Abstract
During embryogenesis and development, the fetus obtains oxygen and nutrients from the mother through placental microcirculation. The placenta is a distinctive organ that develops and differentiates per se, and that organizes fetal growth and maternal condition in the entire course of gestation. Several life-threatening diseases during pregnancy, such as pregnancy-induced hypertension (PIH) and eclampsia, are closely associated with placental dysfunction. Genetic susceptibilities and poor placentation have been investigated intensively to understand the pathophysiology of PIH. It is currently thought that “poor placentation hypothesis”, in which extravillous trophoblasts fail to invade sufficiently the placental bed, explains in part maternal predisposition to this disease. Cumulative studies have suggested that hypoxic micromilieu of fetoplacental site, shear stress of uteroplacental blood flow, and aberrantly secreted proinflammatory substances into maternal circulation synergistically contribute to the progression of PIH. For example, soluble form of vascular endothelial growth factor receptor-1 (sVEGFR-1) and soluble form of CD105 are elevated in circulation of PIH mothers. However, it remains to be poorly understood the pathological events in the placenta during the last half of gestation as maternal systemic disorders get worse. For better understanding and effective therapeutic approaches to PIH, it is important to clarify pathological course of PIH-associated changes in the placenta. In this review, current understanding of placental development and the pathophysiology of PIH placenta are summarized. In addition, recent findings of vasoactive signalings in PIH and rodent PIH models are discussed.
Introduction
The placenta starts organogenesis in the very early stage of embryogenesis, governs fetal growth, and terminates its own fate immediately after delivery. Placental perfusion is maintained by two distinct cardiovascular systems, ie, maternal blood flow and fetal circulation. Therefore, pathophysiology of the placenta is closely associated with both maternal status and fetal development. Among pregnancy-associated complications, eclampsia is an emergency condition for both mother and fetus. To prevent the lethal disaster, it is mandatory to care properly the patients in preeclamptic condition, ie, pregnancy-induced hypertension (PIH). PIH is characterized by blood pressure elevation after 20 weeks of gestation that is often accompanied by proteinuria (CitationNHBPEP 2000). Genetic, immunologic, metabolic susceptibilities and other backgrounds have been investigated to understand the pathogenesis of this disease (CitationHiby et al 2004; Citationvan Dijk et al 2005; CitationHu et al 2006; CitationJohnson et al 2007). Several important findings have contributed to our understanding of maternal genetic predisposition, eg, specific patterns of genetic variant of angiotensinogen gene and quantitative trait loci on some chromosomes including 5q, 10q, and 13q (CitationMorgan et al 1997; CitationKobashi et al 1999; Citationvan Dijk et al 2005; CitationJohnson et al 2007). Both background and progression course of PIH vary among cases, and it is difficult to predict whether the condition is improved in response to treatment or is aggravated and resulted in preterm termination. Currently, the onset of PIH is considered to depend not only on a sole or a few pathological events. It may rather be triggered by a load of predisposing factors that potentially promote circulatory dysfunction (CitationRedman and Sargent 2005).
Wide variety of angiogenic molecules and proteolytic enzymes play critical roles in the establishment of placentation and development of placental circulatory system (CitationAdamson et al 2002; CitationKharfi et al 2003; CitationReynolds et al 2005). For example, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and placenta growth factor (PlGF) are indispensable in the entire course of gestation (CitationReynolds and Redmer 2001; CitationZygmunt et al 2003). In later stages of pregnancy, villous trophoblasts and fetal-side blood vessels of terminal villi (or labyrinth in mouse placenta) form finely-differentiated vascular network to serve a fetus with sufficient amounts of oxygen and substances for exponential fetal growth (CitationReynolds et al 2005). The arterial circulation in the placenta lacks autonomic innervation and is regulated by local signals such as pressure and flow (CitationMyatt 1992). If implantation process is not performed successfully, the placenta suffers from insufficient perfusion and secretes various kinds of proinflammatory molecules that damage maternal endothelial cells (ECs), and in consequence, vascular resistance is increased that further burdens maternal organs with hypertension as well as aggravates fetoplacental milieu (CitationKharfi et al 2003; CitationKarumanchi and Bdolah 2004; CitationRedman and Sargent 2005).
It is thought that systemic inflammatory response and dysfunction of maternal ECs represent the pathological scheme of PIH (CitationGranger et al 2001; CitationKarumanchi and Bdolah 2004; CitationRedman and Sargent 2005). Various proinflammatory cytokines/peptides including serological markers soluble form of VEGF receptor-1 (sVEGFR-1, also named sFlt-1) and soluble form of CD105 (sCD105, also named soluble form of endoglin) are elevated in PIH mothers, and these anti-angiogenic factors are thought to play critical roles in maternal ECs-dysfunction (CitationGranger et al 2001; CitationMaynard et al 2003; CitationVenkatesha et al 2006). On the other hand, it is poorly understood the pathological courses of the placenta and the mechanisms that lead to intrauterine growth restriction (IUGR) of the fetus. With regard to placental dysfunction, since very limited examinations are available for pregnant patients, the analysis of placental circulation is based mainly on Doppler studies and on the pathological investigation of terminated placentas (CitationOhkouchi et al 2000; CitationParretti et al 2003; CitationKraus et al 2004). The dynamics of vasoactive signalings in the placenta during upsurge of blood pressure remain enigmatic.
In this review, placental development and proinflammatory microenvironments of PIH are discussed. First, general structure of the placenta in human and mouse is summarized. Second, current understanding of pathological events of the placenta in PIH is overviewed. Third, the findings obtained from PIH model mice are introduced, and possible pathological events occurred to fetoplacental vasculature under preeclamptic condition are discussed.
Structure of placental vascular network
Both in human and mouse, a normal term placenta is divided largely into three layers (, ): (1) Basal plate (maternal surface) and anchoring villi (most distal extensions of the primary stem villi) that interact directly with maternal endometrium. (2) Terminal villous unit (human) or labyrinth (mouse) where gas/nutrients exchange is taken place actively. (3) Chorionic plate (fetal surface) and stem villi that consist of dense connective tissue containing fetal vessels. Amnion and the underling chorion are membranes that cover chorionic plate, and the umbilical cord collects chorionic arteries and veins on chorionic plate generally in the center part (CitationKraus et al 2004).
Figure 1 Histology of the placenta in human (A) and mouse (B). The photo of whole layer of the placenta is shown in the left. High magnification of each layer is shown in the right.
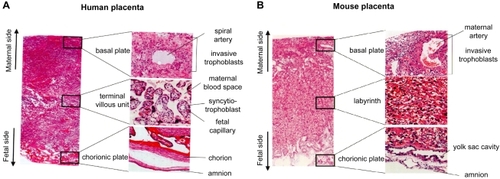
Fundamental structure of the placenta is established during the first half of gestation (CitationReynolds et al 2005). In human placenta (), terminal villus unit (tertiary villus that stems from secondary villus) is composed mainly of fetal-side capillaries lined by ECs, mesenchymal collagen and outlining syncytiotrophoblasts. In earlier stages, cytotrophoblasts lie beneath syncytiotrophoblasts. As the pregnancy progresses, cytotrophoblasts layer become almost undetectable, and fetal capillaries are placed in close proximity to intervillous maternal circulation, probably for the purpose of efficient gas/nutrients exchange (CitationLewis and Benirschke 1997; CitationKraus et al 2004). It is of note, that maternal blood space is lined directly by terminally-differentiated syncytiotrophoblasts, not by ECs (hemochorial interface) (CitationRossant and Cross 2001; CitationKraus et al 2004).
Basic architecture of mouse placenta is almost similar to that of human, but there are some differences at microscopic level (). In the “labyrinth”, both fetal chorionic branches and maternal blood sinusoids are tortuous and exhibit similar dimensions. Blunt-end villi are not detectable. Therefore, it is difficult to distinguish microscopically fetal-side blood flow from that of maternal-side without a help of immunohistochemistry. Between fetal and maternal circulatory interface, there are three trophoblast layers (trichorial); ie, two syncytiotrophoblast layers surrounding fetal-side ECs, and a single mononuclear trophoblast facing to maternal blood sinus (CitationRossant and Cross 2001; CitationGeorgiades et al 2002; CitationAdamson et al 2002).
Fetal weights increase almost twice during the last stage (CitationCunningham et al 1989). On the other hand, the weight of the placenta does not increase significantly in later stages. Instead, vascular networks in terminal villi/labyrinth become further differentiated and they increase functional capacity of fetal-side capillaries as well as of maternal blood sinuses (CitationReynolds et al 2005; CitationFuruya et al 2008) (). In IUGR, however, terminal villous differentiation is often disturbed, and the truncation of distal villi becomes evident (CitationKraus et al 2004) (, center). This pathological course is often accompanied by PIH. Under hypoxic condition, blood flow of fetal body accumulates to the most critical organs such as the brain and the heart. This blood redistribution further reduces placental flow and increases vascular resistance (CitationHecher et al 1995).
Figure 2 Development of vascular networks in the labyrinth between E13 and E19. Both fetal vessels (CD31) and maternal blood sinuses (CK) become complicated as gestation progresses (CitationFuruya et al 2008).
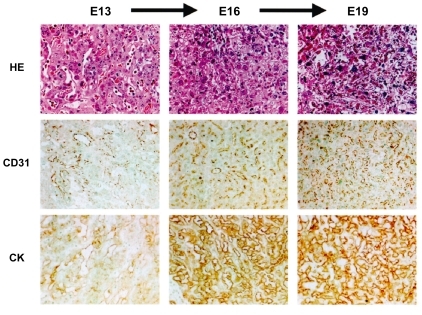
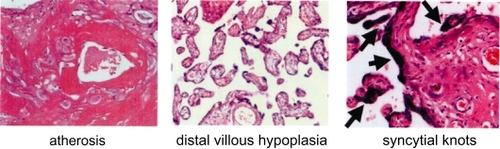
Figure 6 Pathology of human placenta in PIH. Atherosis (left), mural fibrinoid necrosis and the accumulation of macrophages; villous hypoplasia (center), decreased number of villi, with thin, poorly-branched and vascularized villi; syncytial knots (right, arrows), aggregation of syncytiotrophoblastic nuclei.
The early stage of pregnancy: Epithelial-endothelial transformation
It is well known that during implantation, extravillous trophoblasts invade uterine endometrium and also inner third of myometrium (CitationKraus et al 2004; CitationLyall 2005). To establish sufficient blood flow from maternal circulation, invasive trophoblasts undergo ECs-like specialization. Invasive trophoblasts turn to express some ECs markers such as CD31, VE-cadherin, VCAM-1, and αvβ3 integrin (CitationZhou et al 1997, Citation2002). In addition to αvβ3 integrin, these trophoblasts up-regulate the expression of α5β1 and α1β1 integrins and down-regulate the expression of α6β4 integrin (CitationLyall et al 2001). This process is called “epithelial-endothelial transformation” or “pseudo-vasculogenesis” (CitationDamsky and Fisher 1998). A similar process of vascular remodeling has been reported in cancer neovascularization. In several types of malignancies, such as aggressive melanoma, ovarian, prostatic, and breast carcinomas, tumor cells themselves build blood sinus, known as “tumor vasculogenic mimicry” (CitationManiotis et al 1999; CitationShirakawa et al 2001; CitationSood et al 2001; CitationHendrix et al 2003; CitationSharma et al 2002). The tumor cells that form this alternative vascular network express certain ECs markers and embryonic vasculogenesis-related molecules such as VE-cadherin, CD34, and CD105 (CitationSeftor et al 2002; CitationHendrix et al 2003). Cancer invasion is destructive and disorganized, whereas trophoblasts invasion is finely controlled by local proinflammatory microenvironments under physiological condition. Uterine constituents including decidual cells and immune cells govern uteroplacental interaction not only to accept “allogenic” cells, but also to restrict excessive invasion. Pathological feature of impaired epithelial-endothelial transformation in PIH is discussed later.
Proinflammatory microenvironments of implantation site
Microenvironments of the uteroplacental junction are crucial for the process of implantation and fetal development throughout gestation. In response to the upsurge of human chorionic gonadotropin (hCG), endometrium undergoes decidual change (CitationPerrier d’Hauterive et al 2007), and mononuclear cells are accumulated to the decidualized endometrium; ie, distinctive population of uterine immune cells, called natural killer cells (uNK cells, also named decidual natural killer [dNK] cells). uNK cells are CD45+/CD69+/CD56high/CD16− immune cells that represent the majority of leukocytes at the implantation site (CitationVacca et al 2006; CitationMoffett and Loke 2006; CitationMoffett and Hiby 2007). Although the function of uNK cells is not fully characterized, they are thought to play important roles in (1) decidual reaction, (2) remodeling of spiral arteries in the decidua, and (3) immune regulation of trophoblasts invasion (CitationMoffett and Loke 2006; CitationPijnenborg et al 2006).
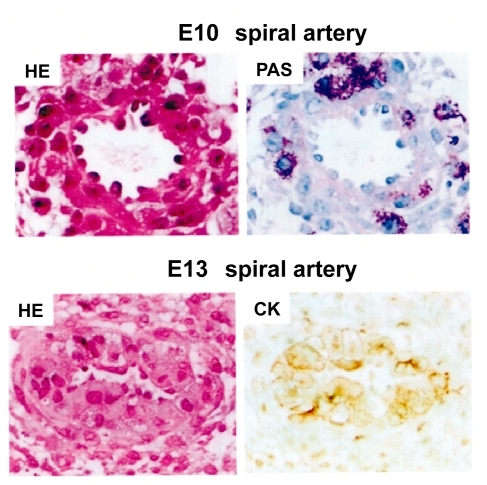
In mouse pregnancy, uNK cells are accumulated to implantation site by 10.5 days of gestation (E10.5), and these uNK cells infiltrate the media of spiral arteries (CitationAdamson et al 2002). These immune cells contribute to the establishment of blood flow from maternal circulation to the placenta in cooperation with invasive trophoblasts by remodeling spiral arteries (). Among various forms of trophoblasts, only extravillous trophoblasts express major histocompatibility complex (MHC) class I molecules (CitationMoffett and Loke 2006). In human pregnancy, these trophoblasts express human leukocyte antigens (HLA)-C, HLA-E, and HLA–G, a unique repertoire of ligands for uNK cell receptors (CitationKing et al 2000a, Citation2000b; CitationMoffett and Loke 2006; Moffett and Hiby 2006). HLA-A and HLA–B, the classical MHC class I molecules with polymorphism that initiate allograft rejection, are not expressed in extravillous trophoblasts. Neither MHC class II is expressed (CitationMoffett and Loke 2006). It is also of note that in fetoplacental site, syncytiotrophoblasts express no MHC antigens on cell surface (CitationMoffett and Loke 2006).
Figure 3 Remodeling of spiral artery between E10 and E13 in normal mouse pregnancy. At E10, PAS-positive uNK cells infiltrate the spiral artery. At E13, ECs are replaced by CK-positive trophoblasts (CitationFuruya et al 2008).
There are more than one pathways that control uNK cells function. uNK cells have both stimulatory and inhibitory surface receptors including NKG2 family (CD94), 2B4 (CD244), and NKp46 (CD335) (CitationKing et al 2000a; CitationVacca et al 2006). For example, the HLA-E in trophoblasts interacts with NKG2A in uNK cells (CitationKing et al 2000a), which may explain in part the mechanism of suppressed uNK cells cytotoxicity against invading trophoblasts. Activating crosstalk between invasive trophoblasts and uNK cells is also required. As mentioned above, polymorphic MHC class I molecules HLA-A and HLA–B are not expressed in extravillous trophoblasts, but another classical class I molecule HLA-C is present at first trimester (CitationKing et al 2000b). Recent studies on immunogenic background of PIH demonstrated that interaction between HLA-C genotypes of trophoblasts and killer-cell immunoglobulin-like receptor (KIR)-family of uNK cells were implicated in unsuccessful pregnancies (CitationHiby et al 2004). CitationHybi and colleagues (2004) found that the combination of fetal HLA-C2 haplotype and maternal KIR-AA genotype resulted in increased risk of PIH. The mechanism is explained as follows; interaction between KIR-A haplotype and HLA-C2 haplotype renders strong inhibitory signal of uNK cells, whereas KIR-B haplotype has an activating receptor for HLA-C haplotype, which increases inflammatory property of uNK cells (CitationHiby et al 2004). Taken together, fine tuning of pro- and antiinflammatory microenvironments of uteroplacental site is mediated by the crosstalk between maternal-side immune cells and fetal-side invasive trophoblasts, and that certain immunogenetic properties may determine the unfavorable process of pseudovasculogenesis.
Predisposing condition of PIH in the early stage of gestation
As mentioned above, it is generally accepted that poor placentation is an important predisposing condition for the pathophysiology of PIH. The combination of maternal KIR-AA genotype in uNK cells and fetal HLA-C2 haplotype in extravillous trophoblasts significantly increased the susceptibility rate of PIH (CitationHiby et al 2004), suggesting that a failure of proper activation of uNK cells leads to insufficient trophoblasts invasion. Narrowed blood canals due to poor placentation make the placenta hypoxic, and in response, a series of proinflammatory factors are released from the placenta that damage maternal circulatory system. This two stage model, ie, poor placentation of early gestational period (stage I) and maternal systemic dysfunction in later period (stage II), has been widely recognized as a mechanism for the development of PIH (CitationRedman and Sargent 2003; CitationRobert and Gammill 2005). However, impaired pseudovasculgenesis is unlikely to be an exclusive cause of the disease, and not a few cases show normal gestational process in spite of restricted placental flow, and vice versa (CitationRedman and Sargent 2003).
Key molecules implicated in the pathophysiology of PIH
There are a wide variety of factors that potentially accelerates vasoconstriction of maternal blood vessels. Neurokinin-B, a family of peptides tachykinins, was at first presented to be a responsible molecule that causes PIH (CitationPage et al 2000). Later studies using large number of samples by different groups, however, have demonstrated controversial results (CitationSchlembach et al 2003). Currently, it seems not to be fully approved the notion that neurokinin-B functions as a causative agent of PIH. Recent studies by Pal et al provided us with a new insight about neurokinin-B in the progression of PIH (CitationPal et al 2006). They demonstrated that neurokinin-B, with the help of thromboxane A2 (TXA2)-like molecule, down-regulated VEGF, VEGFR-1, and VEGFR-2 in cultured ECs, and suppressed angiogenic activities in vitro. Since it is known that the imbalance between TXA2 and prostacyclin PGI2 is contributed to preeclampsia (CitationChen et al 1993; Mills et al 2001), neurokinin-B/TXA2 axis may play some important roles in impaired placental neovascularization. Neurokinin-B is present not only in the maternal sera of PIH but also in those of normal pregnancy, and it is increased as pregnancy progresses (CitationSakamoto et al 2003). Further investigation is necessary to clarify whether and how neurokinin-B/TXA2 suppresses VEGF-mediated signalings and contributes to the pathophysiology of PIH in vivo.
Apart from neurokinin-B, sVEGFR-1 has been noted as an important serum marker of PIH patients (CitationMaynard et al 2003). sVEGFR-1 is an endogenous inhibitor of VEGF and PlGF, and excess sVEGFR-1 causes widespread ECs-dysfunction by interfering with physiological effects of VEGF and PlGF (CitationKendall et al 1993; CitationStepan et al 2006). This phenomenon is observed not only in PIH patients. In cancer patients under antiangiogenic therapies, ie, those who administrated humanized monoclonal antibody bevacizumab specifically targeting VEGF or several receptor tyrosine kinase (RTK) inhibitors targeting VEGF-related pathways, show hypertension, proteinuria and edema (CitationFaivre et al 2006; CitationVeronese et al 2006). The analysis of clinical trials on antiangiogenic therapies has shown that these inhibitors disturb more or less physiological angiogenesis, hematopoiesis, platelet function, and so on (CitationVerheul and Pinedo 2007). In PIH patients, excess sVEGFR-1 seems to be released from the placenta into maternal circulation (CitationCudmore et al 2007).
In addition to sVEGFR-1, several studies confirm that sera of preeclampsia patients show increased level of sCD105, especially in severe cases named HELLP syndrome (Hemolysis, Elevated Liver enzyme, Low Platelets syndrome) (CitationVenkatesha et al 2006). CD105 is a cell surface co-receptor for transforming growth factor-1β (TGF-1β) and TGF-3β, and is expressed in ECs and syncytiotrophoblasts (CitationDuff et al 2003; CitationVenkatesha et al 2006). In cardiovascular system, CD105 is thought to regulate the expression of endothelial nitric oxide synthase (eNOS) and eNOS-dependent vascular tone (CitationJerkic et al 2004; CitationSantibanez et al 2007). In preeclamptic condition, sCD105 probably inhibits TGF-1β signalings of vasculature (CitationVenkatesha et al 2006).
Statistical studies suggest that antiangiogenic milieu in maternal circulation seems to be arranged 2–3 months before preeclamptic symptoms become overt. Increased sVEGFR-1 and sCD105, and decreased PlGF and VEGF are detectable in the middle of second trimester (17–20 weeks of gestation). (CitationLevine et al 2006). Some studies demonstrated that sVEGFR-1, as well as VEGF and PlGF, was produced in isolated trophoblasts from the placenta in vitro, and the level of sVEGFR-1 in culture medium of trophoblasts from preeclamptic patients was higher than that from normal pregnant women (CitationAhmad and Ahmed 2004). It is suggested that VEGF/PlGF axis in villous trophoblasts that face maternal blood flow is responsible for the pathophysiology of PIH. Under other pathophysiological conditions such as diabetic retinopathy and cancer, hypoxia generally stimulates angiogenic signalings, eg, hypoxia-inducible factor (HIF)-1α-mediated transcriptional cascade of proangiogenic molecules including VEGF (CitationSemenza 2003). Currently, it remains poorly understood why hypoxic placenta produces the molecules that suppress angiogenesis in preeclampsia. It is also a subject for future study whether sVEGFR-1 and sCD105 are secreted directly in response to hypoxia or whether some other important regulators, including mechanical stress and vasoactive G protein-coupled receptor (GPCR) cascades, also accelerate the secretion of these anti-angiogenic molecules from the placenta. Recent studies have elucidated the implication of angiotensin II-mediated signalings in excessive production of sVEGFR-1 from the placenta in preeclampsia, which will be discussed later.
Implication of renin-angiotensin system (RAS) in PIH
Renin-angiotensin system (RAS) is one of mastermind signalings that control blood pressure, and this signaling cascade participates not only in vasoconstriction but also in a wide variety of homeostatic activities (CitationLavoie and Sigmund 2003; CitationPaul et al 2006). The expression of renin mRNA was detected in human decidua, basal placenta, macrophages, chorioamniotic membrane and vascular smooth muscle cells (VSMCs) (CitationLentz et al 1989; CitationJikihara et al 1995; CitationKalenga et al 1996; CitationMorgan et al 1998). Angiotensin II receptor type 1 (AT1) was shown to be localized both in villous and extravillous trophoblasts and this AT1 responds to exogenously administrated angiotensin II (CitationLi et al 1998; CitationCooper et al 1999; CitationZhou et al 2007). Functionally, local RAS is thought to participate in the regulation of uteroplacental blood flow, prostaglandin synthesis, estradiol secretion, and so on (CitationKalenga et al 1995; CitationLi et al 1998; CitationNielsen et al 2000).
The circulating level of angiotensin II increases as the pregnancy progresses (CitationZheng et al 2005). It was revealed that in the circulation of PIH mothers RAS was not increased but rather decreased (CitationHanssens et al 1991). Thus RAS has once been considered to be unrelated to the pathophysiology of PIH (CitationHanssens et al 1991; CitationKalenga et al 1996). On the other hand, it is accepted as a classical knowledge that vascular sensitivities to angiotensin II are elevated in preeclamptic women (CitationGant et al 1973). The mechanism of elevated angiotensin II sensitivity in PIH remained unanswered for a few decades.
CitationAbdAlla and colleagues (2001) investigated the presence of AT1-bradykinin B2 heterodimers in the platelets and omental vessels of pregnant women. They demonstrated that bradykinin B2 protein level increased in preeclamptic patients, and that AT1-bradykinin B2 heterodimers accelerated GPCR signal transduction. The results strongly suggest that angiotensin II cascades are enhanced in preeclamptic patients in part by an increase of bradykinin B2 protein which forms heterodimer with AT1 in vivo. The heterodimer formation seems to be present not only in maternal vasculature but also in the placental constituents, because immunohistochemical stainings of bradykinin B2 were reported to be enhanced in the extravillous trophoblasts of preeclampsia cases (CitationCorthorn et al 2006). It is likely that AT1-mediated signalings are augmented by bradykinin B2 at uteroplacental interface which may aggravate fetoplacental microcirculation.
There is an alternative pathway that potentially enhances AT1-mediated vasoconstriction in preeclampsia. Agonistic autoimmune antibody against AT1 (AT1-AA) was detected in the sera of preeclamptic women (CitationWallukat et al 1999). Although later studies have revealed that AT1-AA is also detectable in normotensive pregnant women with IUGR and those without significant complications (CitationWalther et al 2005), the finding provides us with a new insight into preeclampsia in the context of autoimmune disease. In a recent study, AT1 gene was shown to be elevated in the decidua of preeclamptic mothers and AT1-AA was increased in the fetal sera of preeclampsia cases (CitationHerse et al 2007). These results suggest that AT1 cascades in uteroplacental junction are aberrantly activated and that this antibody crosses the placenta and disturb fetal cardiovascular system in preeclampsia. A very recent study by CitationZhou and colleagues (2008) demonstrated that AT1-AA from preeclamptic women accelerated sVEGFR-1 secretion via AT1 expressed in pregnant mice in vivo, and also in human placental villous explants and immortalized human trophoblast cell line in vitro. This study shed a light on the long-standing question; whether and how RAS is implicated in the pathophysiology of PIH. They found that enhanced AT1 cascades disturbed VEGF/PlGF axis thorough calcineurin and nuclear factor of activated T-cells (NFAT) (CitationZhou et al 2008). The mechanism of vascular dysfunction in PIH which involve sVEGFR-1 and AT1 cascades awaits further investigation with special attention, ie, angiogenic crosstalk between GPCR and RTK (CitationHobson et al 2001; CitationWaters et al 2006; CitationFuruya and Yonemitsu 2008). Impaired neovascularization under excess AT1 signalings has been demonstrated in the experiments using PIH model mouse, which will be discussed later.
Preeclamptic mouse based on excessive RAS
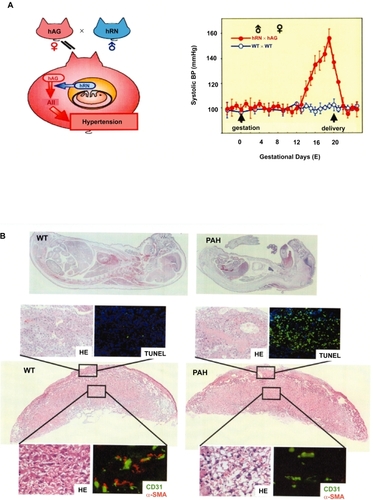
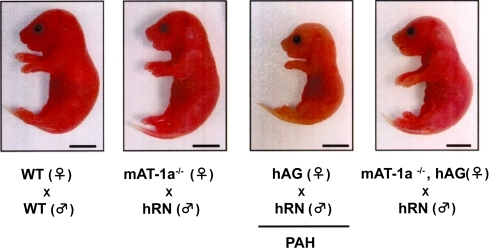
CitationTakimoto and colleagues (1996) generated mice by mating females expressing human angiotensinogen (hAG) with males expressing human renin (hRN), and named them Pregnancy-Associated Hypertension mice (PAH mice) (). In PAH mice, maternal blood pressure starts elevating from 13 days of gestation (E13) until delivery (E19–20) (). Systolic blood pressure at E19 in PAH mother reaches 160 mmHg whereas that in normal pregnant mouse remains around 100 mmHg (CitationTakimoto et al 1996). Blood pressure returns to normal level by 3 days after delivery. This elevation is attributable to the generation of excessive angiotensin I, ie, the precursor of angiotensin II, in the maternal circulation through hRN secretion from fetal side (CitationTakimoto et al 1996). In addition to hypertension, PAH mother shows proteinuria, cardiac hypertrophy and often convulsions. The fetus at term in PAH pregnancy shows severe IUGR, and the mean body weight of PAH fetus at E19 is about 65% of that of wild type (WT) fetus (CitationTakimoto et al 1996; CitationSaito et al 2004; CitationTakimoto-Onishi et al 2005) (). Both biological and physiological data demonstrated that RAS-mediated maternal hypertension beginning in the second half of gestation led PAH mother to the pathological condition that satisfied the criteria of PIH. Similar model was generated in rats (CitationBohlender et al 2000). The hAG-transgenic female rat mated with hRN-transgenic male rat developed hypertension abruptly 10 days before delivery, and sustained this status (approximately 160 mmHg) until delivery. The course of hypertension in this model rat was different from that in PAH mouse; PAH mother showed linear elevation of blood pressure from E13 until delivery.
Figure 4 Schematic representation of PAH (A) Left; Mating females expressing hANG with males expressing hRN results in maternal hypertension. Right; Hypertension starts from E13 until delivery (E20) (CitationTakimoto et al 1996). Pathologic course of PAH pregnancy (B)Top; Whole mounts of WT fetus (left) and PAH fetus (right) at E19. Below; Placental pathology at E19. Maternal site shows diffuse fibrin deposition and apoptosis (TUNEL). In the labyrinth, the majority of ECs (CD31 in green) are poorly covered by pericytes (α-SMA in red). (CitationFuruya et al 2008).
We observed that uNK cells infiltrated the spiral arteries sufficiently and that trophoblasts replaced spiral arteries in the uteroplacental site of PAH placenta earlier than E13 (CitationFuruya et al 2008). The results suggest that epithelial-endothelial transformation is probably taken place properly in PAH as well as in normal one. Interestingly, in a recent study using the placenta of preeclamptic rat as mentioned above, endovascular trophoblast invasion in hypertensive transgenic rat was also evident, and it was shown that the invasion was rather deeper than that in normal rat (CitationGeusens et al 2008). This finding in rat model might reflect the variability of species-specific regulation of local RAS. The precise mechanism of enhanced trophoblast invasion needs further investigation.
In spite of sufficient invasion at an early stage, maternal circulatory sVEGFR-1 in PAH mouse is significantly increased at E19. The finding was consistent with that of another group, in which infusion of angiotensin II significantly increased circulating levels of sVEGFR-1 in pregnant mice (CitationZhou et al 2007). CitationZhou and colleagues (2008) have further shown that the inhibition of AT1 signalings by administration of losartan or FK506 resulted in reduced sVEGFR-1. Taken together, it is highly suggested that maternal sVEGFR-1 can be elevated not only by poor placentation but also by AT1 activation in which angiotensin II, AT1-AA, and AT1-bradykinin B2 heterodimers are potentially implicated.
AT1 plays critical roles in the development of IUGR
Increased angiotensin II is generally not the case in human pre-eclampsia. Therefore, in the context of “pathogenesis”, PAH mice may not be relevant to human PIH. On the other hand, as mentioned above, it is now widely recognized the aberrantly activated AT1 signalings in PIH patients. To clarify the significance of AT1 signalings in PAH, Saito et al investigated the effects of AT1 blockade in this model (CitationSaito et al 2004). They generated hAG+/+ mother that lacked AT1a (hAG+/+/mAT1a−/−). In hAG+/+/mAT1a−/−female, hypertension did not occur when mated with hRN+/+ male. Furthermore, fetal condition was significantly improved (). Administration of AT1-antagonists to hAG+/+ mother also improved both maternal and fetal conditions, supporting the notion that AT1-mediated signalings play critical roles in PAH model (CitationSaito et al 2004). The study clearly proved that AT1-mediated signalings in maternal circulation contributed to the pathophysiology of fetal IUGR.
Figure 5 The effect of mAT1a blockade in PAH model. PAH fetus is shown to be significantly small in size (center right). In the fetus from hANG+/+mAT-1a−/− female mated with hRN+/+ male, the growth is markedly improved (right) (CitationSaito et al 2004).
Although AT1 and VEGFRs play pivotal roles in the pathophysiology of IUGR, it should be noted that vessel receptors are activated not only by biochemical substances. Signalings in vascular cells are also potentially controlled by shear stress and hemodynamic load (Li et al 1999; CitationKalluri 2003). Mechanical force is known to act on several sensors including platelet-derived growth factor receptor (PDGFR)-β, integrins and ion channels in the vascular cells, and to modulate cellular morphology (CitationLi and Xu 2000). Thus, it is suggested that in PIH patients, once blood pressure starts elevating, shear stress per se accelerates unfavorable signalings in maternal circulation which contributes to placental dysfunction and IUGR development.
The process of impaired neovascularization after the onset of hypertension
Pathological examination of human PIH placenta reveals diffuse fibrin deposition and acute atherosis in uteroplacental sites, ie, mural fibrinoid necrosis of spiral arteries (, left). With regard to fetoplacental circulatory unit, terminal villi are poorly differentiated, so called distal villous hypoplasia (, center). Syncytial knots, ie, aggregation of syncytiotrophoblastic nuclei, are also noted (, right). It is generally thought that such changes in PIH reflect placental hypoxia due to shallow invasion of extravillous trophoblasts at initial stage. However, it has not been answered whether acute atherosis and impaired neovascularization also occur by other predisposing factors, eg, accelerated AT1 signaling.
As mentioned above, PAH placenta shows normal-looking pseudovasculogenesis at initial stage; ie, sufficient uNK cells infiltration and appropriate trophoblasts invasion (CitationFuruya et al 2008). It may not be surprising because at this stage the level of hRN produced from the placenta is not enough to act on hAG of maternal circulation (CitationTakimoto et al 1996). We investigated placental pathology, and found significant atherosis with apoptotic change in uteroplacental site of PAH at E19. Time course analysis showed impaired fetoplacental vascular development and maturation after the onset of hypertension. Microvessel densities were significantly low, and fetal-derived ECs were lacking for appropriate pericytes coverage and basement membrane support () (CitationFuruya et al 2008). These findings indicate that placental neovascularization is potentially suppressed under maternal hypertension without the history of poor placentation. In the analysis of vasoactive molecules, some vasoactive genes were down-regulated within 24 hrs after the onset of hypertension. Then the molecules that mediate ECs-pericytes interaction were hampered at the middle stage of hypertension (CitationFuruya et al 2008). These results suggest that the fate of pathological neovascularization might be designated by early responsive genes and that following disturbance of the signalings for vessel maturation might result in abnormal fetoplacental vasculature in the terminal stage. Further studies are necessary to clarify whether the series of angiogenic disturbances are taken place in time-dependent manner one after another, or the dysfunction of each molecule is determined by the level of shear stress and/or AT1 signaling strength.
Conclusions and future prospects
As described in this review, AT1-mediated signalings are aberrantly activated in many, if not all, PIH cases. It is necessary to understand the roles of AT1 in pregnancy complications in a wide range of view. Various GPCRs including AT1 are reported to transactivate epidermal growth factor receptor (EGFR) in vitro (CitationPrenzel et al 1999; CitationOlivares-Reyes et al 2005), thus it might not be surprising that AT1 and other GPCRs potentially cross-talk with VEGF/PlGF-mediated RTK signalings and contribute to sVEGFR-1 production (CitationFuruya et al 2005; CitationMilstien and Spiegel 2006; CitationDorsam and Gutkind 2007; CitationZhou et al 2008). Some GPCRs such as neurokinin-B and endothelin-1 are likely to play critical roles in the pathophysiology of PIH (CitationPage et al 2000; CitationAjne et al 2003), thus they may also be involved in RTK-mediated neovascularization in the placenta.
Currently, the pathophysiology of PIH should also be investigated from artificial immunogenic milieu that had been unexpected in old era. Because of improved therapeutic approaches to infertility, backgrounds of PIH and IUGR have become more and more complicated. Obstetricians may face PIH cases in which the third genetic factor is involved, ie, gestational surrogacy and oocyte donation pregnancy. It is reported that the risk of PIH is increased up to 30% in oocyte donation pregnancy (CitationSalha et al 1999). Although maternal conditions vary among cases, it is very likely that the frequency of PIH in the women after infertility treatment will be increased.
For better understanding and management of PIH, it is necessary to investigate not only human cases but also rodent PIH models in which the mechanism of placental dysfunction and IUGR under maternal hypertension can be monitored in detail. It is also desirable to analyze therapeutic effects of targeting molecules involved in various angiogenic pathways of the placenta. If the mechanism of placental dysfunction in PIH and other IUGR-associated complications is fully elucidated, it will certainly provide more precise disease-specific strategies, and contribute to more effective and safer therapies in future.
Acknowledgements
The protocol to obtain tissue samples was approved by the review boards of Sapporo General Hospital and Chiba University Hospital. The authors thank to Drs T Takenouchi, H Hareyama, H Usui, M Shozu, and Y Nakatani for providing human samples, and the members of Fukamizu laboratory and Dr Y Yonemitsu for discussion. This work is supported by Grant-in-Aid for Scientific Research (S) (AF), Grant-in-Aid for Exploratory Research (AF), and Grant-in-Aid for Scientific Research (C) (MF) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- AbdAllaSLotherHel MassieryA2001Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsivenessNat Med71003911533702
- AdamsonSLLuYWhiteleyKJ2002Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placentaDev Biol2503587312376109
- AhmadSAhmedA2004Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsiaCirc Res958849115472115
- AjneGWolffKFyhrquistF2003Endothelin converting enzyme (ECE) activity in normal pregnancy and preeclampsiaHypertens Pregnancy222152414572358
- BohlenderJGantenDLuftFC2000Rats transgenic for human renin and human angiotensinogen as a model for gestational hypertensionJ Am Soc Nephrol1120566111053481
- ChenGWilsonRCummingG1993Production of prostacyclin and thromboxane A2 in mononuclear cells from preeclamptic womenAm J Obstet Gynecol1691106118238168
- CooperACRobinsonGVinsonGP1999The localization and expression of the renin-angiotensin system in the human placenta throughout pregnancyPlacenta204677410419812
- CorthornJGermainAAChacónC2006Expression of kallikrein, bradykinin b2 receptor, and endothelial nitric oxide synthase in placenta in normal gestation, preeclampsia, and placenta accretaEndocrine29491916943589
- CudmoreMAhmadSAl-AniB2007Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1Circulation11517899717389265
- CunningFGMacDonaldPCGantNF1989The morphological and functional development of the fetusCunningFGMacDonaldPCGantNF18th edWilliams Obstetrics. Prentice-Hall International Inc87128
- DamskyCHFisherSJ1998Trophoblast pseudo-vasculogenesis: faking it with endothelial adhesion receptorsCurr Opin Cell Biol1066069818178
- DorsamRTGutkindJS2007G-protein-coupled receptors and cancerNat Rev Cancer7799417251915
- DuffSELiCGarlandJM2003CD105 is important for angiogenesis: evidence and potential applicationsFASEB J179849212773481
- DuleyLHenderson-SmartDKnightM2001Antiplatelet drugs for prevention of pre-eclampsia and its consequences: systematic reviewBMJ3223293311159655
- FaivreSDelbaldoCVeraK2006Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancerJ Clin Oncol24253516314617
- FuruyaMNishiyamaMKasuyaY2005Pathophysiology of tumor neovascularizationVasc Health Risk Manag12779017315600
- FuruyaMIshidaJInabaS2008Impaired placental neovascularization in mice with pregnancy-associated hypertensionLab Invest884162918268474
- FuruyaMYonemitsuY2008Cancer neovascularization and proinflammatory microenvironmentsCurr Cancer Drug Targets82536518537549
- GantNFDaleyGLChandS1973A study of angiotensin II pressor response throughout primigravid pregnancyJ Clin Invest52268294355997
- GeorgiadesPFerguson-SmithACBurtonGJ2002Comparative developmental anatomy of the murine and human definitive placentaePlacenta2331911869088
- GeusensNVerlohrenSLuytenC2008Endovascular trophoblast invasion, spiral artery remodelling and uteroplacental haemodynamics in a transgenic rat model of pre-eclampsiaPlacenta296142318502502
- GrangerJPAlexanderBTBennettWA2001Pathophysiology of pregnancy-induced hypertensionAm J Hypertens14178S85S11411754
- HanssensMKeirseMJSpitzB1991Angiotensin II levels in hypertensive and normotensive pregnanciesBr J Obstet Gynecol9815561
- HecherKCampbellSDoyleP1995Assessment of fetal compromise by Doppler ultrasound investigation of the fetal circulation. Arterial, intracardiac, and venous blood flow velocity studiesCirculation91129387805194
- HendrixMJSeftorEAHessAR2003Vasculogenic mimicry and tumour-cell plasticity: lessons from melanomaNat Rev Cancer34112112778131
- HerseFDechendRHarsemNK2007Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsiaHypertension496041117261642
- HibySEWalkerJJO’shaughnessyKM2004Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive successJ Exp Med2009576515477349
- HobsonJPRosenfeldtHMBarakLS2001Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motilityScience2911800311230698
- HuYDutzJPMacCalmanCD2006Decidual NK cells alter in vitro first trimester extravillous cytotrophoblast migration: a role for IFN-gammaJ Immunol17785223017142750
- JerkicMRivas-ElenaJVPrietoM2004Endoglin regulates nitric oxide-dependent vasodilatationFASEB J186091114734648
- JikiharaHPoisnerAMHirschR1995Human uterine decidual macrophages express reninJ Clin Endocrinol Metab80127377714100
- JohnsonMPFitzpatrickEDyerTD2007Identification of two novel quantitative trait loci for pre-eclampsia susceptibility on chromosomes 5q and 13q using a variance components-based linkage approachMol Hum Reprod1361717085769
- KalengaMKDe GasparoMThomasK1995Angiotensin-II stimulates estradiol secretion from human placental explants through AT1 receptor activationJ Clin Endocrinol Metab80123377714093
- KalengaMKThomasKde GasparoM1996Determination of renin, angiotensin converting enzyme and angiotensin II levels in human placenta, chorion and amnion from women with pregnancy induced hypertensionClin Endocrinol (Oxf)44429338706309
- KalluriR2003Basement membranes: structure, assembly and role in tumour angiogenesisNat Rev Cancer34223312778132
- KarumanchiSABdolahY2004Hypoxia and sFlt-1 in preeclampsia: the “chicken-and-egg” questionEndocrinology1454835715489315
- KendallRLThomasKA1993Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptorProc Natl Acad Sci U S A901070598248162
- KharfiAGiguèreYSapinV2003Trophoblastic remodeling in normal and preeclamptic pregnancies: implication of cytokinesClin Biochem363233112849862
- KingAAllanDSBowenM2000HLA-E is expressed on trophoblast and interacts with CD94/NKG2 receptors on decidual NK cellsEur J Immunol3016233110898498
- KingABurrowsTDHibySE2000Surface expression of HLA-C antigen by human extravillous trophoblastPlacenta213768710833373
- KobashiGHataAShidoK1999Association of a variant of the angiotensinogen gene with pure type of hypertension in pregnancy in the Japanese: implication of a racial difference and significance of an age factorAm J Med Genet86232610482871
- KrausFTSobinLHTockerJT2004Anatomy, structure and functionKrausFTSobinLHTockerJTPlacental PathologyWashington, DCAFIP122
- LenzTSealeyJEAugustP1989Tissue levels of active and total renin, angiotensinogen, human chorionic gonadotropin, estradiol, and progesterone in human placentas from different methods of deliveryJ Clin Endocrinol Metab693172659618
- LevineRJLamCQianC2006Soluble endoglin and other circulating antiangiogenic factors in preeclampsiaN Engl J Med355992100516957146
- LavoieJLSigmundCD2003Minireview: overview of the renin-angiotensin system – an endocrine and paracrine systemEndocrinology14421798312746271
- LewisSHBenirschkeK1997The placentaSternbergSSHistology for Pathologist2nd edNew YorkLippincott-Raven96194
- LiXShamsMZhuJ1998Cellular localization of AT1 receptor mRNA and protein in normal placenta and its reduced expression in intrauterine growth restriction. Angiotensin II stimulates the release of vasorelaxantsJ Clin Invest101442549435317
- LiCXuQ2000Mechanical stress-initiated signal transductions in vascular smooth muscle cellsCell Signal124354510989277
- LyallFSimpsonHBulmerJN2001Transforming growth factor-beta expression in human placenta and placental bed in third trimester normal pregnancy, preeclampsia, and fetal growth restrictionAm J Pathol15918273811696443
- LyallF2005Priming and remodelling of human placental bed spiral arteries during pregnancy – a reviewPlacenta26Suppl AS31615837064
- ManiotisAJFolbergRHessA1999Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicryAm J Pathol1557395210487832
- MaynardSEMinJYMerchanJ2003Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsiaJ Clin Invest1116495812618519
- MillsJLDerSimonianRRaymondE1999Prostacyclin and thromboxane changes predating clinical onset of preeclampsia: a multicenter prospective studyJAMA2823566210432033
- MilstienSSpiegelS2006Targeting sphingosine-1-phosphate: a novel avenue for cancer therapeuticsCancer Cell91485016530698
- MoffettALokeC2006Immunology of placentation in eutherian mammalsNat Rev Immunol65849416868549
- MoffettAHibySE2007How Does the maternal immune system contribute to the development of pre-eclampsia?Placenta28Suppl AS51617292469
- MorganTCravenCNelsonL1997Angiotensinogen T235 expression is elevated in decidual spiral arteriesJ Clin Invest1001406159294106
- MorganTCravenCWardK1998Human spiral artery renin-angiotensin systemHypertension3268379774363
- MyattL1992Control of vascular resistance in the human placentaPlacenta12329411438081
- NielsenAHSchauserKHPoulsenK2000Current topic: the uteroplacental renin-angiotensin systemPlacenta214687710940196
- [NHBPEP] National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy2000Report of the National High Blood Pressure Education Program Working Group on High Blood Pressure in PregnancyAm J Osbtet Gynecol183S1S22
- OhkuchiAMinakamiHSatoI2000Predicting the risk of pre-eclampsia and a small-for-gestational-age infant by quantitative assessment of the diastolic notch in uterine artery flow velocity waveforms in unselected womenUltrasound Osbtet Gynecol161718
- Olivares-ReyesJAShahBHHernández-ArandaJ2005Agonist-induced interactions between angiotensin AT1 and epidermal growth factor receptorsMol Pharmacol683566415905421
- RobertsJMGammillHS2005Preeclampsia: recent insightsHypertension461243916230510
- PageNMWoodsRJGardinerSM2000Excessive placental secretion of neurokinin B during the third trimester causes pre-eclampsiaNature40579780010866201
- PalSWuJMurrayJK2006An antiangiogenic neurokinin-B/throm-boxane A2 regulatory axisJ Cell Biol1710475817000881
- ParrettiEMealliFMagriniA2003Cross-sectional and longitudinal evaluation of uterine artery Doppler velocimetry for the prediction of pre-eclampsia in normotensive women with specific risk factorsUltrasound Osbtet Gynecol221605
- PaulMPoyan MehrA2006Physiol Rev8674780316816138
- Perrier d’HauteriveSBerndtSTsampalasM2007Dialogue between blastocyst hCG and endometrial LH/hCG receptor: which role in implantation?Gynecol Osbtet Invest6415660
- PijnenborgRVercruysseLHanssensM2006The uterine spiral arteries in human pregnancy: facts and controversiesPlacenta279395816490251
- PrenzelNZwickEDaubH1999EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGFNature402884810622253
- RedmanCWSargentIL2003Pre-eclampsia, the placenta and the maternal systemic inflammatory response – a reviewPlacentaSuppl AS21712842410
- RedmanCWSargentIL2005Latest advances in understanding preeclampsiaScience3081592415947178
- ReynoldsLPRedmerDA2001Angiogenesis in the placentaBiol Reprod6410334011259247
- ReynoldsLPBorowiczPPVonnahmeKA2005Animal models of placental angiogenesisPlacenta2668970816226119
- RossantJCrossJC2001Placental development: lessons from mouse mutantsNat Rev Genet25384811433360
- SaitoTIshidaJTakimoto-OhnishiE2004An essential role for angiotensin II type 1a receptor in pregnancy-associated hypertension with intrauterine growth retardationFASEB J183889014688210
- SakamotoROsadaHIitsukaY2003Profile of neurokinin B concentrations in maternal and cord blood in normal pregnancyClin Endocrinol58597600
- SalhaOSharmaVDadaT1999The influence of donated gametes on the incidence of hypertensive disorders of pregnancyHum Reprod1422687310469693
- SantibanezJFLetamendiaAPerez-BarriocanalF2007Endoglin increases eNOS expression by modulating Smad2 protein levels and Smad2-dependent TGF-beta signalingJ Cell Physiol2104566817058229
- SchlembachDScaleraFFischerT2003Neurokinin B peptide serum levels are higher in normotensive pregnant women than in preeclamptic pregnant womenAm J Osbtet Gynecol189141822
- SeftorEAMeltzerPSSchattemanGC2002Expression of multiple molecular phenotypes by aggressive melanoma tumor cells: role in vasculogenic mimicryCrit Rev Oncol Hematol44172712398997
- SemenzaGL2003Targeting HIF-1 for cancer therapyNat Rev Cancer37213213130303
- SharmaNSeftorRESeftorEA2002Prostatic tumor cell plasticity involves cooperative interactions of distinct phenotypic subpopulations: role in vasculogenic mimicryProstate5018920111813211
- ShirakawaKTsudaHHeikeY2001Absence of endothelial cells, central necrosis, and fibrosis are associated with aggressive inflammatory breast cancerCancer Res614455111212228
- SoodAKSeftorEAFletcherMS2001Molecular determinants of ovarian cancer plasticityAm J Pathol15812798811290546
- StepanHFaberRDornhöferN2006New insights into the biology of preeclampsiaBiol Reprod74772616421233
- TakimotoEIshidaJSugiyamaF1996Hypertension induced in pregnant mice by placental renin and maternal angiotensinogenScience27499588875944
- Takimoto-OhnishiESaitoTIshidaJ2005Differential roles of renin and angiotensinogen in the feto-maternal interface to the development of complications of pregnancyMol. Endocrinol1913617215695374
- VaccaPPietraGFalcoM2006Analysis of natural killer cells isolated from human decidua: Evidence that 2B4 (CD244) functions as an inhibitory receptor and blocks NK-cell functionBlood10840788516931625
- van DijkMMuldersJPoutsmaA2005Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene familyNat Genet37514915806103
- VenkateshaSToporsianMLamC2006Soluble endoglin contributes to the pathogenesis of preeclampsiaNat Med12642916751767
- VerheulHMPinedoHM2007Possible molecular mechanisms involved in the toxicity of angiogenesis inhibitionNat Rev Cancer74758517522716
- VeroneseMLMosenkisAFlahertyKT2006Mechanisms of hypertension associated with BAY 43–9006J Clin Oncol241363916446323
- WallukatGHomuthVFischerT1999Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptorJ Clin Invest1039455210194466
- WaltherTWallukatGJankA2005Angiotensin II type 1 receptor agonistic antibodies reflect fundamental alterations in the uteroplacental vasculatureHypertension461275916260641
- WatersCMLongJGorshkovaI2006Cell migration activated by platelet-derived growth factor receptor is blocked by an inverse agonist of the sphingosine 1-phosphate receptor-1FASEB J205091116319133
- ZhengJBirdIMChenDB2005Angiotensin II regulation of ovine fetoplacental artery endothelial functions: interactions with nitric oxideJ Physiol565596915790666
- ZhouCCAhmadSMiT2007Angiotensin II induces soluble fms-Like tyrosine kinase-1 release via calcineurin signaling pathway in pregnancyCirc Res100889517158338
- ZhouCCAhmadSMiT2008Autoantibody from women with preeclampsia induces soluble Fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of activated T-cells signalingHypertension511010918259044
- ZhouYFisherSJJanatpourM1997Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion?J Clin Invest992139519151786
- ZhouYMcMasterMWooK2002Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndromeAm J Pathol16014052311943725
- ZygmuntMHerrFMünstedtK2003Angiogenesis and vasculogenesis in pregnancyEur J Obstet Gynecol Reprod Biol110 Suppl1S101812965086