729
- KuhlmannCRWLiFLuddersDWDose-dependent activation of Ca2+-activated K+ channels by ethanol contributes to improved endothelial cell functionsAlcohol Clin Exp Res20042810051115252286
- KumarKVDasUNAre free radicals involved in the pathobiology of human essential hypertension?Free Rad Res Comm1993195966
- LiaoYKwanSShaughnessySCritical evaluation of adult treatment panel III criteria in identifying insulin resistance with dyslipidemiaDiabet Care20042797883
- LieberCSMechanism of ethanol induced hepatic injuryPharmac Ther199046141
- LosonczyKGHarrisTBHavlikRJVitamin E and vitamin C supplement use and risk of all-cause and coronary heart disease mortality in older persons: the Established Populations for Epidemiologic Studies of the ElderlyAm J Clin Nutr19966419068694019
- MacknessBDaviesGKTurkieWParaoxonase status in coronary heart disease. Are activity and concentration more important than genotype?Arterioscler Thromb Vasc Biol2001211451711557671
- MaggiEMarchesiERavettaVLow-density lipoprotein oxidation in essential hypertensionJ Hypertens1993111103118258675
- ManciaGFacchettiRBombelliMRelationship of office, home, and ambulatory blood pressure to blood glucose and lipid variables in the PAMELA populationHypertens20054510727
- MayesPAMartinDWRodwellVWMayesPACarbohydrate metabolismHarper's Review of Biochemistry 19th ed1983Los Altos, CALange Medical Pub16187
- MerrittRGurugeBLMillerDDModerate alcohol feeding attenuates postinjury vascular cell proliferation in rabbit angioplasty modelJ Cardiovasc Pharmacol19973019259268217
- MidaouiAEElimadiAWuLLipoic acid prevents hypertension, hyperglycemia, and the increase in heart mitochondrial superoxide productionAm J Hypertens200316173912620694
- MitidieriFde MeisLEthanol has different effects on Ca (2+)–transport ATPases of muscle, brain and blood plateletsBiochem J199531273378554513
- MizutaniKIkedaKTsudaKInhibitor for advanced glycation end products formation attenuates hypertension and oxidative damage in genetic hypertensive ratsJ Hypertens20022016071412172323
- MorcosMBorceaVIsermannBEffect of α-lipoic acid on the progression of endothelial cell damage and albuminuria in patients with diabetes mellitus: an exploratory studyDiab Res Clin Pract20015217583
- MorganPEDeanRTDaviesMJInactivation of cellular enzymes by carbonyls and protein-bound glycation/glycoxidation productsArch Biochem Biophys20024032596912139975
- NagaiRHayashiCMXiaLIdentification in human atherosclerotic lesions of GA-pyridine, a novel structure derived from glycoaldehyde-modified proteinsJ Biol Chem2002277489051212377783
- NagarajRHSarkarPMallyAEffect of pyridoxamine on chemical modification of proteins by carbonyls in diabetic rats: characterization of a major product from the reaction of pyridoxamine and methylglyoxalArch Biochem Biophys20024021101912051689
- NeutelJMWhy lowering blood pressure is not enough: the hypertension syndrome and the clinical context of cardiovascular risk reductionHeart Dis20002370411728284
- OkuboYSuwazonoYKobayashiEAlcohol consumption and blood pressure change: 5 year follow-up study of the association in normotensive workersJ Hum Hypertens2001153677211439310
- OshimaTYoungEWSystemic and cellular calcium metabolism and hypertensionSemin Nephrol1995154965038588109
- OyaTHattoriNMizunoYMethylglyoxal modification of proteinJ Biol Chem19992741849250210373458
- PalinskiWKoschinskyTButlerSWImmunological evidence for the presence of advanced glycosylation end products in atherosclerotic lesions of euglycemic rabbitsArterioscler Thromb Vasc Biol199515571827749871
- ParkYSKohYHTakahashiMIdentification of the binding site of methylglyoxal on glutathione peroxidase: Methylglyoxal inhibits glutathione peroxidase activity via binding to glutathione binding sites Arg 184 and 185Free Rad Res20033720511
- PepineCJHandbergEMThe vascular biology of hypertension and atherosclerosis and intervention with calcium antagonists and angiotensin-converting enzyme inhibitorsClin Cardiol200124V1511712769
- PianoMRThe cardiovascular effects of alcohol: the good and the bad. How low-risk drinking differs from high-risk drinkingAJN2005105879115995407
- PitsavosCMakrilakisKPanagiotakosDBThe J-shape effect of alcohol intake on the risk of developing acute coronary syndromes in diabetic subjects: the CARDIO 2000 II StudyDiabet Med200522243815717869
- PortaluppiFBoariBManfrediniROxidative stress in essential hypertensionCurr Pharmaceut Design20041016958
- RaoMNMarmillotPGongMLight, but not heavy alcohol drinking, stimulates paraoxonase by upregulating liver mRNA in rats and humansMetabol200352128794
- ReavenGMInsulin resistance/compensatory hyperinsulinemia, essential hypertension, and cardiovascular diseaseJ Clin Endocrin Metab2003882399403
- ResnickLMIonic basis of hypertension, insulin resistance, vascular disease, and related disorders. The mechanism of “syndrome X”Am J Hyperten19936123S134S
- ReynoldsKLewisLBNolenJDLAlcohol consumption and risk of strokeJAMA20032895798812578491
- RodwellVWMartinDWRodwellVWMayesPACatabolism of the carbon skeletons of amino acidsHarper's Review of Biochemistry 19th ed1983Stamford, COAppleton & Lange283306
- RojasARomaySGonzalezDRegulation of endothelial nitric oxide synthase expression by albumin-derived advanced glycosylation end productsCirc Res200086e50e5410679490
- RossRAtherosclerosis – An inflammatory diseaseN Engl J Med1999340115269887164
- SaadMFRewersMSelbyJInsulin resistance and hypertension. The insulin resistance atherosclerosis studyHypertens200443132431
- SakaguchiTYanSFYanSDCentral role of RAGE-dependent neointimal expansion in arterial restenosisJ Clin Invest20031119597212671045
- ScharfsteinJSKeaneyJFSlivkaAIn vivo transfer of nitric oxide between a plasma protein-bound reservoir and low molecular weight thiolsJ Clin Invest199494143297929818
- SchauensteinEEsterbauerHZollnerHLagnadoJRAldehydes in biological systemsAldehydes in biological systems, their natural occurrence and biological activities1977LondonPion Limited17
- SchmidtAMYanSDYanSFThe multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responsesJ Clin Invest20011089495511581294
- ScivittaroVGanzMBWeissMFAGEs induce oxidative stress and activate protein kinase C-βII in neonatal mesangial cellsAm J Physiol Renal Physiol2000278F6768310751230
- SechiLAGriffinCAGiacchettiGAbnormalities of insulin receptors in spontaneously hypertensive ratsHypertens19962795561
- ShamsiFAPartalASadyCImmunological evidence for methylglyoxal-derived modifications in vivoJ Biol Chem19982736928369506998
- ShinozakiKKashiwagiAMasadaMMolecular mechanism of impaired endothelial function associated with insulin resistanceCurr Drug Targets Cardiov Haemat Dis20044111
- ShinozakiKSuzukiMIkebuchiMDemonstration of insulin resistance in coronary artery disease documented with angiographyDiabetes Care199619178720524
- SimaAStancuCModified lipoproteins accumulate in human coronary atheromaJ Cell Mol Med200261101112003674
- SinghRBNiazMARastogiSSEffect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery diseaseJ Human Hypertens199913203810204818
- SmitAJLutgersHLThe clinical relevance of advanced glycation endproducts (AGE) and recent developments in pharmaceutics to reduce AGE accumulationCurr Med Chem20041127678415544475
- StampferMJHennekensCHMansonJEVitamin E consumption and the risk of coronary disease in womenN Engl J Med1993328144498479463
- StephensNGParsonsASchofieldPMRandomized controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS)Lancet199634778168622332
- StittAWFrizzellNThorpeSRAdvanced glycation and advanced lipoxidation: possible role in initiation and progression of diabetic retinopathyCurr Pharm Design200410334960
- StockerRKeaneyJFRole of oxidative modifications in atherosclerosisPhysiol Rev200484138147815383655
- SuhIShatenBJCutlerJAAlcohol use and mortality from coronary heart disease: The role of high-density lipoprotein cholesterolAnn Intern Med199211688171580443
- TaddeiSGhiadoniLVirdisAMechanisms of endothelial dysfunction: Clinical significance and preventative non-pharmacological therapeutic strategiesCurr Pharmaceut Design200392385402
- TakeuchiMYamagishiSTAGE (toxic AGEs) hypothesis in various diseasesMed Hypoth20046344952
- TegosTJKalodikiESabetaiMMThe genesis of atherosclerosis and risk factors: A reviewAngiology200152899811228092
- ThornalleyPWolffSCrabbeJThe autoxidation of glyceraldehyde and other simple monosaccharides under physiological conditions catalysed by buffer ionsBiochim Biophys Acta1984797276876365176
- ThornalleyPJBattahSAhmedNQuantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometryBiochem J20033755819212885296
- ThornalleyPJThe glyoxalase system in health and diseaseMol Aspects Med1993142873718277832
- ThorpeSRBaynesJWMaillard reaction products in tissue proteins: New products and new perspectivesAmino Acids2003252758114661090
- ThunMJPetoRLopezADAlcohol consumption and mortality among middle-aged and elderly U.S. adultsN Engl J Med19973371705149392695
- TouyzRMSchiffrinELReactive oxygen species in vascular biology: implications for hypertensionHistochem Cell Biol20041223395215338229
- TrovatiMAnfossiGMassuccoPInsulin stimulates nitric oxide synthesis in human platelets and, through nitric oxide, increases platelet concentrations of both guanosine-3′, 5′-cyclic monophosphate and adenosine-3′, 5′-cyclic monophosphateDiabetes19974674299133539
- UzunHKarterYAydinSOxidative stress in white coat hypertension; role of paraoxonaseJ Hum Hypertens200418523814985779
- VasdevSFordCALongerichLAldehyde induced hypertension in rats: Prevention by N-acetylcysteineArtery199823103610846614
- VasdevSFordCALongerichLAntihypertensive effect of low ethanol intake in spontaneously hypertensive ratsMol Cell Biochem1999200859210569187
- VasdevSFordCAParaiSDietary alpha-lipoic acid supplementation lowers blood pressure in spontaneously hypertensive ratsJ Hypertens2000a185677310826559
- VasdevSFordCAParaiSDietary lipoic acid supplementation prevents fructose-induced hypertension in ratsNutr Metab Cardiovasc Dis2000b103394611302009
- VasdevSGillVLongerichLGuptaSKSingalPKAgrawalRole of methylglyoxal in essential hypertension2005New Delhi, IndiaAnamaya Pub7288
- VasdevSLongerichLSingalPNutrition and hypertensionNutr Res20022211123
- VenkovCDMyersPRTannerMAEthanol increases endothelial nitric oxide production through modulation of nitric oxide synthase expressionThromb Haemost1999816384210235453
- VerbekePPerichonMFriguetBInhibition of nitric oxide synthase activity by early and advanced glycation end products in cultured rabbit proximal tubular epithelial cellsBiochim Biophys Acta200015024819411068190
- VlachopoulosCTsekouraDTsiamisEEffect of alcohol on endothelial function in health subjectsVasc Med20038263515125487
- VlasakovaZPelikanovaTKarasovaLInsulin secretion, sensitivity, and metabolic profile of young healthy offspring of hypertensive patientsMetab20045346975
- WangXDesaiKClausenJTIncreased methylglyoxal and advanced glycation end products in kidney from spontaneously hypertensive ratsKid Internat200466231521
- WangYMarshallSMThompsonMGCardiovascular risk in patients with end-stage renal disease: a potential role for advanced glycation end productsContrib Nephrol20051491687415876841
- WautierMPChappeyOCordaSActivation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGEAm J Physiol Endocrinol Metab2001280E6859411287350
- WitztumJLSteinbergDRole of oxidized low density lipoprotein in atherogenesisJ Clin Invest1991881785921752940
- WongRKMPettitAIQuinnPAAdvanced glycation end products stimulate an enhanced neutrophil respiratory burst mediated through the activation of cytosolic phospholipase A2 and generation of arachidonic acidCirc2003108185864
- World Health OrganizationWorld Health Report. Chapter 6: Neglected global epidemics: three growing threats [online]2003a115 Accessed on 8 February 2006. URL: http://www.who.int/entity/whr/2003/en/Chapter6-en.pdf
- World Health OrganizationGlobal strategy on diet, physical activity and health: chronic disease risk factors2003b Accessed on 8 February 2006. URL: http://www.who.int/dietphysicalactivity/publications/facts/riskfactors/en/
- WuLJuurlinkBHJIncreased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cellsHypertens20023980914
- YoshidaRShiojiIKishidaAModerate alcohol consumption reduces urinary 8-hydroxydeoxyguanosine by inducing of uric acidIndust Health2001393229
- ZaidiNFLagenaurCFAbramsonJJReactive disulfides trigger Ca2+ release from sarcoplasmic reticulum via an oxidation reactionJ Biol Chem198926421725362532212