Abstract
The pathogenesis that is primarily responsible for Alzheimer’s disease (AD) and cerebrovascular accidents (CVA) appears to involve chronic hypoperfusion. We studied the ultrastructural features of vascular lesions and mitochondria in brain vascular wall cells from human AD biopsy samples and two transgenic mouse models of AD, yeast artificial chromosome (YAC) and C57B6/SJL Tg (+), which overexpress human amyloid beta precursor protein (AβPP). In situ hybridization using probes for normal and 5 kb deleted human and mouse mitochondrial DNA (mtDNA) was performed along with immunocytochemistry using antibodies against the Aβ peptide processed from AβPP, 8-hydroxy-2’-guanosine (8OHG), and cytochrome c oxidase (COX). More amyloid deposition, oxidative stress markers as well as mitochondrial DNA deletions and structural abnormalities were present in the vascular walls of the human AD samples and the AβPP-YAC and C57B6/SJL Tg (+) transgenic mice compared to age-matched controls. Ultrastructural damage in perivascular cells highly correlated with endothelial lesions in all samples. Therefore, pharmacological interventions, directed at correcting the chronic hypoperfusion state, may change the natural course of the development of dementing neurodegeneration.
Introduction
Vascular endothelial cells, neurons and glia are all able to synthesize, store and release reactive oxygen species (ROS) and vasoactive substances in response to certain stimuli, especially those produced by chronic hypoxia/hypoperfusion (for review see CitationAliev et al 2003b). The contribution of these substances to the pathophysiology of stroke, cerebrovascular disease or CVAs and AD is extremely important. It has been suggested that hypoperfusion can be an initiator of AD (CitationAliev et al 2002b, Citation2003a, Citation2003b, Citation2007; Citationde la Torre 2002a). This idea is based on a positive correlation between AD and cardiovascular diseases. ROS are generated at sites of injury and/or inflammation. We hypothesize that the cellular and molecular mechanisms, by which hypoperfusion-induced ROS-accumulation impairs endothelial barrier function and promotes leukocyte adhesion, induce alterations in normal vascular function and result in the development of AD (CitationAliev et al 2003b).
Sustained hypoperfusion promoting oxidative stress of brain tissues could also stimulate secondary damage via the overexpression of inducible and neuronal specific nitric oxide synthase (iNOS and nNOS, respectively) and endothelin-1 (ET-1) in brain cells (CitationAliev et al 2000; Citationde la Torre 2002a; CitationAliyev et al 2004; Citationde la Torre and Aliev 2005). The continuous accumulation of oxidative stress products, such as peroxynitrite accumulation (via the overexpression of the iNOS and/or nNOS), may be secondary as well as an accelerating factor for the damage and compromise of the blood brain barrier (BBB) in hypoxia/hypoperfusion or AD (CitationAliyev et al 2004).
One of the main effects of chronic hypoperfusion induced vascular abnormality in AD appears to be tissue oxygen deficiency. Chronic cerebral hypoperfusion induces reduction of tissue oxygen delivery that causes the development of cognitive impairments such as AD (CitationKumar et al 1990; CitationFriston and Frackowiak 1991; CitationDe Jong et al 1997; Citationde la Torre 1997, Citation2002a; CitationAliev et al 2007). CitationDe la Torre (2000) proposes that advanced aging with a comorbid condition, such as a vascular risk factor that further decreases cerebral perfusion, promotes a critically attained threshold of cerebral hypoperfusion (CATCH). With time, CATCH induces brain capillary degeneration and suboptimal delivery of energy substrates to neuronal tissue (Citationde la Torre 2000). Because glucose is the main fuel of brain cells, its impaired delivery together with a deficient delivery of oxygen, compromises neuronal stability because the supplies for aerobic glycolysis fail to meet the brain tissue demand. The outcome of CATCH is a metabolic cascade that involves, among other things, mitochondrial dysfunction, oxidative stress, decreased adenosine triphosphate (ATP) production and increased calcium entry, abnormal protein synthesis, cell ionic pump deficiency, signal transduction defects, and neurotransmission failure. These events contribute to the progressive cognitive decline characteristic of patients with AD, as well as regional anatomic pathology consisting of synaptic loss, senile plaques (SP), neurofibrillary tangles (NFT), tissue atrophy, and neurodegeneration. CATCH encompasses the clinical heterogenic pattern that characterizes AD and provides compelling evidence that any of a multitude of different etiopathophysiologic vascular risk factors, in the presence of advanced aging, can lead to AD (Citationde la Torre 2000, Citation2002b; Citationde la Torre and Aliev 2005; CitationAliev et al 2007).
The aim of the present study was to investigate the features of the vascular lesions in human AD and two transgenic mouse models of AD, lines overexpressing a mutated form of the amyloid beta precursor protein (AβPP). We compared the ultrastructural features of each and identified common changes relative to age-matched controls that may give clues to underlying mechanisms affecting brain neuronal function.
Materials and methods
All experimental procedures were performed in accordance with University of Texas San Antonio and Case Western Reserve University guidelines for the use of human biopsy and post-mortem tissues and the care and use of laboratory animals for research. Human AD brain biopsy and postmortem tissues for ultrastructural studies were taken as described earlier and processed for future electron microscopic (EM) and in situ hybridization studies (CitationHirai et al 2001; CitationAliev et al 2002a, Citation2002b, Citation2003a, Citation2007). Mouse brain samples were obtained from a normal, nontransgenic line (Py8.9-Hemi [wild type]) and 2 transgenic lines, AβPP-YAC and C57B6/SJL Tg (+), were obtained from Bruce CitationLamb and colleagues (1999). Animals were 22–26 months old (n = 6 for each group). All animals received a standard laboratory diet ad libitum. Mice, under terminal anesthesia were perfusion fixed via the heart as described previously (CitationAliev et al 2002a, Citation2003a; CitationAliyev et al 2005). Brain samples were processed for future analysis by electron microscope and in situ hybridization for cytological detection of mtDNA on the electron microscopic level. In situ hybridization was performed using wild type and Δ5kb deleted human and mouse specific probes as described recently (CitationAliev et al 2003a; CitationAliev 2007). Finally, all the sections were exposed to OsO4 for 1 h at RT, rinsed, dehydrated and flat embedded in Spurr’s embedding media. Ultrathin sections were stained with uranylacetate and lead citrate and viewed in a JEOL 100CX or Jeol 1230 EX electron microscope at 80 kV.
Results and discussion
Endothelial cells (EC) and perivascular cells of human brain microvessels from age-matched control cases showed no visible changes in their ultrastructure. Mitochondria in the EC were intact (data not shown).
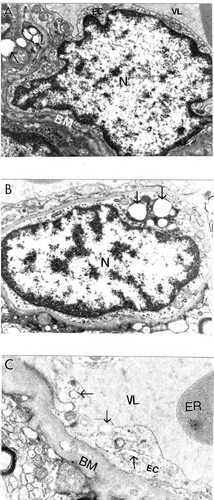
Conversely, cortical microvessels from AD brain biopsies were characterized by different degrees of damage, and heterogeneous lesions (). In some areas microvessels showed multiple lesions such as the presence of a cluster of mitochondria-derived lysosomes and necrotic changes in the ultrastructure of the vascular EC and perivascular cells (). Very often capillary endothelium cells showed the presence of “giant” sized lipid vacuoles in their matrices. Transformation of the mitochondria to mitochondria-derived lysosomes was generalized to all brain cellular compartments (). In addition, EC occupied only a small part of the vessel wall. Perivascular cells showed the presence of a large number of the mitochondria-derived vacuoles in their matrices (). Sometimes microvascular endothelium, at the early stages of AD, did not show any damage in their ultrastructure. However, the luminal plasma membrane of this EC sharply protruded into the vessel lumen, indicating the effect of hypoperfusion before any visible ultrastructural damage ().
Figure 1 Ultrastructural characteristics of brain microvessels from human Alzheimer disease brain biopsies. A: Microvessels with minimal changes did not show any particular pathology in the ultrastructure of the vascular endothelium; however, perivascular cells show the presence of vacuolar degenerative structures in the matrix (indicated by single arrow). B: Vascular endothelium and perivascular cells of damaged microvessels display ultrastructural lesions in their cytoplasmic organelles especially mitochondria. Completely damaged matrices appeared to be permanent features of all of these cells (indicated by single arrow). Original magnification ×20,000 (A and B). C: Vascular endothelium with non-reversible damage have completely damaged mitochondria and destructive changes in the membranous structures. The basal membrane (BM) is very thick and occupies a large area of the vascular wall. Original magnification ×12,000.
The ultrastructural features of vascular lesions and mitochondria changes in neuronal cell bodies in transgenic and non-transgenic age-matched control mice were analyzed following perfusion fixation (CitationAliev et al 2000, Citation2003a, Citation2005). Ultrastructural changes in the brain samples of AβPP-YAC, but not age-matched control mice, coexisted with different degrees of amyloid deposition (CitationAliev 2002; CitationAliev et al 2002a).
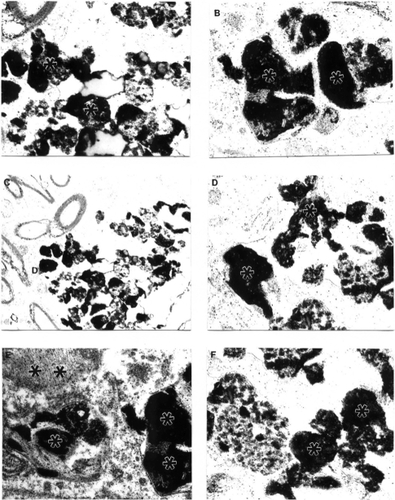
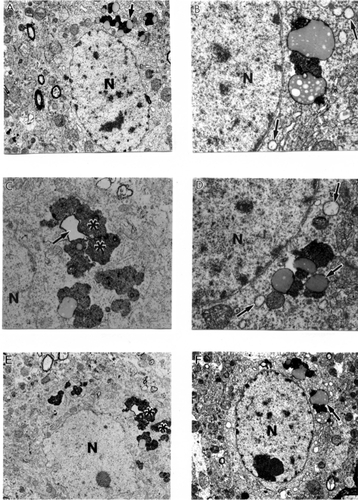
Very often, clusters of Aβ positive immunoreactivity were observed in the neuronal cell bodies of perivascular paired cortical neurons (CitationAliev et al 2000; CitationAliev 2002). EM immunocytochemistry revealed different sizes of fibrils and extracellular types of amyloid deposits in the brain tissues (). The amyloid depositions were associated with the formation of parietal helical filamental (PHF) structures (), which is a permanent feature of neuronal lesions in AD brains (CitationAliev et al 2000, Citation2003a).
Figure 2 Features of amyloid deposition in a neuronal cell body and the extracellular matrix in AβPP-YAC aged transgenic mouse brain (PAP pre-embedding immunocytochemistry). Amyloid depositions are associated with the formation of PHF-like structures (double asterisk in figure E). Original magnification A–D ×20,000. E–F ×25,000.
Brain microvessels showed immunopositive staining for Aβ and were characterized by the presence of large-sized lipid-laden vacuoles in the matrices of EC and perivascular cells (). These changes were generalized to cortical microvessels as in AD samples (CitationAliev 2002). The ultrastructural abnormalities of vascular wall cells were dependent on the presence of Aβ deposits around the microvessels (-). In contrast with these observations, age-matched control mouse brain vessels did not show any particular changes in the ultrastructure of vascular EC at different levels of microcirculation. Only sometimes did a very small number of lipid droplets appear in the matrices of perivascular cells (CitationAliev 2002; CitationAliev et al 2003a). This data clearly indicates that disruption of BBB function in vascular EC may be a major factor in lipid accumulation and amyloid deposition during the development of AD-like pathology in AβPP-YAC mice without cholesterol feeding (CitationAliev 2002; CitationShi et al 2002b; CitationAliev et al 2003a).
Figure 3 Features of APP immunostaining in frontal cortical microvessels in aged AβPP-YAC mice determined by PAP pre-embedding immunocytochemistry. A, B and C are without counter staining. D is with counter staining. Asterisks indicate amyloid deposition. Original magnification ×12,000.
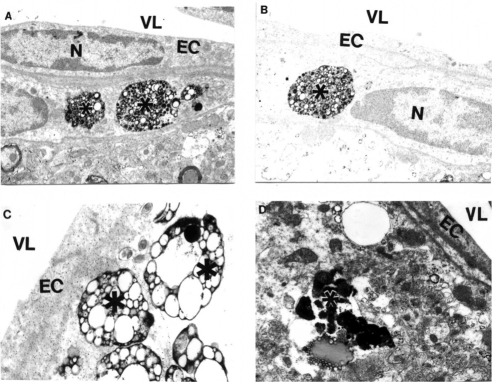
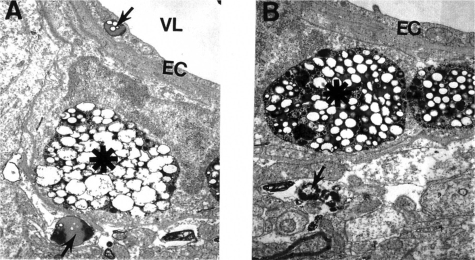
Figure 4 Electron microscopic subcellular features of brain cortical microvessels from 24-month old AβPP-YAC mice. A: Vascular endothelial cells display lipid vacuoles in their cytoplasmic matrices (single arrow). Perivascular cells have “giant”- sized lipid/amyloid granules in their cytoplasmic matrices (indicated by asterisk). Original magnification: ×12,000. B: Microvessels with amyloid “angiopathy” are characterized by the accumulation of large-sized amyloid/lipid granules in the matrices of perivascular cells (indicated by asterisk). Vacuolar structures with lipid filled amyloid deposition (single arrow) are also present in the matrices of perivascular cells. Original magnification: ×12,000.
Ultrastructural immunocytochemical analyses using peroxidase-anti-peroxidase (PAP) and/or colloidal gold probes indicated that the damaged vascular walls in AβPP-YAC transgenic mice possess atherosclerotic lesions (), while control and non-damaged vessels from AβPP-YAC mice do not show Aβ immunopositive staining (data not shown).
Age-matched control mice showed no particular changes in their neuronal ultrastructure (). Cortical neuronal cell bodies in AβPP-YAC mice were characterized by different degrees of ultrastructural alterations in their mitochondrial structures () similar to human AD samples (CitationAliev et al 1999, Citation2000). Giant and electron- dense (ED) mitochondria appeared to be permanent features of the neuronal abnormality and was associated with the AβPP overexpression (CitationAliev et al 2000, Citation2003a). We also found that mitochondrial lesions in neuronal cell bodies appeared to be associated with the absence of microtubules and with lipofuscin formation. The same observations were seen in the populations of vulnerable neurons in AD brain biopsies indicating neuronal cytoskeletal abnormalities (CitationCash et al 2003).
Figure 5 Age-matched control, non-transgenic mice did not show any particular changes in their neuronal ultrastructure. Lipofuscin was present in some neurons (indicate by asterisk in figure A). Original magnification of A and B: ×5,000, and ×20,000, respectively.
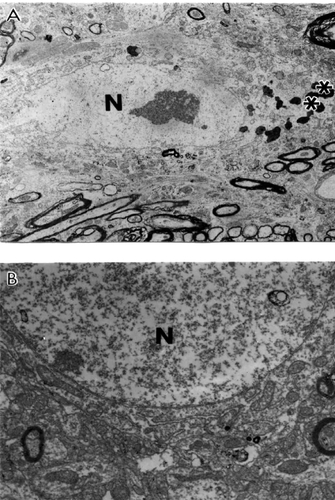
Figure 6 Ultrastructural characteristics of the neuronal damage in AβPP-YAC transgenic mouse hippocampus. Mitochondrial lesions were associated with lipofuscin formation in the neuronal cell body (asterisk) and mitochondria appeared to be a major substrate for lipofuscin formation (single arrows). Original magnification: A, E, and F ×5,000. B, C, and D ×20,000 respectively.
We looked at ultrastructural features of brain tissue from C57B6/SJL Tg (+) transgenic mice overexpressing AβPP. We demonstrated that the binding of basic fibroblast growth factor (bFGF) and serum amyloid P (SAP) to Aβ is a marker of a lack of BBB integrity in this mouse model. Adjacent sections of brain were stained with 4G8, a monoclonal antibody to the Aβ peptide (aa17–24), followed by 48.1, a monoclonal antibody against bFGF. The binding of bFGF in this mouse line is similar to that of AD cases in which bFGF binds specifically to Aβ neuritic plaques and the basement membrane of cerebral microvessels (CitationShi et al 1999, Citation2002a). In addition, the cores of amyloid plaques were intensely stained with 4G8, and bFGF binding is co-localized with amyloid immunoreactivity as visualized by polyclonal antiserum to amyloid. Our ultrastructural study indicated that bFGF immunostaining in aged C57B6/SJL Tg (+) mice was associated with damaged, but not normal neurons (CitationAliev et al 2000, Citation2002a, Citation2003a, Citation2007). Moreover, this was associated with different degrees of Aβ immunostaining in the neuronal cell body and vascular wall cells (CitationAliev 2002; CitationAliev et al 2003a; CitationAliyev et al 2005). The degree of mitochondrial abnormality, such as ED mitochondria-derived lysosomes and lipofuscin formation appeared to be features of damaged neurons in 24-month old aged C57B6/SJL Tg (+) but not age-matched control mice (). Vascular abnormality was associated with the selective damage to cortical neurons suggesting a direct relationship between vascular abnormality, BBB breakdown, neuronal loss and amyloid depositions during the maturation of AD-like pathology (CitationAliev 2002; CitationAliev et al 2003a).
Figure 7 Ultrastructural characteristics of cortical neurons in C57B6/SJL transgenic mouse (Tg +) brain. The main characteristic of neuronal damage appeared to be the transformation of completely damaged mitochondria (mitochondria without any residues of mitochondrial cristae; indicated by single arrows) into the lipofuscin granules, characterised by their cluster type localization in the neuronal cell body. Original magnification: A and B ×5,000; C and D ×20,000, respectively.
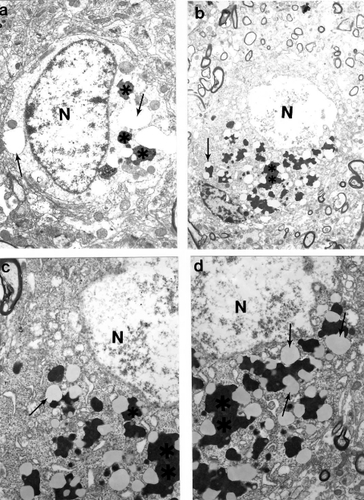
Previously we demonstrated that the ultrastructural features of cortical neurons from AD brain biopsies are characterized by the selective localization of mitochondria abnormalities to the cell bodies (CitationAliev et al 1999, Citation2000; CitationHirai et al 2001; CitationAliev 2002; CitationShi et al 2002b). The majority of the neurons, which were closely associated with lesioned vessels, possessed different degrees of ultrastructural abnormality. Partially and completely damaged mitochondria were associated with lipofuscin formation and mitochondria appeared to be a major substrate for this process (-). In many cases, neuronal cell bodies showed an absence of cellular organelles. Different stages of mitochondrial abnormality, such as formation of mitochondria-derived lysosomes and lipofuscin, were seen in damaged but not in normal neurons (-). The mitochondria-derived lysosomes and lipofuscin deposits of varied density and sizes were permanent features of the neuronal abnormality (CitationAliev et al 2003a). Mitochondrial lesions and lipofuscinogenesis were also present in glial cells of the brain parenchyma (CitationAliev et al 2003a).
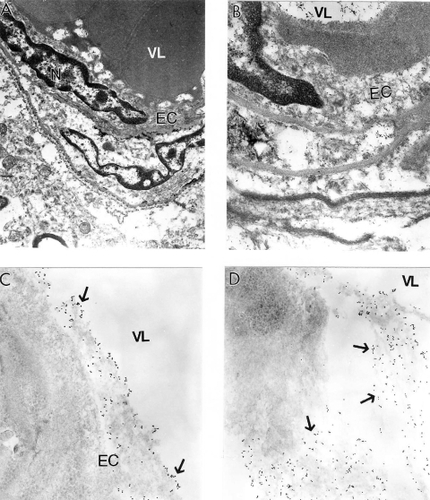
Quantitative morphometric measurements of the percentage of the different types of mitochondria (normal, partially damaged, and completely damaged) indicate that age-matched control groups have a significantly higher percentage of normal mitochondria compared with AD cases (CitationHirai et al 2001). Cytological in situ hybridization studies using probes for human normal and 5 kB deleted mtDNA found that mtDNA signals were associated with severely damaged or mitochondria-derived lysosomal structures (). However, the areas containing lipofuscin did not show any mtDNA containing positive signals. On the ultrastructural level, clusters of 5 kb deleted mtDNA containing gold particles (17 nm) were localized in mitochondria-derived lysosomal structures, indicating that mtDNA deletions and turnover occur at the late stages of the mitochondrial lesion formation. Similarly, areas of neuronal cell bodies containing normal mtDNA and lipofuscin were free from any 5 kb deleted mtDNA positive signals (CitationHirai et al 2001; CitationAliev et al 2002a). In contrast to this observation, hippocampal neuronal mitochondria of age-matched control subjects did not show mtDNA positive signals containing gold particles in their matrices.
Figure 8 Electron microscopic determination of mitochondrial DNA signals visualized by using wild type and chimeric 5 kb deleted mtDNA probes in a human postmortem Alzheimer’s disease (AD) brain (A–B) and 24-month old AβPP-YAC transgenic mouse brain (C–D). A and B: AD brain microvessels endothelium and perivascular cells show clusters of wild type mtDNA containing positive signals visualized by indirect colloidal gold techniques (indicated by the dark dots). Original magnification: A and B ×10,000 and ×25,000, respectively. C–D: AβPP-YAC transgenic mice show the presence of clusters of chimeric 5 kb deleted mitochondria DNA positive signals throughout the matrices of vascular and perivascular cells (single arrows). Original magnification: C and D ×30,000 and ×20,000, respectively.
Our detailed immunocytochemical analysis also demonstrated that the mitochondrial abnormalities in neurons are associated with increased markers of lipid peroxidation (CitationMoreira et al 2007). Moreover, lipid peroxidation markers were associated with RNA oxidation (staining by 8OHG). Clusters of 8OHG containing immunopositive gold particles (17 nm) were localized to the matrices of completely and/or partially damaged mitochondria (CitationNunomura et al 2001; CitationAliev et al 2002, Citation2002a). Large quantities of immunopositive gold particles were also associated with the cytoplasmic matrix. In addition, the cytoplasmic matrices, especially of the regions where damaged mitochondria or mitochondria-lipofuscin were present, showed clusters of 8OHG containing immunopositive gold particles (CitationAliev et al 2002, Citation2002a). Our quantitative study indicated that the extent of oxidative damage (eg, 8OHG staining) is highly dependent on the degree of mitochondrial abnormalities (CitationNunomura et al 2001; CitationAliev et al 2003a). Based on the evidence presented in this study we theorize the hypothetical timeline of the damage in the brain during maturation of AD is much more complex than the previously proposed hypothetical schematic (CitationAliev et al 2003b). With the onset of AD, normal neurons develop numerous forms of oxidative damage including nitration (nitrotyrosine), lipid peroxide adducts (lipid peroxidation), and nucleic acid oxidation prior to the formation of pre-neurofibrillary tangles (pre-NFT) that induce nonreversible damage to neurons and therefore finally failure of neurotransmission that occurs in AD brain.
The detailed analysis of 8OHG immunostaining demonstrated that only vulnerable neurons show immunopositive staining for 8OHG in AD, but not in age-matched controls. By using ultrastructural analysis we have found that 8OHG immunostaining was selectively associated in vulnerable neurons and microvessels of AD brain (CitationAliev et al 2002b; CitationAliyev et al 2005). The 8OHG immunogold labeling (17 nm) was seen throughout the cytoplasm, including the damaged mitochondria or electron dense abnormal mitochondria (CitationHirai et al 2001; CitationNunomura et al 2001; CitationAliev et al 2002b; CitationMoreira et al 2007). However, we did not find 8OHG in normal mitochondria or in lipofuscin. The capillary EC and perivascular pericytes showed a high intensity of 8OHG immunostaining (CitationAliev et al 2002b).
In this study, the main location of the mtDNA was in damaged mitochondria and mitochondria derived lysosomes, but not in lipofuscin in all brain cellular compartments including the vascular wall cells (). Endothelial cells (EC) of vessels with atherosclerotic lesions and nearby perivascular cells contain clusters of normal and deleted mtDNA positive signals (). These observations emphasize the key role of hypoperfusion, mitochondrial abnormality and oxidative stress in the pathogenesis of vascular and nonvascular cells lesions during the development of AD-like pathology in AβPP-YAC mice (CitationAliev et al 2002a; CitationAliyev et al 2005). Many of these characteristics overlap the neuropathology of human AD (CitationAliev et al 2002a).
In situ hybridization analysis with mouse mtDNA probes found abundant deleted mtDNA in AβPP-YAC mice compared to age-matched controls (CitationAliev et al 2000, Citation2002a, Citation2003a). Moreover, the majority of deleted mtDNA was found in mitochondria-derived lysosomes () in regions closely associated with lipofuscin, suggesting that proliferation, deletion and duplication of mtDNA occur in mitochondria, many of which have been fused with lysosomes (CitationAliev et al 2000). These finding suggest that abnormalities in mitochondrial structures and mtDNA are features of damaged neurons in AβPP-YAC mice, and may also play a key role in the pathogenesis of AD (CitationAliev et al 2000). We found that neurons in AD samples were dominated by abnormal mitochondria as compared with the control group (CitationAliyev et al 2005). By in situ hybridization analyses with a chimeric cDNA probe to the 5 kb common deletion (CitationHirai et al 2001), we found that deleted mtDNA is increased at least 3 fold for the AD cases compared to the controls (CitationHirai et al 2001). In situ hybridization detected mtDNA proliferation, deletion and duplication in abnormal mitochondria, many of which were fused with lysosomes, indicating they were being turned over (see ).
A growing body of evidence suggests that there are direct links between heart disease, hypertension, stroke and neurodegeneration (CitationRoher et al 2003; Citationde la Torre 2006; CitationSkoog and Gustafson 2006). The relationship between atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer’s disease has been well documented (CitationRoher et al 2003; CitationKalback et al 2004). Hypertension has been suggested to be an initiator of the ischemic pathway leading to AD (Aliev et al 2003, Citation2007; Citationde la Torre 2006; CitationSkoog et al 2006). Recently, studies by CitationChoi and colleagues (2007) showed that preconditioning with chronic cerebral hypoperfusion reduces a focal cerebral ischemic injury and increases apurinic/apyrimidinic endonuclease/redox factor-1 and matrix metalloproteinase-2 expression. Another report by CitationAnnaházi and colleagues (2007) demonstrated that the pre- and post-treatment with alpha-tocopherol attenuates hippocampal neuronal damage in experimental cerebral hypoperfusion. Moreover, circulating CD34 positive T-cells are able to provide an index of cerebrovascular function (CitationTaguchi et al 2004). The beneficial effect of carotid endarterectomy on the cerebral blood flow and cerebral blood volume improvement was studied by SPECT (CitationLushmanov et al 1997).
Based on this literature evidence and our present study we believed that managing AD as a vascular disease will open a new window of insight not only for better understanding of the etiopathogenesis but also new and more effective treatment strategies.
We theorize that the oxidative stress markers seen in the human AD and AD mouse models selectively affect the population of vulnerable neurons, vascular EC and perivascular cells. We believe that hypoperfusion-induced oxidative stress plays a key role in the pathogenesis of vascular and nonvascular cell lesions during the maturation of AD.
Conclusion
Our data for the first time demonstrates that a chronic injury stimulus induces the hypoperfusion seen in the microcirculation of vulnerable brain regions. Determining the mechanisms behind these imbalances may provide crucial information in the development of new, more effective therapies for the treatment of cerebrovascular diseases, including AD. Future studies must seek to answer the following questions: (1) What are the major factors altering and/or controlling cerebral blood flow during the progression of chronic hypoperfusion and/or the development of atherosclerotic changes in brain microvessels? (2) What are the roles of endothelial content vasoactive substances (especially nitric oxide [NO] and endothelin-1 [ET-1]) during the development of this pathophysiology? (3) Does acute or chronic hypoperfusion with concomitant oxidative stress accelerate the vascular and neuronal lesions during normal aging and/or when brain is exposed to chronic hypoperfusion induced factors? Resolving these issues will allow for novel therapeutic approaches that will modify the natural history of these chronic age onset disorders in the 21st century.
Acknowledgements
Supported by the Philip Morris USA Research Management Group and Alzheimer’s Association. The authors report no conflict of interest.
References
- AlievGIs non-genetic Alzheimer’s disease a vascular disorder with neurodegenerative consequences?J Alzheimer’s Dis2002451316
- AlievGLiuJXuKApoE4 in mice induces age-dependent brain hypoperfusion, neuronal, glial and microvascular damage, and cognitive impairment, which can be prevented by feeding Acetyl-L-Carnitine and R-Lipoic Acid8th International Conference on Alzheimer’s and Parkinson’s Diseases AD/PD (Salzburg, Austria)2007Proceeding AD/PD ConferenceMonduzzi Editore, Bologna, Italyin press
- AlievGObrenovichMESeyidovaDExploring ischemia-induced vascular lesions and potential pharmacological intervention strategiesHistol Histopathol2005202617315578444
- AlievGSeyidovaDLambBTMitochondria and vascular lesions as a central target for the development of Alzheimer’s disease and Alzheimer disease-like pathology in transgenic miceNeurol Res2003a256657414503022
- AlievGSeyidovaDNealMLAtherosclerotic lesions and mitochondria DNA deletions in brain microvessels as a central target for the development of human AD and AD-like pathology in aged transgenic miceAnn N Y Acad Sci2002a977456412480733
- AlievGShiJPerryGNeuronal mitochondrial abnormalities in yeast artificial chromosome (YAC) transgenic mice overexpressing amyloid precursor protein (APP)Society for Neuroscience Abstracts Book200030
- AlievGSmithMAObrenovichMERole of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Alzheimer diseaseNeurotox Res2003b549150414715433
- AlievGSmithMASeyidovDThe role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer’s diseaseBrain Pathol2002b12213511770899
- AlievGSmithMAVintersHMitochondria abnormalities mark vulnerable neurons in Alzheimer’s diseaseJ Neuropathol Exp Neurol199958511
- AliyevAChenSGSeyidovaDMitochondria DNA deletions in atherosclerotic hypoperfused brain microvessels as a primary target for the development of Alzheimer’s diseaseJ Neurol Sci2005229–23028592
- AliyevASeyidovaDRzayevNIs nitric oxide a key target in the pathogenesis of brain lesions during the development of Alzheimer’s disease?Neurol Res2004265475315265272
- AnnaháziAMracskóESüleZPre-treatment and post-treatment with alpha-tocopherol attenuates hippocampal neuronal damage in experimental cerebral hypoperfusionEur J Pharmacol20075712–3120817597609
- CashADAlievGSiedlakSLMicrotubule reduction in Alzheimer’s disease and aging is independent of tau filament formationAm J Pathol20031621623712707046
- ChoiSAKimEHLeeJYPreconditioning with chronic cerebral hypoperfusion reduces a focal cerebral ischemic injury and increases apurinic/apyrimidinic endonuc-lease/redox factor-1 and matrix metalloproteinase-2 expressionCurr Neurovasc Res20074899717504207
- De JongGIDe VosRASteurENCerebrovascular hypoperfusion: a risk factor for Alzheimer’s disease? Animal model and postmortem human studiesAnn N Y Acad Sci199782656749329681
- de la TorreJCHemodynamic consequences of deformed microvessels in the brain in Alzheimer’s diseaseAnn N Y Acad Sci199782675919329682
- de la TorreJCCritically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesisNeurobiol Aging2000213314210867218
- de la TorreJCAlzheimer’s disease: how does it start?J Alzheimer’s Dis2002a4497512
- de la TorreJCAlzheimer disease as a vascular disorder: nosological evidenceStroke2002b3311526211935076
- de la TorreJCHow do heart disease and stroke become risk factors for Alzheimer’s disease?Neurol Res200666374416945216
- de la TorreJCAlievGInhibition of vascular nitric oxide after rat chronic brain hypoperfusion: spatial memory and immunocytochemical changesJ Cereb Blood Flow Metab2005256637215703700
- FristonKJFrackowiakRSCerebral function in aging and Alzheimer’s disease: the role of PETElectroencephalogr Clin Neurophysiol Suppl199142355651915028
- HiraiKAlievGNunomuraAMitochondrial abnormalities in Alzheimer’s diseaseJ Neurosci20012130172311312286
- LambBABardelKAKulnaneLSAmyloid production and deposition in mutant amyloid precursor protein and presenilin-1 yeast artificial chromosome transgenic miceNat Neurosci19992695710412057
- KumarASchapiroMBHaxbyJVCerebral metabolic and cognitive studies in dementia with frontal lobe behavioral featuresJ Psychiatr Res199024971092213642
- KalbackWEshCCastañoEMAtherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer’s diseaseNeurol Res2004265253915265270
- LishmanovYShveraIUssovWThe effect of carotid endarterectomy on cerebral blood flow and cerebral blood volume studied by SPECTJ Neuroradiol199724155629297936
- MoreiraPISiedlakSLWangXAutophagocytosis of mitochondria is prominent in Alzheimer diseaseJ Neuropathol Exp Neurol2007665253217549012
- NunomuraAPerryGAlievGOxidative damage is the earliest event in Alzheimer diseaseJ Neuropathol Exp Neurol2001607596711487050
- RoherAEEshCKokjohnTACircle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s diseaseArterioscler Thromb Vasc Biol20032320556214512367
- ShiJPerryGAlievGSerum amyloid P is not present in amyloid beta deposits of a transgenic animal modelNeuro Report199910322932
- ShiJPerryGBerridgeMSLabeling of cerebral amyloid beta deposits in vivo using intranasal basic fibroblast growth factor and serum amyloid P component in miceJ Nucl Med2002a4310445112163630
- ShiJSeyidovaDPerryGNeuronal mitochondria abnormalities in a transgenic mouse model overexpressing amyloid betaSociety for Neuroscience2002b29520
- SkoogIGustafsonDUpdate on hypertension and Alzheimer’s diseaseNeurol Res2006286051116945211
- TaguchiAMatsuyamaTMoriwakiHCirculating CD34-positive cells provide an index of cerebrovascular functionCirculation20041092972515184275