Abstract
Since the pioneering work of John Gofman in the 1950s, our understanding of high density lipoprotein cholesterol (HDL-C) and its relationship to coronary heart disease (CHD) has grown substantially. Numerous clinical trials since the Framingham Study in 1977 have demonstrated an inverse relationship between HDL-C and one’s risk of developing CHD. Over the past two decades, preclinical research has gained further insight into the nature of HDL-C metabolism, specifically regarding the ability of HDL-C to promote reverse cholesterol transport (RCT). Recent attempts to harness HDL’s ability to enhance RCT have revealed the complexity of HDL-C metabolism. This review provides a detailed update on HDL-C as an evolving therapeutic target in the management of cardiovascular disease.
Introduction
In the early 1900s, a German chemist named Adolph Windaus determined atheromatous plaque from human aortas contained 20-fold higher concentrations of cholesterol than normal aortas. Soon after, the Russian pathologist Nikolai Anitschov strengthened this observation by feeding rabbits a high-cholesterol diet and creating the first animal model of atherosclerosis (CitationKhavkin et al 1975; CitationVance et al 2000). In 1955, a biophysicist named John Gofman used ultracentrifugation to separate plasma lipoproteins by density and correlated risk of myocardial infarction (MI) with elevated low-density lipoprotein cholesterol (LDL-C) levels. Henceforth, the ‘lipid hypothesis’ proposed that elevated LDL-C, elevated triacylglycerols (TG), and low levels of high density lipoprotein cholesterol (HDL-C) were causally associated with an increased risk of coronary heart disease (CHD) (CitationSoloff 1998).
In 1977, the Framingham Study showed that elevated LDL-C and reduced HDL-C levels independently predict one’s risk for developing cardiovascular disease (CitationGordon et al 1977). Since the first Adult Treatment Panel (ATP) recommendations in 1988 [CitationExpert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II) 1993], guidelines have increasingly focused on aggressive management of elevated LDL-C in populations at risk for CHD. According to the National Cholesterol Education Panel (NCEP) ATP III guidelines published in 2001, patients with established CHD, non-coronary atherosclerosis, diabetes mellitus, or greater than two major cardiac risk factors with a calculated Framingham risk score of greater than 20% should have an LDL-C goal of less than 100 mg/dL [CitationExpert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) 2001]. Subsequently, the Heart Protection Study (HPS) (CitationMRC/BHF Heart Protection Study 1999) and Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) (CitationCannon et al 2004) trial reported incremental 22% and 16% reductions, respectively, in the risk of cardiovascular events with LDL-C levels lowered below 100 mg/dL. Based on these data, an “optional” target of therapy focusing on aggressive LDL-C lowering less than 70 mg/dL for patients with known coronary disease or CHD equivalent status has been proposed (CitationGrundy et al 2004).
However, despite lowering LDL-C to levels recommended by the NCEP in 90% of treated patients, an overall reduction of only 20%–35% in the rate of cardiovascular events has been observed in randomized trials (CitationShah et al 2002). Moreover, nearly 15% of patients with acute myocardial infarction (MI) have LDL-C levels less than 100 mg/dL at presentation (CitationForrester et al 2005), suggesting that previously cited LDL-C targets remain too high or the benefit of very aggressive LDL-C lowering is quite limited. The Treating to New Targets (TNT) trial (CitationLaRosa et al 2005) showed a 2.2% absolute and a 22% relative risk reduction for major cardiovascular events in patients receiving high dose compared to low dose atorvastatin. The mean LDL-C levels were 77 mg/dL in the high dose statin group versus 100 mg/dL in the low dose statin group. Thus, while great emphasis has been placed on the importance of LDL-C lowering in CVD risk reduction, there is growing interest directed at raising HDL-C levels for further risk reduction.
At present, no approved therapies increase HDL-C levels by any comparable magnitude to therapies designed to lower LDL-C levels. This review provides a detailed update on HDL-C as a therapeutic target for CVD risk reduction.
HDL-C and coronary heart disease
Substantial epidemiologic evidence suggests a negative linear correlation between HDL-C levels and the incidence of CHD. First proposed by CitationBarr and colleagues in 1951 (CitationBarr et al 1951), an inverse relationship between HDL-C and cardiovascular disease was not well established until the Framingham study in the 1970s (CitationGordon et al 1997). This analysis of 2815 men and women aged 49–82 years identified HDL-C as a powerful risk factor inversely associated with the incidence of CHD. Similarly, in the early 1980s, the Prospective Cardiovascular Munster (PROCAM) (CitationKannel 1983) study evaluated 4559 male participants aged 40–64 years and found a strong negative linear correlation between the incidence of CAD and HDL-C levels (CHD risk ratio of 4.0 for HDL-C <25 mg/dL versus 1.0 for HDL-C >65 mg/dL, p < 0.001).
Data from four studies (Framingham Heart Study, the Lipid Research Clinic Prevalence Mortality Follow-up Study, Lipid Research Clinic Primary Prevention Trial, and Multiple Risk Factor Intervention Trial) estimate a 2% reduction in cardiovascular risk for every 1 mg/dL increase in serum HDL-C (Gordon et al 1989; CitationCastelli et al 1992; CitationMultiple Risk Factor Intervention Trial 1982). Individuals with low HDL-C (<40 mg/dL in men and <50 mg/dL in women) are at increased risk of CVD (CitationMiller et al 1977), restenosis following coronary balloon angioplasty (CitationShah and Amin 1992) and cardiovascular death (CitationWilson et al 1988).
One of the first trials demonstrating the benefit of raising HDL-C and lowering TG levels in individuals with low baseline HDL-C levels was the Helsinki Heart Study (HHS) (CitationManninen et al 1992), in which 4081 men with dyslipidemia were randomized to receive gemfibrozil (600 mg twice daily) or placebo. At 5 years of follow-up, gemfibrozil therapy increased HDL-C levels by 11% and reduced total cholesterol (TC), LDL-C, and TG levels by 10%, 11%, and 35% respectively. Gemfibrozil therapy reduced the primary end point of cardiac death or non-fatal MI by 34% (27.3 vs 41.4/1,000, p < 0.02) with the greatest reduction found in patients with low HDL-C and high TG at baseline (CitationManttari et al 1990).
The Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) (CitationDowns et al 1998) compared treatment with lovastatin versus placebo for the prevention of a first major coronary event in adults with average TC and LDL-C levels, but low baseline levels of HDL-C. Lovastatin decreased both TC and LDL-C levels by 18% and 25%, respectively, while increasing HDL-C levels by 6%. After more than 5 years of follow-up, the absolute risk in the primary composite end point of fatal or non-fatal MI, unstable angina, or sudden cardiac death was reduced in absolute terms by 2.2% in men and 1.2% in women with a relative risk reduction of 37%. This study was the first primary prevention study to show that individuals with HDL-C <40 mg/dL received the greatest benefit, suggesting the lower cutpoint HDL-C of 35 mg/dL in ATP II should be raised to 40 mg/dL in ATP III. These clinical trials confirm an increased risk associated with low serum levels of HDL-C and the beneficial effects of pharmacotherapy in adults with low HDL-C in primary prevention.
Patients with manifest CHD also benefit from raising HDL-C and lowering TG. The Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial (VA-HIT) compared treatment with gemfibrozil versus placebo in more than 2500 men with established CHD, average LDL-C levels (<140 mg/dL), and low HDL-C levels (<40 mg/dL). After a mean follow-up of 5 years, gemfibrozil decreased TG levels by 31% and increased HDL-C levels by 6%, while levels of LDL-C remained quantitatively unchanged; there was a relative risk reduction of 22% (17.3% vs 21.7% p < 0.006) in CHD death and non-fatal MI in the treatment group. Gemfibrozil therapy was associated with a 24% relative risk reduction in the composite end point of nonfatal MI, stroke, and CHD death (p < 0.001) (CitationRubins et al 2001).
The Scandanavian Simvastatin Survival Study Group (4S) was a large, randomized, placebo-controlled trial evaluating simvastatin (20–40 mg/day) in 4444 men and women aged 35–70 years over a median follow-up period of 5.4 years. Simvastatin therapy decrease TC and LDL-C (25% and 35%, respectively) and increased HDL-C by 8% compared to placebo. Simvastatin treatment resulted in a 30% relative risk reduction in overall mortality (8.2% vs 11.5%, p = 0.0003) and reduced non-fatal MI, ischemic heart disease death, and coronary revascularization (CitationScandinavian Simvastatin Survival Study Group 1994).
The Bezafibrate Infarction Prevention Study (BIPS) was a blinded, placebo-controlled trial of bezafibrate therapy in 3122 patients with previous MI or angina pectoris and baseline LDL-C <180 mg/dL, HDL-C <45 mg/dL, and TG <300 mg/dL. At 5–7 years of follow-up, bezafibrate increased HDL-C more than 15% and decreased TG levels by 25%. The overall relative reduction in the primary end point of 9% was not statistically significant; however, a 40% relative risk reduction was observed in patients with baseline TG >200 mg/dL, suggesting fibrates may be beneficial in patients with known CAD and elevated TG levels (CitationKaplinsky 1998).
Based on the epidemiologic data available, the NCEP ATP III guidelines raised the cut-point for low HDL-C levels from 35 mg/dL to 40 mg/dL, thereby identifying a larger number of adults at risk for developing CHD. Current guidelines define “high” HDL-C levels as above 60 mg/dL (CitationGrundy et al 2004). The definition of “optimal” HDL-C will likely undergo further modification as data becomes available.
HDL-C metabolism and reverse cholesterol transport (RCT)
By transporting excess cholesterol from peripheral cells to the liver for excretion in a process known as reverse cholesterol transport (RCT), HDL-C may retard the progression of atherosclerosis. In animal studies, exogenous infusions of HDL-C or apolipoprotein A-1 (Apo AI), the major apolipoprotein associated with HDL-C, prevents atherosclerosis from developing or progressing (CitationBadimon et al 1990; CitationDuverger et al 1996). Similar findings have been reported in animal models of Apo AI gene over-expression (CitationDimayuga et al 1999). Harnessing this unique ability of HDL-C requires further insight into the basic mechanisms of HDL-C metabolism.
HDL-C metabolism involves numerous enzymes and unique nuclear transcription regulatory proteins intimately linked to other lipoproteins. HDL-C is a macromolecule containing lipids and proteins that transport water-insoluble fats in blood. A phospholipid (PL) monolayer containing free cholesterol (FC) and apolipoproteins (Apo) surrounds a non-polar lipid core containing FC and TG. Apo AI and AII are the major protein components of HDL-C. Apo AI is produced by the liver and intestines and constitutes 70% of HDL-C protein content. Apo AII is produced only by the liver and constitutes 20% of HDL-C protein content. While Apo AI is ubiquitously associated with HDL-C, Apo AII is found in about 60% of HDL-C molecules (CitationLewis et al 2005). Apolipoproteins serve as receptor ligands and enzymatic catalysts for all circulating liporoteins.
Clinical observations of premature CHD in patients with inherited disorders of metabolism resulting in low circulating HDL-C levels, such as homozygous Apo AI/CIII deficiency and hypoalphaproteinemia support the association between low serum HDL-C levels and atherosclerosis (CitationForte TM 1984). Conversely, individuals with inherited defects resulting in low serum HDL-C levels have variable or no premature CHD (eg, Tangier disease, Apo AI Milano, and familial lecithin:cholesterol acyltransferase (LCAT) deficiency), highlighting the complexity of HDL-C metabolism (CitationMiller et al 1990; CitationElkhalil et al 1997). Characterized in 1968 by Glomset, RCT can be divided into four phases (CitationGlomset 1968) ().
Figure 1 HDL-C mediated reverse cholesterol transport. Reverse cholesterol transport (RCT) can be divided into four phases. 1) transfer of free cholesterol (FC) to pre-b HDL via ABCA1, 2) esterification of surface-associated FC by the enzyme Lecithin:acyl CoA Transferase (LCAT), 3) transfer of FC and triglycerides (TG) between HDL-C and Apo B-containing lipoproteins mediated by the enzyme cholesteryl ester transfer protein (CETP), and 4) uptake by the scavenger receptor B1 (SR-B1) and catabolism of mature HDL-C into bile or small HDL-C particles by hepatic lipase (HL). Apo B-containing lipoproteins can be acquired by the LDL-receptor (LDLr) for hepatic catabolism.
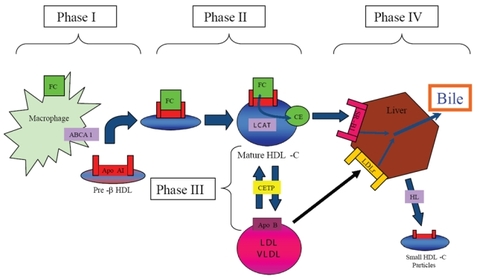
Phase one: nascent HDL-C acquires free cholesterol
The nascent form of circulating HDL-C rich in Apo AI, termed discoidal pre-β HDL, removes FC and PL from peripheral cells throughout the body by interacting with a membrane associated protein ubiquitously expressed in peripheral tissues, known as ATP-binding cassette transporter 1 (ABCA1). Pre-β HDL is rich in Apo AI and serves as a template for the generation of lipid-rich HDL-C (CitationSviridov et al 2002). Pre-β HDL is generated by either de novo secretion from hepatocytes or the intestinal mucosa, direct dissociation from chylomicrons and very low density lipoprotein (VLDL) mediated by lipoprotein lipase (LL), or as a by-product of HDL-C particle interconversion (CitationKwiterovich 1998).
Once generated, pre-β HDL receives PL and FC from peripheral cells by associating with the surface protein ABCA1 (CitationOram and Lawn 2001), which is expressed by the liver and intestinal mucosa. Patients with Tangier Disease, an autosomal recessive disorder characterized by two non-functional ABCA1 alleles and extremely low levels of HDL-C, exemplify the significance of ABCA1 in HDL-C metabolism (CitationBodzioch et al 1999). Heterozygous individuals with a partial reduction in functional ABCA1 have a corresponding 50% decrease in serum HDL-C levels (CitationMarcil et al 1999).
Animal models further support the critical role of ABCA1 in RCT. ABCA1-deficient mice generated by targeted gene ablation in DBA-1J embryonic stem cells demonstrated a 99.5% and 99.8% reduction in serum HDL-C and Apo AI levels, respectively. In this model, loss of ABCA1 correlated with increased accumulation of lipid-laden macrophages, an integral component of atherosclerotic plaque (CitationMcNeish et al 2000). Conversely, overexpression of ABCA1 in transgenic mice is associated with increased TC, HDL-C levels, and Apo AI with enhanced cholesterol efflux and reduced levels of atherogenesis (CitationBrewer et al 2004). Finally, crossing transgenic mice overexpressing ABCA1 with athero-susceptible transgenic mice, such as LDL receptor (LDLr) or Apolipoprotein E knockout (KO) models, reduced atheromatous progression (CitationJoyce et al 2003).
ABCG1 is another member of the ATP-binding cassette family that promotes efflux of PL and FC from macrophages to mature HDL-C rather than pre-β HDL (CitationKennedy et al 2005) Macrophages deficient in ABCG1 also have impaired FC efflux and accumulate excess cholesterol (CitationOut R 2006). Taken together, these data suggest that both ABCA1 and ABCG1 are potential therapeutic targets to raise HDL-C and promote RCT.
Transcription of both ABCA1 and ABCG1 is regulated by members of a steroid superfamily of nuclear receptors known as the Liver X receptor/Retinoid X receptor (LXR/RXR) heterodimer. When activated by oxysterols from FC this heterodimer stimulates ABCA1 and ABCG1 gene expression, thereby enhancing cholesterol efflux (CitationVaughan and Oram 2005; CitationVenkateswaran et al 2000). The heterodimer is also regulated by the activity of peroxisome proliferator-activated receptors (PPAR) α and γ, which are closely linked to insulin resistance and the metabolic syndrome (CitationAnderson et al 2004). PPAR-α and PPAR-γ agonists have been shown to upregulate LXR and ABCA1 expression and promote macrophage cholesterol efflux (CitationSchmitz et al 2002; CitationChawla et al 2001). Synthetic LXR agonists also promote cholesterol efflux in vivo and promote regression of atherosclerosis in mice (CitationBruemmer and Law 2005).
Phase two: lecithin: acyl CoA transferase (LCAT) esterifies free cholesterol
As FC is acquired from peripheral tissues, pre-β HDL matures from an incipient discoidal form into a larger spherical molecule. Newly acquired FC undergoes esterfication to form cholesteryl esters (CE) which migrate to the center of the discoidal pre-beta HDL molecule. The spherical morphology of mature α-HDL-C promotes further HDL-C metabolism and cholesterol efflux (CitationWang and Briggs 2004). Cholesterol esterification prevents transfer of FC back to the periphery, thereby potentiating further RCT.
Mediating this necessary step in HDL-C maturation is the enzyme LCAT, which is synthesized by the liver. Circulating LCAT esterifies lecithin and FC on both HDL-C and Apo B lipoproteins. The phospholipid component of HDL-C appears to mediate binding to LCAT, while the apolipoprotein component activates the enzyme (CitationFurbee et al 2002). Recent evidence suggests that increased glycation of Apo A1 in subjects with diabetes progressively decreases the rate of LCAT-mediated cholesterol esterification (CitationNobecourt et al 2007).
Familial LCAT deficiency (FLD) and fish-eye disease (FED) are distinct inherited syndromes associated with absent or impaired LCAT expression, respectively. Low HDL-C levels and corneal opacification characterize both diseases, while individuals with FLD also suffer from anemia and renal failure. Variable degrees of premature atherosclerosis have been observed in these populations (CitationFunke et al 1991). Conversely, over expression of human LCAT in transgenic animal models correlates with a 7-fold increase in serum HDL-C, increases in Apo AI levels, and a marked reduction in atheromatous plaque burden (CitationFrancone et al 1990). These findings suggest that LCAT over expression may serve as a novel therapeutic target in the prevention and treatment of CHD.
Phase three: cholesterol ester transfer protein (CETP) mediates exchange of cholesterol esters between HDL-C and Apo B lipoproteins
The enzyme cholesteryl ester transfer protein (CETP) exchanges CE from HDL-C for TG in LDL-C and VLDL. Newly acquired CE in VLDL and LDL-C is then taken up by the hepatic LDLr for excretion as bile (CitationMorton and Greene 1997). Found predominantly in the liver, spleen, and adipose tissue, expression of CETP is enhanced by hypercholesterolemic diets via activation of a sterol regulatory protein and through binding of oxysterols to the LXR/RXR receptor (CitationDe Grooth et al 2004).
Heritable CETP deficiencies in the Japanese population are associated with significant increases in large HDL-C particles and smaller LDL-C particles with a lower affinity for LDL-C receptors (CitationInazu et al 1990). Heterozygotes with a 40% decrease in CETP levels had a mean increase in HDL-C of 30% and no significant change in LDL-C levels, while homozygotes with complete loss of CETP levels had a greater than 100% increase in HDL-C and 40% decrease in LDL-C and Apo B levels (CitationInazu et al 1990; CitationKoizumi et al 1991). Despite these findings the correlation between low CETP levels and reduced CHD risk remained elusive as heterozygotes deficient in CETP continue to manifest atherosclerotic coronary disease (CitationHirano et al 1995).
Two population studies have associated high CETP levels with an increased risk of CHD. A nested case control study known as the European Prospective Investigation into Cancer and nutrition (EPIC)-Norfolk cohort study suggested an increased CHD risk in patients with elevated TG and elevated CETP levels (CitationBoekholdt et al 2004). Data from the Regression Growth Evaluation Statin Study (REGRESS) (CitationKlerkx et al 2004) study also associated high CETP levels and rapid progression of established CHD in men. In this study, treatment with pravastatin significantly improved lipid and angiographic parameters in patients with high baseline CETP independent of baseline lipids, suggesting plasma CETP levels may be a marker of response to statin therapy.
The role of CETP in modulating atherogenesis is complicated by dual pro-atherogenic and anti-atherogenic effects of transferring CE and TG between lipoproteins (). Potential proatherogenic properties of CETP activity include: 1) TG-laden HDL-C particles may undergo enhanced renal excretion thereby reducing total circulating HDL-C levels and RCT, 2) CE-laden VLDL and LDL-C particles may be acquired by peripheral macrophages and promote atherosclerosis, and 3) CETP also transfers TG from VLDL to LDL-C thereby generating small atherogenic LDL particles (CitationHirano et al 2000; CitationBarter et al 2003).
Figure 2 Dual nature of CETP activity. By shuttling cholesteryl esters (CE) and triglycerides (TG) between HDL-C and Apo B-associated lipoproteins, the enzyme cholesteryl ester transfer protein (CETP) creates substrate for both pro-atherogenic and anti-atherogenic pathways.
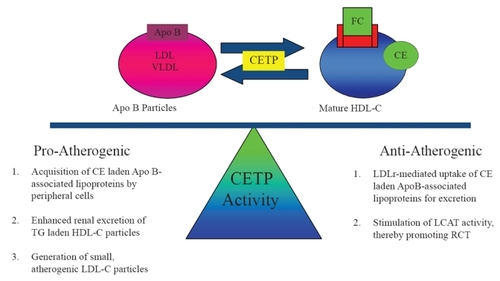
However, complete abolition of CETP activity results in large, cholesterol-laden, dysfunctional HDL-C with reduced cholesterol efflux RCT capacity (CitationYamashita et al 1988; CitationSakai et al 1991; CitationIkewaki et al 1995). Furthermore, CETP activity may be anti-atherogenic if CE-laden lipoproteins are bound by the LDLr for hepatic uptake and excretion. CETP activity may promote RCT by stimulating LCAT activity and regenerating pre-β HDL (CitationBrewer et al 2004). For this reason, partial inhibition of CETP activity has been the focus of novel therapeutic strategies employing CETP inhibition as discussed below.
Phase four: HDL-C catabolism
As CE accumulate in its central core, pre-β HDL-C matures into larger HDL-C particles known as HDL-3 and HDL-2. These larger molecules undergo hepatic catabolism and excretion in bile. HDL-C catabolism is mediated by 4 mechanisms: 1) hepatic uptake of larger HDL-C particles via hepatic scavenger receptor B1 (SR-B1) receptors for excretion as bile, 2) metabolism of mature HDL-C by hepatic lipase (HL) to smaller particles devoid of lipid and rich in Apo AI, 3) renal uptake of smaller HDL-C particles mediated by apo-E receptors such as cubulin, or 4) LDLr-mediated hepatic uptake of LDL-C and VLDL-C after acquiring CE via CETP activity (CitationMoestrup and Koz 2000; CitationLewis 2006).
Pleiotropic effects of HDL-C: beyond RCT
The vasoprotective properties of HDL-C extend beyond its ability to initiate RCT (). Substantial evidence supports the contention that HDL-C and Apo AI prevent oxidative damage, inhibit systemic inflammation, promote vascular reactivity and integrity, and prevent thrombosis (CitationHayek et al 1995; CitationWatson 1995; CitationNavab et al 1996; Bonnefant-Rousselot et al 1999; CitationMineo et al 2006).
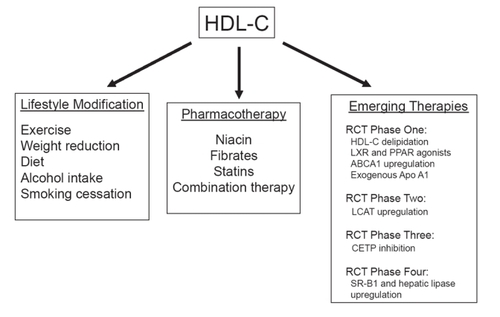
Figure 3 Approaches to enhance HDL-C levels and activity.
Antioxidant effects of HDL-C
The major anti-oxidant effects of HDL-C are mediated by two associated enzymes paroxonase (PON) and platelet-activating factor acetylhydrolase (PAFAH) (CitationGraham et al 1997). PON, an arylesterase enzyme carried by Apo AI, inhibits oxidation of LDL-C (CitationMackness et al 2000). LDLr-null mice lacking PON are susceptible to organophosphate toxicity and manifest accelerated atherosclerosis (CitationShih et al 1998). In epidemiologic studies, genetic polymorphisms of the PON gene may be associated with an increased risk of CHD (CitationHegele 1999).
Platelet activating factor (PAF) is a potent phospholipid released by activated endothelial cells. PAF promotes cell adhesion, platelet aggregation, and vascular permeability. HDL-C inhibits PAF production by endothelial cells in dose-dependent manner (CitationSugatani et al 1996) via hydrolysis of acetyl residues mediated by PAFAH. Genetic polymorphisms of the enzyme PAFAH have been associated with an increased risk of acute MI (CitationLiu et al 2006). PAFAH also degrades oxidized LDL-C and inhibit oxidation of LDL-C (CitationMackness et al 2004; CitationArakawa et al 2005).
Anti-inflammatory effects of HDL-C
The role of inflammation in atherogenesis has been well-established by a number of studies demonstrating accumulation of macrophages derived from circulating monocytes in atheromatous plaques. Anti-inflammatory effects of HDL-C include: 1) neutralization of lipopolysaccharide-induced tumor necrosis factor alpha (TNF-α) release, 2) inhibition of complement activation, 3) inhibition of vascular cell adhesion molecules (VCAM) and monocyte chemotactic protein (MCP-1), which are known to mediate monocyte-endothelial cell interaction, and 4) induced expression of the anti-inflammatory cytokine transforming growth factor-beta 2 by HDL-3 (CitationDimayuga et al 1999; CitationShah et al 2001; CitationCalabresi et al 2003; CitationBarter et al 2004; CitationNorata et al 2005).
Effects of HDL-C on endothelial function and integrity
In patients with known CAD, elevation of HDL-C levels via pharmacologic therapy improves endothelial function (CitationO’Connell et al 2001). In patients with CHD, HDL-C levels correlate positively with coronary vasomotor tone (CitationZeiher et al 1994). In vitro, HDL-C enhances endothelial nitric oxide synthase (eNOS) activity (CitationKuvin et al 2002). The mechanism of HDL-C mediated eNOS activation remains unknown, however may involve an interaction between endothelial SR-B1 and Apo AI (CitationYuhanna et al 2001). In a murine model of myocardial infarction, exogenous administration of human HDL-C increases myocardial perfusion as measured by 99mTc-MIBI uptake via NOS-dependent mechanisms (CitationLevkau et al 2004).
HDL-C also enhances endothelial integrity. HDL-C inhibits endothelial apoptosis induced by TNF-α in a dose-dependent manner by inhibiting caspase 3 activity (CitationSugano et al 2000). HDL-associated lysosphingolipids also suppress mitochondrial pathways of apoptosis by activating the anti-apoptotic serine/threonine protein kinase, Akt (CitationNofer et al 2001). HDL-C also promotes endothelial cell proliferation via a mechanism involving increased phospholipase C activity (CitationDarbon et al 1986; CitationHonda et al 1999).
Antithrombotic effects of HDL-C
HDL-C is also associated with anti-thrombotic and profibrinolytic effects. HDL-C inhibits platelet aggregation by blocking thromboxane-A2 (TXA2) and PAF activity, while stimulating nitric oxide (NO) and PGI2 synthesis (CitationSaku et al 1985; CitationNaqvi et al 1999). In the Atherosclerosis Risk in Communities (ARIC) study, HDL-C levels inversely correlated with circulating von Willebrand factor (vWF) levels, suggesting that HDL-C may prevent synthesis of this pro-thrombotic protein. HDL-C also enhances the anti-thrombotic activity of protein C and protein S (CitationGriffin et al 1999). HDL-C may also attenuate the activity of tissue factor, a potent stimulant of the extrinsic coagulant pathway (CitationCarson 1981).
Taken together, the pleiotropic effects of HDL-C suggest that its use either as a supplement or an infusion may not be limited to chronic therapy, but may eventually play a role in the acute management of vascular disorders. To date, exogenous infusions of synthetic HDL-C have been effective in animal models of atherosclerosis (CitationShah et al 2001), restenosis after angioplasty (CitationAmeli et al 1994), vascular thrombosis (Li et al 1999), myocardial ischemia-reperfusion injury (CitationCalabresi et al 2003), and septic shock (CitationMcDonald et al 2003). Approaches to raise HDL-C levels and subsequently promote RCT include lifestyle modifications, standard pharmacologic therapy, and several emerging therapeutics based on metabolic targets involved in RCT ().
Approaches to raising HDL-C levels: lifestyle modifictions
Exercise and weight loss
Numerous studies associate excess body weight with higher TC, LDL-C, and TG levels and lower HDL-C levels. Active weight loss improves HDL-C levels, while decreasing LDL-C levels (8% decrease for every 1 kg of weight lost). A meta-analysis of 70 studies examining the effects of weight reduction on lipid profiles published between 1966 and 1989 demonstrated a 1 mg/dL increase in HDL-C for every 3 kg of weight lost (CitationDattilo et al 1992). A one-year randomized controlled study evaluating weight loss on plasma lipid profiles in 131 overweight sedentary men demonstrated a significant increase in plasma HDL-C levels (44 mg/dL with exercise, 47 mg/dL with diet, versus 40 mg/dL in controls; p < 0.01), while LDL-C levels remained unchanged (Wood et al 1998).
As a means to reducing weight, regular aerobic exercise increases HDL-C by 10%–20% on average in sedentary adults (CitationWilliams 1997). Previous studies report an increase in HDL-C levels by 1 mg/dL for every 4 to 5 miles run per week (ie, 49 mg/dL with 5 miles (8 km) run per week, 51 mg/dL with 9 miles (15) run per week, 53 mg/dL with 12 miles (20 km) run per week, and 57 mg/dL with 31 miles (50 km) run per week; p < 0.001 versus non-runners) (CitationKokkinos et al 1995). While exercise quantity and intensity differ between studies, the duration of aerobic exercise rather than intensity appears to have a greater impact on HDL-C levels (CitationDurstine et al 2001).
Variable changes in HDL-C have been observed in response to exercise. Some individuals significantly increase HDL-C levels after 8 weeks of regular aerobic exercise (running), while other individuals may not manifest changes in HDL-C for nearly 2 years (CitationDurstine et al 2001). In another study, no significant change in HDL-C was observed in adults with low HDL-C and moderately elevated LDL-C after 6 weeks of walking or jogging 10 miles (16 km) per week (CitationStefanick et al 1998). Moreover, women appear to experience greater improvement in HDL-C with cardiac rehabilitation than men (CitationSavage et al 2004). In general, HDL-C increases with exercise supporting the recommendation of a program of regular, brisk aerobic exercise program most days of the week (CitationUS Department of Health and Human Services 1999). The mechanisms, by which exercise and reduced weight increase HDL-C likely involves enchanced lipoprotein lipase (LL) activity, increased RCT, and increased levels of pre-β HDL (CitationGupta et al 1993; CitationSviridov et al 2003).
Dietary modifications
Major dietary influences on HDL-C levels include total fat intake (independent of fat type), trans – fatty acids, and alcohol intake (CitationThornton et al 1983; CitationRossner and Bjor 1987; CitationLichtenstein 1999). A low saturated fat diet lowers both LDL-C and HDL-C. In a recent study, 11 healthy volunteers were randomized to either a low fat diet (19% fat) or a high fat diet (50% fat) (CitationMeksawan et al 2004). Individuals consuming a low fat diet had a significantly (p < 0.05) lower HDL-2 subpopulation (54 ± 3 vs 63 ± 3 mg/dL) and Apo AI (118 ± 4 vs 127 ± 3 mg/dL) compared with subjects consuming a high fat diet. In addition, reduced dietary fat has been shown to significantly decrease HDL-C2 fractions, which has been linked to antiatherogenic effects of HDL-C (CitationBerglund et al 1999). This suggests that low fat diets may adversely affect the most antiatherogenic HDL subpopulation. However, a simultaneous decrease in LDL-C with low fat diets appears to be more clinically important than the reduction in HDL-C levels.
High consumption of n-3 polyunsaturated fatty acids observed in Native Chukot Peninsula residents is associated with higher HDL-C/Apo AI ratios and increased cholesterol efflux from cellular membranes to HDL-C (CitationGerasimova et al 1991). Consumption of foods high in n-3 polyunsaturated fats (cold-water fish, some shellfish, as well as flax seed, canola, soybean oils and walnuts) increase HDL-C. However, the ability of n-3 polyunsaturated fats to raise HDL-C maybe influenced by TG levels. Dietary modification with omega-3 fatty acids (fish oil), such as eicosapentaenoic acid and docosahexaenoic acid, leads to significant reductions in VLDL-C (25%–30%) and triglyceride levels, yet exerts only a modest effect on HDL-C levels (0 to 3% increase) in patients with TG levels above approximately 175 mg/dL (CitationKris-Etherton et al 2002). This suggests that optimization of an individuals TG must occur before a clinically significant increase in HDL-C in response to a diet high in n-3 polyunsaturated fats is observed.
Moderate alcohol consumption has been shown to elevate HDL-C levels (CitationEllison RC 2004). Mechanisms by which alcohol consumption increases HDL-C may involve changes in Apo AI synthesis and transportation, inhibition of CETP activity and stimulation of early steps in RCT (CitationVan der Gaag et al 2001). A meta-analysis of 25 studies found that consumption of 30 g of alcohol per day increases HDL-C by about 4 mg/dl, irrespective of the type of alcohol consumed. With weighted regression, this represents a 0.133 mg/dL increase in HDL-C per gram of alcohol consumed per day, an 8% increase from pre-treatment levels (CitationRimm et al 1999). Similarly, in a review of 340 MI patients presenting with MI, alcohol consumption was strongly associated with increased HDL-C and a significantly reduced relative risk of MI in the two highest consumption categories (≥1 drink/day and 3 drinks/day) (CitationGaziano et al 1993). Mild to moderate alcohol consumption (1–2 alcoholic beverages several days a week) is reasonable for those individuals with low HDL-C. Caution should be used, however, when recommending alcohol consumption as a therapeutic mechanism in populations at risk for alcohol abuse.
A recent study suggests that the greatest improvement in HDL-C for both men and women in response to weight loss, exercise, and alcohol consumption was seen in individuals within the highest percentiles of HDL-C at baseline, with lower levels of baseline HDL-C being more resistant to lifestyle modifications (CitationWilliams 2004). Separating the effect of one lifestyle modification from another on HDL-C is difficult. For instance, increases in HDL-C with exercise may be due to the resultant weight loss or gene-environmental interactions (Miller et al 2003).
Smoking cessation
Cigarette smoking is associated with lower HDL-C levels (CitationCraig et al 1989). The mechanisms by which cigarette smoking lowers HDL-C remain unclear. Cigarette smokers have significantly lower LCAT activity (CitationImamura et al 2002) and exhibit changes in lipid transfer proteins and CETP activity compared to non-smokers (CitationFreeman et al 1998; CitationMero et al 1998). In a meta-analysis of 29 studies HDL-C levels were significantly increased after smoking cessation, more so in women and to a greater extent in individuals with higher baseline HDL-C (>1.2 mmol/L) (CitationMaeda et al 2003). Moffatt and colleagues demonstrated women smokers have 15%–20% lower HDL-C levels compared to non-smokers (p < 0.05); the HDL-C values improved to normal levels within 30–60 days of smoking cessation (CitationMoffatt 1988). Using a comprehensive approach to smoking cessation (wellbutrin/nicotine-replacement/counseling) remains an important recommendation for those individuals with low HDL-C. Despite the complexity of lifestyle modifications on HDL-C, patients with low HDL-C should be encouraged to become physically active on a regular basis, stop smoking, obtain stable weight reduction with a BMI <25, and increase consumption of polyunsaturated and monounsaturated fats.
Approaches to raising HDL-C levels: standard pharmacotherapy
While dietary and lifestyle modifications can raise HDL-C levels, their effect on cardiovascular outcomes may result from beneficial effects on non-HDL-C lipid components such as LDL-C. At present, standard pharmacotherapy to raise HDL-C levels includes niacin, fibrates, and statins.
Niacin
Since 1955, the B-vitamin niacin (nicotinic acid) has been used in the treatment of dyslipidemia (CitationAltschul et al 1955). Niacin is the most useful pharmacologic therapy for raising HDL-C levels; it has been shown to increase HDL-C by 35%, while lowering TG levels by 20%–50% and LDL-C levels by 5%–25% (CitationSzapary and Rader 2004). Niacin raises HDL-C levels by reducing the fractional catabolic rate of Apo AI containing HDL-C particles, decreasing hepatic removal of lipoprotein A-I (LpA-I) (a cardioprotective subfraction of HDL-C without Apo AII), and inhibiting removal of Apo AI without affecting HDL cholesterol ester (CitationJin et al 1997); resulting in an increase of Apo AI enriched, pre-β HDL particles (CitationGanji et al 2003). Using carotid intima-medial thickness (CIMT) as a measure of subclinical atherosclerosis, a randomized, placebo-controlled study of extended release niacin in addition to statin therapy in 167 patients with known CAD and low serum HDL-C (<45 mg/dL) showed a significantly reduced rate of IMT progression in individuals without insulin resistance (p = 0.026) (CitationTaylor et al 2004). An upcoming study known as ARBITER 6-HALTS (HDL and LDL Treatment Strategies) will randomize 400 subjects with coronary heart disease to HDL-C (extended-release niacin) and LDL-C (ezetimibe) focused strategies of lipid therapy and will measure changes in mean CIMT after 14 months (CitationDevine et al 2007).
Fibrates
Fibric acid derivatives (fibrates) reduce CHD risk in patients with baseline LDL:HDL-C ratios of >5.0 (CitationHuttunen 1991). Fibrates slow the progression of coronary atherosclerosis and reduce coronary events (CitationEricsson et al 1996; CitationFrick et al 1997). Fibrates induce a 5%–20% increase in HDL-C, with generally modest reductions in LDL-C and a pronounced reduction in triglyceride-rich lipoproteins (CitationDespres 2001).
Fibrate therapy increases HDL-C levels by activating PPARα and by enhancing expression of Apo AI and AII, LL, and ABCA1, which collectively enhance RCT (CitationTilly-Kiesi et al 1992). By inducing LL activity, fibrates also increase hepatic fatty acid uptake, enhance removal of LDL particles, and reduce lipid exchange between VLDL and HDL (CitationStaels et al 1998). The hypotriglyceridemic effects of fibrate therapy result from enhanced LL activity and inhibition of Apo CIII gene expression by fibrate-mediated PPARα activation (CitationStaels et al 1995; CitationMotojima et al 1997).
Depending on baseline lipid profiles and the potency of individual fibrates, variable effects on HDL-metabolism have been observed. Despite a greater than 15% increase in HDL-C levels with bezafibrate therapy, the Bezafibrate Infarction Prevention (BIP) failed to demonstrate a significant reduction in the primary composite end point of fatal or nonfatal MI or sudden death (CitationGoldbourt et al 1993). In contrast, in the VA-HIT study, gemfibrozil increased HDL-C on average by 7.5% with a 2% reduction in risk correlated with every 1% increase in HDL-C (Rubenset al 2001). Similarly, in the Lopid Coronary Angiography Trial (LOCAT), gemfibrozil slowed progression of coronary atherosclerosis and the formation of bypass graft lesions.
Statins
Statins inhibit HMG-CoA reductase, the rate-limiting step in cholesterol biosynthesis, resulting in increased LDLr density with decreases in LDL, IDL, and VLDL particle synthesis (CitationFarnier 1998; CitationSegrest et al 2000). Subsequent reductions in LDL-C and TG concentrations are the primary antiatherogenic properties of statins (CitationHarper and Jacobsen 1999). Statins reduce LDL-C levels by 25%–35% at low to moderate doses, which correlates with a reduction in relative risk of coronary events by 25%–37% (CitationSacks et al 1996; CitationWest of Scotland Coronary Prevention Group 1996; CitationLong-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group 1998).
Statins increase HDL-C by 5%–15% and decrease TG levels by 7%–30% (CitationBelalcazar et al 1998). The mechanism of statin-induced increases in HDL-C remains incompletely understood. Some studies suggest increased HDL-C results from a decreased fractional catabolic rate of Apo AI and an increased production of Apo AI induced by inhibiting HMG-Co A reductase (CitationSchaefer et al 1999). Statins have also been shown to increase Apo AI levels by inhibiting the Rho-A kinase signal transduction pathway, resulting in activation of PPARα (CitationMartin et al 2001).
Statins may also reduce hepatic lipase activity, resulting in enhanced synthesis of mature HDL-C. Another potential mechanism for increased HDL-C levels in response to statin therapy is by inhibiting CETP activity. A study of patients with the “B1” variant of the CETP gene showed high levels of baseline CETP activity and low levels of HDL-C with corresponding progression of atherosclerosis. Treatment with pravastatin abolished the progression in atheromatous burden and non-significantly increased HDL-C levels in patients with the “B1” variant (CitationKuivenhoven et al 1998).
Of the available statins, simvastatin, rosuvastatin, and fluvastatin more effectively raise HDL-C levels compared with atorvastatin at doses that lead to similar reductions in LDL-C. In a 36-week, multicenter, double-blind, dose titration study, 826 patients with LDL-C >160 mg/dL and triglyceride <350 mg/dL were randomized to receive titrated doses of simvastatin (maximum 40 mg/day) or atorvastatin (maximum 40 mg/day) over 6–12 weeks. Significantly greater increases in HDL-C and Apo AI with simvastatin compared to atorvastatin (HDL-C: 9% vs 7% p < 0.001; Apo AI: 6% vs 3%, p < 0.001) were observed (CitationKastelein et al 2000). Independent clinical studies have shown that 40 mg/day of simvastatin increase HDL-C by approximately 7%–9% versus a 4%–5% increase with 20 mg/day of atorvastatin (CitationHeinonen et al 1996; CitationCrouse et al 1999). Generally niacin is more effective than statins alone in raising HDL-C levels.
Combination therapy
Combination therapy using statins with niacin or fibrates has been evaluated in a number of small clinical trials (CitationDavidson 2002). The HDL Atherosclerosis Treatment Study (HATS) studied the combination of statin plus extended-release niacin in 160 patients with known CAD, low serum HDL-C levels (<35 mg/dL in males and <40 mg/dL in females), LDL-C levels <145 mg/dL, and TG levels <400 mg/dL. After 3 years of follow-up, combination therapy increased HDL-C levels by 26%, reduced LDL-C levels by 42%, induced regression of the average coronary stenosis by 0.4% (p < 0.001) and reduced cardiovascular events by greater than 60% (CitationBrown et al 2001).
Reflecting the ability of statins and fibrates to synergistically activate PPARα, statin-fibrate combinations have been evaluated in a number of clinical trials (CitationEllen and McPherson 1998; CitationPapadakis et al 1999; CitationFarnier et al 2000; CitationAthyros et al 2002; CitationVega et al 2003;). A multi-center, double-blind trial of 333 patients using fluvastatin compared to bezafibrate versus a combination of the two agents for 24 weeks showed a significant decrease in LDL-C levels in treatment groups receiving fluvastatin compared to bezafibrate alone. Furthermore, HDL-C levels were significantly elevated with bezafibrate alone or in combination with fluvastatin compared with fluvastatin alone (p < 0.001). The combination of fluvastatin and bezafibrate was well tolerated and significantly increased HDL-C (22%), decreased LDL-C (24%), and decreased TG levels (38%) compared with fluvastatin alone (p < 0.001) (CitationPauciullio et al 2000).
Recently, the Comparative Effects on Lipid Levels (COMPELL) trial determined the relative efficacy of combination therapy with a statin and niacin or ezetimibe compared with a statin alone over 12 weeks. Coadministration of niacin (500 mg up-titrated to 2000 mg) with atorvastatin (20–40 mg, n = 60) or rosuvastatin (10–20 mg, n = 65) decreased LDL-C by 56% and 51% and increased HDL-C by 22% and 24% respectively (p = NS). While simvastatin plus ezetimibe decreased LDL-C by 57%, HDL-C only increased by 10% compared to baseline. Rosuvastatin (10–40 mg) monotherapy decreased LDL-C by 53% and raised HDL-C by 7% (CitationJones 2006). Future studies involving rosuvastatin/fenofibrate combination therapy and the recently announced combination of rosuvastatin with a next generation fenofibrate (ABT-335) will provide further insight into the efficacy of dual-targeted therapy.
Approaches to raising HDL-C levels: emerging therapeutics
Based on preclinical data, multiple strategies to enhance the beneficial effects of HDL-C are being considered. HDL-C delipidation therapy (CitationKostner et al 2002), exogenous Apo AI mimetics (CitationNavab et al 2004), CETP inhibition (CitationBrousseau et al 2004), LXR/RXR agonists (CitationBrewer et al 2004), selective and non-selective PPAR agonists (CitationOliver et al 2001; CitationSchmitz et al 2002), and drugs targeting HDL-C catabolism (CitationMezdour et al 1997; CitationJansen et al 2004) are among some of the novel emerging therapies harnessing the anti-atherogenic, anti-oxidant, anti-inflammatory, and pro-endothelial functions of HDL-C.
HDL-C delipidation therapy
Selective HDL-C delipidation therapy utilizes plasmapheresis whereby extracted plasma is mixed with a delipidating agent and separated into an inorganic and organic phase. The organic component contains a high concentration of delipidated HDL, similar to lipid-poor pre-β HDL produced by the liver, which is then returned to the circulation. In a series of animal studies delipidation therapy has been shown to markedly increase circulating pre-β HDL levels and subsequently increase ABCA1-mediated cholesterol efflux from peripheral cells without exerting a significant effect on LDL-C metabolism Animal studies evaluating delipidation therapy followed by intravascular ultrasound assessment of vascular plaque progression or regression are ongoing (CitationShah 2007).
Exogenous administration of Apo AI and Apo AI mimetics
Exogenous administration of Apo AI directly enhances RCT via the ABCA1 pathway (CitationZhang et al 2003; CitationNavab et al 2004; CitationArakawa et al 2004). Treating normal human LDL-C with exogenous Apo AI in vitro reduces levels of oxidized lipids by 50%–60% and prevents monocyte chemotactic activity, a primary step in atherogenesis (CitationPoon et al 1997). Exogenous administration of Apo AI-associated lecithin discs reduces the ability of LDL to induce monocyte chemotaxis, increases concentrations of pre-β HDL, and stimulates RCT in human subjects (CitationNanjee et al 2001). Apo AI infusions also modulate phospholipids transfer protein (PLTP), LCAT, and CETP activity, all of which potentially contribute to RCT (CitationKujiraoka et al 2003).
Recently, Tardiff and colleagues administered 4 weekly infusions of a mixture of human wild-type Apo AI and soybean phosphatidylcholine (CSL-111; 40 mg/kg or 80 mg/kg) or volume-matched placebo to 183 patients presenting with an acute coronary syndrome as part of the Effect of reconstituted HDL on Atherosclerosis – Safety and Efficacy (ERASE) study. Two weeks after the last infusion, intravascular ultrasound (IVUS) and quantitative coronary angiographic (QCA) measurements were compared to baseline. Patients receiving CSL-111 experienced a −3.4% change in atheroma volume (p = 0.48 vs placebo; p < 0.001 vs baseline) with an absolute change of −5.3 mm3 (p = 0.39 vs placebo; p < 0.001 vs baseline). Notable, transient liver function abnormalities were observed in the CSL-111 group versus placebo. One patient developed a 100-fold increase in ALT levels in the high dose (80 mg/kg) infusion group (CitationTardif et al 2007) While the primary endpoint of the study was negative, the data presented suggest a potential benefit for inducing plaque regression. This study highlights the complexity surrounding exogenous Apo AI therapy.
Based on observations in a family with low HDL-C and a lack of atherosclerotic disease from Limone sul Garda, Italy, a variant form of Apo AI, known as Apo AI Milano (AIM), was identified in 1980 (CitationFranceschini et al 1980). A cysteinearginine substitution at position 173 in the amino acid sequence allows the mutant protein to form disulfide bonds with other Apo AI molecules and Apo AII. AIM homodimers and heterodimers may enhance cholesterol efflux thereby augmenting RCT (CitationChiesa et al 2002).
Administration of recombinant Apo AI Milano (rAIM) reduces plaque cross sectional area compared to saline-placebo by up to 40% in rabbit carotid models of atherosclerosis (CitationAmeli et al 1994; CitationIbanez 2007). Similar results have been demonstrated in balloon injured arteries in hypercholesterolemic rabbits, Apo E-deficient mice, and in transgenic mouse models of Apo AI over-expression (CitationRubin et al 1991; CitationShah et al 1998). Exogenous HDL-C or Apo AI administration also enhance fecal steroid excretion, increase serum pre-β HDL, and enhance RCT in humans (CitationWestman et al 1995; CitationEriksson et al 1999).
In 2003 a landmark study using rAIM (ETC-216) quantified coronary plaque volume as a response to pharmacologic intervention with intravascular ultrasound (IVUS). This study evaluated the effect of exogenous administration of ETC-216 on coronary atherosclerosis in patients with acute coronary syndromes. ETC-216 reduced total atheroma volume by 1.3% (39.7–38.4) and 0.7% (37.2–36.6) in the moderate and high dose treatment groups respectively, while a 0.14% (34.8–34.9) increase was noted in the placebo group (CitationNissen et al 2003). This “proof-of-concept” study demonstrated the ability of Apo AI mimetic peptides to halt progression and potentially induce regression of atheromatous plaque.
A series of Apo AI mimetic peptides are currently under investigation. ETC-642 is a second generation Apo AI synthetic peptide containing three charged residues in a 22 amino-acid sequence, rendering the peptide more hydrophobic (Navab et al 2005). Within hours of treatment with ETC-642 increased HDL-C serum levels have been observed in rabbit models. Increased CE content in HDL-C indicates concomitant LCAT activation by ETC-642. This rapid elevation of HDL-C levels suggests a possible future role for Apo AI mimetic peptides in the management of acute coronary syndromes or in the setting of ischemia-reperfusion injury (CitationMarchesi et al 2004).
Another Apo AI mimetic peptide known as D4F reduces atherosclerosis in mouse models (CitationGarber et al 2001). Peptide D4F contains 18 amino acids in a class A amphipathic helix with polar and non-polar faces yielding high lipid affinity (CitationDatta et al 2001). D4F enhances the anti-inflammatory properties of HDL-C, reduces LDL-mediated monocyte chemotaxis, reduces macrophage migration into atheromatous plaques, and reduces atherosclerosis in Apo E KO mice alone or in combination with statin therapy. Both oral and intraperitoneal administration of D4F significantly reduced evolving atherosclerotic lesions in vein grafts but not established atherosclerotic lesions in the aortic sinus, suggesting specific types of atherosclerotic lesions may modulate the beneficial effects of Apo AI mimetic peptides (CitationLi et al 2004).
Taking advantage of the amphipathic helical structure common to apolipoproteins, numerous Apo AI mimetic peptides are being developed. Unique helical configuration with opposing hydrophobic and hydrophilic faces enhances interaction between lipid surfaces and apolipoproteins for the removal of membrane bound cholesterol. Some novel agents under development include: ETC-588 (large unilamellar vesicles – LUV), ETC-1001 (small molecule investigational product), helical peptides (Esperion 24218), and trimeric Apo-A (Proteopharma/Borean pharma) (CitationNavab et al 2006).
Nuclear regulation of RCT: LXR and PPAR agonists
Liver X-receptors (LXR) are nuclear receptors that sense excess intracellular cholesterol (CitationWang and Briggs 2004). Hydroxylated cholesterol stimulates LXR-mediated transcription of ABCA1, which subsequently enhances RCT from peripheral tissues (CitationLund et al 2006). Two types of LXR receptors exist, LXRα and LXRβ. LXRα has been identified in liver, intestine, macrophages and adipose tissues, while LXRβ is ubiquitously expressed similar to ABCA1 (CitationLala et al 2005).
LXR agonists prevent development of atherosclerosis by modulating metabolic and inflammatory gene expression in rodent models. Non-selective LXR agonists increase ABCA1 synthesis with a gradual increase in HDL-C serum levels (CitationLund et al 2006). In a mouse LXRα knockout model, treatment with a non-selective LXR agonist increased HDL-C by day 7 with a less significant increase in hepatic TG content (CitationJoseph et al 2002). Similarly, treating LDLr KO mice with the LXR ligand, T-0901317, reduced atherosclerotic lesion development without affecting plasma total cholesterol levels (CitationTerasaka et al 2003). Recently administration of the LXR agonist GW3965 to mice increased the rate of RCT from macrophages to feces in vivo (CitationNaik et al 2006).
A major concern associated with LXR agonists is the development of hepatic steatosis. Since LXR agonists induce genes that stimulate lipogenesis, including the sterol response element binding protein (SREBP1-c) and fatty acid synthetase (FAS). The induction of these genes in the liver cause increased hepatic triglyceride synthesis, hypertriglyceridemia, and hepatic steatosis. Current research has focused on selective LXR modulators that may circumvent this adverse effect on hepatic function (CitationMiao et al 2004).
First identified in rodent models of fibrate-induced hepatic peroxisome proliferation, peroxisome proliferators activated receptors (PPARs) are another family of nuclear receptors closely linked to HDL-C metabolism (CitationEverett et al 2000). Acting as synthetic ligands for PPARα activation, fibrates increase circulating levels of HDL-C, enhance RCT and reduce vascular inflammation and thrombogenicity (CitationBarbier et al 2002; CitationGervois et al 2007). PPARα agonists enhance gene expression of SR BI, Apo AI, Apo AII, LPL, and ABCA1 (CitationToth 2005). Statins also enhance PPARα activity and may enhance cholesterol efflux (CitationMartin et al 2001; CitationInoue et al 2002).
Agents for the management of Type 2 diabetes such as thiazolidinediones are known PPARγ agonists and enhance ABCA1 mediated RCT and increase HDL-C levels in primates (CitationOliver et al 2001). PPARα and γ mediate activation of the LXR/RXR heterodimer, which in turn regulates cholesterol efflux via ABCA1 and ABCG1 activation (CitationSchmitz et al 2002). Unfortunately, the development of novel PPAR agonists to date, particularly PPARγand PPARα/γ, have been halted due to preclinical and clinical adverse effects (CitationRubenstrunk et al 2007).
Cholesterol exchange transfer protein (CETP) inhibition
The complex relationship between CETP activity and atherosclerotic disease has been illustrated by several recent studies evaluating CETP inhibition therapy. In the past, antisense oligodeoxynucleotides and antibodies against CETP increased HDL-C levels and reduced aortic atherosclerotic burden in cholesterol-fed rabbits (CitationSugano et al 1998; CitationRittershaus et al 2000). In a phase II, randomized, placebo-controlled trial, 148 statin-naïve patients with mild hyperlipidemia were treated with a CETP inhibitor known as JTT-705. In this study, a 37% reduction in CETP activity correlated with a 34% increase in HDL levels and 7% decrease in LDL levels (CitationDe Grooth et al 2002).
Recently, the CETP inhibitor, torcetrapib, has been extensively studied evaluated in a number of human trials. In a phase I, multi-dose study, torcetrapib was administered to 40 normolipidemic individuals in doses of 10, 30, 60, and 120 mg per day and 120 mg twice daily. Significant increases in serum HDL-C levels ranging from 16% to 91% (10 mg daily versus 120 mg twice daily) with a decrease in LDL-C from 7% to 42% (60 mg daily versus 120 mg twice daily) without changing TC levels was observed. At the highest dosing regimen, Apo AI and Apo E increased by 27% and 66% respectively, while Apo B decreased by 26% (CitationClark et al 2004).
Another single-blinded study compared torcetrapib alone at variable doses versus torcetrapib in combination with 20 mg of atorvastatin for one month. HDL-C levels increased by 46%, 61%, and 106%, while LDL-C levels decreased by 17%, 7.5%, and 17% after treatment with 120 mg daily, 120 mg daily plus 20 mg atorvastatin, and 120 mg twice daily respectively. No major adverse events were reported (CitationBrousseau et al 2004).
Based on these promising findings, torcetrapib was evaluated in a large international clinical trial known as the Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events (ILLUMINATE). In this study, 15,000 patients at high risk for CHD received torcetrapib with atorvastatin versus atorvastatin alone. In December 2006, this study was prematurely terminated due to an excess of deaths in the torcetrapib/atorvastatin versus atorvastatin groups (81 vs 51, respectively). Additionally, patients receiving torcetrapib had a higher incidence of heart failure, angina, and revascularization procedures. The adverse outcomes of the ILLUMINATE study may have been related to off-target effects of torcetrapib, such as an increase in systolic blood pressure (limited to 1–2 mmHg) or low levels of CETP inhibition and reduced RCT (CitationHoney 2007). Notably, inherited deficiencies of CETP and other CETP inhibitor formulations did not increase systolic blood pressure, suggesting that the chemical structure of torcetrapib itself may have contributed to poor outcomes (CitationTall et al 2007).
Two separate studies evaluated the impact of torcetrapib on atherosclerotic progression. The Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation (ILLUSTRATE) studied 1,188 patients with coronary disease in a prospective, randomized, double-blinded fashion. Patients initially received atorvastatin monotherapy until LDL-C levels were below 100 mg/dL, after which time they were randomized to either atorvastatin (10–80 mg daily) monotherapy or atorvastatin plus 60mg torcetrapib daily. After 24 months, the torcetrapib/atorvastatin group had a 61% relative increase in HDL-C and 20% decrease in LDL-C. However, no significant reduction in percent atheroma volume was observed between atorvastatin monotherapy and torcetrapib combination therapy (0.19% vs 0.12%, respectively). Torcetrapib therapy was again associated with a mean 4.6 mmHg increase in systolic blood pressure (CitationNissen et al 2007).
Further substantiating these findings, the Rating of Atherosclerotic Disease Change by Imaging with a New CETP Inhibitor (RADIANCE 1 and 2) trials, demonstrated no further reduction of atherosclerosis progression in carotid intima-media thickness after 24 months of therapy with atorvastatin versus atorvastatin/torcetrapib combination (Bots et al 2007; Kastelein et al 2007).
Summary
A considerable body of evidence supports the correlation between HDL-C levels and cardiovascular risk. However, trials evaluating HDL-C targeted therapies are limited, in part due to a lack of pharmacologic agents specifically designed to raise HDL-C and our limited ability to measure HDL-C effectiveness. As a result, there is not enough data to support guidelines recommending aggressive increases in HDL-C levels. With this in mind, evaluating the clinical efficacy of emerging HDL-C targeted therapies will be of paramount importance.
Given the complexity of HDL-C metabolism, serum levels of HDL-C may not be an adequate indicator of efficacy. At present, plasma HDL-C measurements have a ± 10% margin of error, which could lead to errors in measurement of up to 4 mg/dL (CitationFriedewald et al 2007). The functional properties of circulating HDL-C levels, the kinetics of HDL-C metabolism, and the variable effects of HDL-C subfractions on atherogenesis are ignored by current laboratory measures of HDL-C (CitationForrester et al 2005). While measuring HDL-C subfractions are not recommended at present, recent data suggests that increased Apo AI plasma levels and Apo AI:Apo B ratios correlate with a reduced risk of myocardial infarction and stroke (CitationQureshi et al 2002).
As a crude marker of RCT, measuring sterol content in fecal matter indirectly reflects the amount of cholesterol excreted by the liver as bile. Emerging therapies such as Apo AI mimetics (CitationEriksson et al 1999) and LXR agonists (CitationPlosch et al 2002) increase fecal sterol excretion (FSE), while CETP inhibition with torcetrapib fails to affect fecal sterol content (CitationBrousseau et al 2005). The clinical significance of FSE as a marker of RCT remains uncertain.
At present, volumetric IVUS in combination with standard HDL-C measurement is our best measure of plaque stabilization or regression. Novel approaches using high-speed multislice CT, cardiac MRI, and intracoronary MRI may replace IVUS. Clinical trials using both surrogate endpoints such as volumetric IVUS and carotid IMT in combination with hard clinical endpoints may be required to evaluate the efficacy of emerging HDL-C based therapies.
During the next 10 years, many clinical trials will evaluate the effects of HDL-modification therapy. While the past decade has focused on lowering levels of LDL-C and non-HDL-C to reduce atherosclerotic progression and CVD events, the next decade may prove that HDL-focused therapy induces regression of luminal atherosclerosis and improves CVD outcomes. We eagerly await the completion of the ongoing clinical trials employing a variety of novel strategies to raise HDL-C and possibly halt the progression of atherosclerotic vascular disease.
References
- AltschulRHofferAStephenJDInfluence of Nicotinic Acid on Serum Cholesterol in ManArch Biochem Biophys1955545589
- AmeliSHultgardh-NilssonACercekBRecombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbitsCirculation1994901935417923682
- AndersonSPDunnCLaughterAOverlapping transcriptional programs regulated by the nuclear receptors peroxisome proliferator-activated receptor alpha, retinoid X receptor, and liver X receptor in mouse liverMol Pharmacol20046614405215371561
- ArakawaRHayashiMRemaleyATPhosphorylation and stabilization of ATP binding cassette transporter A1 by synthetic amphiphilic helical peptidesJ Biol Chem2004279862172014701850
- ArakawaHQianJYBaatarDLocal expression of platelet-activating factor-acetylhydrolase reduces accumulation of oxidized lipoproteins and inhibits inflammation, shear stress-induced thrombosis, and neointima formation in balloon-injured carotid arteries in nonhyperlipidemic rabbitsCirculation20051113302915956136
- AthyrosVGPapageorgiouAADemitriadesDSAtorvastatin versus four statin-fibrate combinations in patients with familial combined hyperlipidaemiaJ Cardiovasc Risk2002933911984215
- BarbierOTorraIPDuguayYPleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosisArterioscler Thromb Vasc Biol2002227172612006382
- BarrDPRussEMEderHAProtein-lipid relationships in human plasmaAm J Med19511148014885223
- BarterPJBrewerHBChapmanMJCholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosisArterioscler Thromb Vasc Biol200323160712588754
- BarterPJNichollsSRyeKAAntiinflammatory properties of HDLCirc Res200487647215486323
- BadimonJJBadimonLFusterVRegression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbitJ Clin Invest1990851234412318976
- BelalcazarLMBallantyneCMDefining specific goals of therapy in treating dyslipidemia in the patient with low high-density lipoprotein cholesterolProg Cardiovasc Dis199841151749790415
- BerglundLOliverEHFontanezNHDL-subpopulation patterns in response to reductions in dietary total and saturated fat intakes in healthy subjectsAm J Clin Nutr199970992100010584043
- BoekholdtSMKuivenhovenJAWarehamNJPlasma levels of cholesteryl ester transfer protein and the risk of future coronary artery disease in apparently healthy men and women: the prospective EPIC (European Prospective Investigation into Cancer and nutrition)-Norfolk population studyCirculation200411014182315337694
- BodziochMOrsoEKluckenJThe gene encoding ATP-binding cassette transporter 1 is mutated in Tangier diseaseNat Genet1999223475110431237
- Bonnefont-RousselotDTherondPBeaudeuxJLHigh density lipoproteins (HDL) and the oxidative hypothesis of atherosclerosisClin Chem Lab Med1999379394810616747
- BrewerHBJrRemaleyATNeufeldEBRegulation of plasma high-density lipoprotein levels by the ABCA1 transporter and the emerging role of high-density lipoprotein in the treatment of cardiovascular diseaseArterioscler Thromb Vasc Biol20042417556015319263
- BrousseauMEDiffenderferMRMillarJSEffects of cholesteryl ester transfer protein inhibition on high-density lipoprotein subspecies, apolipoprotein A-I metabolism, and fecal sterol excretionArterioscler Thromb Vasc Biol20052510576415761191
- BrousseauMSchaeferEJWolfeMLEffects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterolN Engl J Med200435015051515071125
- BrownBGZhaoXQChaitASimvastatin and niacin, anti-oxidant vitamins, or the combination for the prevention of coronary diseaseN Engl J Med200134515839211757504
- BruemmerDLawRELiver x receptors: potential novel targets in cardiovascular diseasesCurr Drug Targets Cardiovasc Haematol Disord200555334016503873
- CalabresiLGomaraschiMFranceschiniGEndothelial protection by high-density lipoproteins: from bench to bedsideArterioscler Thromb Vasc Biol20032317243112969988
- CannonCPBraunwaldEMcCabeCHIntensive versus moderate lipid lowering with statins after acute coronary syndromesN Engl J Med2004350149550415007110
- CarsonSDPlasma high density lipoproteins inhibit the activation of coagulation factor X by factor VIIa and tissue factorFEBS Lett198113237406795061
- CastelliWPAndersonKWilsonPWLipids and risk of coronary heart disease. The Framingham StudyAnn Epidemiol199222381342260
- ChawlaABoisvertWALeeCHA PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesisMol Cell200171617111172721
- ChiesaGMonteggiaEMarchesiMRecombinant apolipoprotein A-I (Milano) infusion into rabbit carotid artery rapidly removes lipid from fatty streaksCirc Res2002909748012016263
- ClarkRWSutfinTARuggeriRBRaising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: an initial multidose study of torcetrapibArterioscler Thromb Vasc Biol200424490714739125
- CraigWYPalomakiGEHaddowJECigarette smoking and serum lipid and lipoprotein concentrations: an analysis of published dataBMJ198929878482496857
- CrouseJRFrohlichJOseLEffects of high doses of simvastatin and atorvastatin on high-density lipoprotein cholesterol and apolipoprotein A-IAm J Cardiol19998314767A710335764
- DarbonJMTournierJFTauberJPPossible role of protein phosphorylation in the mitogenic effect of high density lipoproteins on cultured vascular endothelial cellsJ Biol Chem198626180023486870
- DattaGChaddhaMHamaSEffects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptideJ Lipid Res200142109610411441137
- DattiloAMKris-EthertonPMEffects of weight reduction on blood lipids and lipoproteins: a meta-analysisAm J Clin Nutr19925632081386186
- DavidsonMHCombination therapy for dyslipidemia: safety and regulatory considerationsAm J Cardiol20029050K60K
- De GroothGJKlerkxAHStroesESA review of CETP and its relation to atherosclerosisJ Lipid Res20044519677415342674
- De GroothGJKuivenhovenJAStalenhoefAFEfficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans: a randomized phase II dose-response studyCirculation200210521596511994249
- DespresJPIncreasing high-density lipoprotein cholesterol: an update on fenofibrateAm J Cardiol20018830N6N11423054
- DevinePJTurcoMATaylorAJDesign and Rationale of the ARBITER 6 Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol)-6-HDL and LDL Treatment Strategies in Atherosclerosis (HALTS)Cardiovasc Drugs Ther200721221517404825
- DimayugaPZhuJOguchiSReconstituted HDL containing human apolipoprotein A-1 reduces VCAM-1 expression and neointima formation following periadventitial cuff-induced carotid injury in apoE null miceBiochem Biophys Res Commun1999264465810529386
- DownsJRClearfieldMWeisSPrimary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention StudyJAMA199827916152229613910
- DurstineJLGrandjeanPWDavisPGBlood lipid and lipoprotein adaptations to exercise: a quantitative analysisSports Med20013110336211735685
- DuvergerNKruthHEmmanuelFInhibition of atherosclerosis development in cholesterol-fed human apolipoprotein A-I-transgenic rabbitsCirculation19969471378772693
- ElkhalilLMajdZBakirRFish-eye disease: structural and in vivo metabolic abnormalities of high-density lipoproteinsMetabolism199746474839160810
- EllenRLMcPhersonRLong-term efficacy and safety of fenofibrate and a statin in the treatment of combined hyperlipidemiaAm J Cardiol19988160B65B
- EllisonRCZhangYQureshiMMLifestyle determinants of high-density lipoprotein cholesterol: the National Heart, Lung, and Blood Institute Family Heart StudyAm Heart J20041475293514999205
- EricssonCGHamstenANilssonJAngiographic assessment of effects of bezafibrate on progression of coronary artery disease in young male postinfarction patientsLancet1996347849538622389
- ErikssonMCarlsonLAMiettinenTAStimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I. Potential reverse cholesterol transport in humansCirculation1999100594810441095
- EverettLGalliACrabbDThe role of hepatic peroxisome proliferator-activated receptors (PPARs) in health and diseaseLiver200020191910902968
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in AdultsSummary of the second report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II)JAMA19932693015238501844
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in AdultsExecutive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III)JAMA200128524869711368702
- FarnierMCerivastatin in the treatment of mixed hyperlipidemia: the RIGHT study. The Cerivastatin Study Group. Cerivastatin Gemfibrozil Hyperlipidemia TreatmentAm J Cardiol19988247J51J
- FarnierMPortalJJMaigretPEfficacy of atorvastatin compared with simvastatin in patients with hypercholesterolemiaJ Cardiovasc Pharmacol Ther20005273210687671
- ForresterJSMakkarRShahPKIncreasing high-density lipoprotein cholesterol in dyslipidemia by cholesteryl ester transfer protein inhibition: an update for cliniciansCirculation200511118475415824213
- ForteTMNicholsAVKraussRMFamilial apolipoprotein AI and apolipoprotein CIII deficiency. Subclass distribution, composition, and morphology of lipoproteins in a disorder associated with premature atherosclerosisJ Clin Invest1984741601136501564
- FranceschiniGSirtoriCRCapursoA2A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian familyJ Clin Invest1980668929007430351
- FranconeOLFieldingCJFieldingPEDistribution of cell-derived cholesterol among plasma lipoproteins: a comparison of three techniquesJ Lipid Res19903121952002090713
- FreemanDJCaslakeMJGriffinBAThe effect of smoking on post-heparin lipoprotein and hepatic lipase, cholesteryl ester transfer protein and lecithin:cholesterol acyl transferase activities in human plasmaEur J Clin Invest199828584919726040
- FrickMHSyvanneMNieminenMSPrevention of the angiographic progression of coronary and vein-graft atherosclerosis by gemfibrozil after coronary bypass surgery in men with low levels of HDL cholesterol. Lopid Coronary Angiography Trial (LOCAT) Study GroupCirculation1997962137439337181
- FriedewaldVEBrewerHBGrundySMThe Editor’s Roundtable: High-Density Lipoprotein CholesterolAm J Cardiol200799169870517560878
- FunkeHvon EckardsteinAPritchardPHA molecular defect causing fish eye disease: an amino acid exchange in lecithin-cholesterol acyltransferase (LCAT) leads to the selective loss of alpha-LCAT activityProc Natl Acad Sci199188485592052566
- FurbeeJWJrFranconeOParksJSIn vivo contribution of LCAT to apolipoprotein B lipoprotein cholesteryl esters in LDL receptor and apolipoprotein E knockout miceJ Lipid Res2002434283711893779
- GanjiSHKamannaVSKashyapMLNiacin and cholesterol: role in cardiovascular disease (review)J Nutr Biochem20031429830512873710
- GarberDWDattaGChaddhaMA new synthetic class A amphipathic peptide analogue protects mice from diet-induced atherosclerosisJ Lipid Res2001425455211290826
- GazianoJMBuringJEBreslowJLModerate alcohol intake, increased levels of high-density lipoprotein and its subfractions, and decreased risk of myocardial infarctionN Engl J Med19933291829348247033
- GerasimovaEPerovaNOzerovaIThe effect of dietary n-3 polyunsaturated fatty acids on HDL cholesterol in Chukot residents vs muscovitesLipids19912626151907706
- GervoisPFruchartJCStaelsBDrug Insight: mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptorsNat Clin Pract Endocrinol Metab200731455617237841
- GlomsetJAThe plasma lecithin:cholesterol acyltransferase reactionJ Lipid Res19689155674868699
- GoldbourtUBeharSReicher-ReissHRationale and design of a secondary prevention trial of increasing serum high-density lipoprotein cholesterol and reducing triglycerides in patients with clinically manifest atherosclerotic heart disease (the Bezafibrate Infarction Prevention Trial)Am J Cardiol1993719099158465780
- GordonDJProbstfieldJLGarrisonRJHigh-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studiesCirculation1977798152642759
- GordonTCastelliWPHjortlandMCHigh density lipoprotein as a protective factor against coronary heart disease. The Framingham StudyAm J Med62570714193398
- GrahamAHassallDGRafiqueSEvidence for a paraoxonase-independent inhibition of low-density lipoprotein oxidation by high-density lipoproteinAtherosclerosis19971351932049430369
- GriffinJHKojimaKBankaCLHigh-density lipoprotein enhancement of anticoagulant activities of plasma protein S and activated protein CJ Clin Invest1999103219279916134
- GrundySMCleemanJIMerzCNImplications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III GuidelinesJ Am Coll Cardiol2004447203215358046
- GuptaAKRossEAMyersJNIncreased reverse cholesterol transport in athletesMetabolism199342684908510511
- JoyceCBrewerBHFreemanLStudy of ABCA1 function in transgenic miceArterioscler Thromb Vasc Biol2003239657112615681
- HarperCRJacobsonTANew perspectives on the management of low levels of high-density lipoprotein cholesterolArch Intern Med199915910495710335681
- HayekTOiknineJDanknerGHDL apolipoprotein A-I attenuates oxidative modification of low density lipoprotein: studies in transgenic miceEur J Clin Chem Clin Biochem19953372158608194
- HegeleRAParaoxonase genes and diseaseAnn Med1999312172410442677
- HeinonenTMSteinEWeissSRThe lipid-lowering effects of atorvastatin, a new HMG-CoA reductase inhibitor: results of a randomized, double-masked studyClin Ther199618853638930429
- HiranoKYamashitaSKugaYAtherosclerotic disease in marked hyperalphalipoproteinemia. Combined reduction of cholesteryl ester transfer protein and hepatic triglyceride lipaseArterioscler Thromb Vasc Biol1995151849567583564
- HiranoKYamashitaSMatsuzawaYPros and cons of inhibiting cholesteryl ester transfer proteinCurr Opin Lipidol2000115899611086331
- HondaHMLeitingerNFrankelMHigh-density lipoprotein increases intracellular calcium levels by releasing calcium from internal stores in human endothelial cellsAtherosclerosis1999229930610217358
- HoneyKDrug designed to raise HDL levels falls downJ Clin Invest200711728217273546
- HuttunenJThe Helsinki Heart Study: central findings and clinical implicationsAnn Med19912315592069792
- IbanezBShort-term treatment with Apo AI Milano indues atherosclerotic plaque regression and signs of plaque stabilization: in vivo MRI study2007Paper presented at: Annual Meeting of the American College of CardiologyMarch 24-27New Orleans, LA
- IkewakiKNishiwakiMSakamotoTIncreased catabolic rate of low density lipoproteins in humans with cholesteryl ester transfer protein deficiencyJ Clin Invest1995961573817657828
- ImamuraHTeshimaKMiyamotoNCigarette smoking, high-density lipoprotein cholesterol subfractions, and lecithin: cholesterol acyltransferase in young womenMetabolism2002511313612370852
- InazuABrownMLHeslerCBIncreased high-density lipoprotein levels caused by a common cholesterylester transfer protein gene mutationN Engl J Med1990323123482215607
- InoueIItohFAoyagiSFibrate and statin synergistically increase the transcriptional activities of PPARalpha/RXRalpha and decrease the transactivation of NFkappaBBiochem Biophys Res Commun2002290131911779144
- JansenHHepatic lipase: friend or foe and under what circumstances?Curr Atheroscler Rep20046343715296699
- JinFYKamannaVSKashyapMLNiacin decreases removal of high-density lipoprotein apolipoprotein A-I but not cholesterol ester by Hep G2 cells. Implication for reverse cholesterol transportArterioscler Thromb Vasc Biol199717202089351367
- JonesP2006International Symposium on AtherosclerosisJune 18-22Rome, Italy
- JosephSBMcKilliginEPeiLSynthetic LXR ligand inhibits the development of atherosclerosis in miceProc Natl Acad Sci USA2002997604912032330
- KannelWBHigh-density lipoproteins: epidemiologic profile and risks of coronary artery diseaseAm J Cardiol1983529B12B
- KaplinskyE1998Data presented at European Society of Cardiology MeetingVienna, Austria
- KasteleinJJIsaacsohnJLOseLComparison of effects of simvastatin versus atorvastatin on high-density lipoprotein cholesterol and apolipoprotein A-I levelsAm J Cardiol200086221310913488
- KennedyMABarreraGCNakamuraKABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulationCell Metab200511213116054053
- KhavkinTNNikolai Nikolaevich AnitschkowBeitr Pathol197515630112130900
- KlerkxAHde GroothGJZwindermanAHCholesteryl ester transfer protein concentration is associated with progression of atherosclerosis and response to pravastatin in men with coronary artery disease (REGRESS)Eur J Clin Invest20043421814984434
- KoizumiJInazuAYagiKSerum lipoprotein lipid concentration and composition in homozygous and heterozygous patients with cholesteryl ester transfer protein deficiencyAtherosclerosis199190189961759989
- KokkinosPFHollandJCNarayanPMiles run per week and high-density lipoprotein cholesterol levels in healthy, middle-aged men. A dose-response relationshipArch Intern Med1995155415207848025
- KostnerKMSmithJLSmithBEPresentation. A novel extracorporeal plasma delipidation procedure for the treatment of atherosclerosis200273rd Eur Athero Soc Meeting
- Kris-EthertonPMHarrisWSAppelLJFish consumption, fish oil, omega-3 fatty acids, and cardiovascular diseaseCirculation200210627475712438303
- KuivenhovenJAJukemaJWZwindermanAHThe role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The Regression Growth Evaluation Statin Study GroupN Engl J Med199833886939420339
- KujiraokaTNanjeeMNOkaTEffects of intravenous apolipoprotein A-I/phosphatidylcholine discs on LCAT, PLTP, and CETP in plasma and peripheral lymph in humansArterioscler Thromb Vasc Biol2003231653912893687
- KuvinJTRametMEPatelARA novel mechanism for the beneficial vascular effects of high-density lipoprotein cholesterol: enhanced vasorelaxation and increased endothelial nitric oxide synthase expressionAm Heart J20021441657212094204
- KwiterovichPOJrThe antiatherogenic role of high-density lipoprotein cholesterolAm J Cardiol19988213Q21Q9671001
- LalaDSThe liver X receptorsCurr Opin Investig Drugs2005693443
- LaRosaJCGrundySMWatersDDIntensive Lipid Lowering with Atorvastatin in Patients with Stable Coronary DiseaseN Engl J Med200535214253515755765
- LevkauBHermannSTheilmeierSHigh-density lipoprotein stimulates myocardial perfusion in vivoCirculation20041103355915545521
- LewisGFDeterminants of plasma HDL concentrations and reverse cholesterol transportCurr Opin Cardiol2006213455216755204
- LewisGFRaderDJNew insights into the regulation of HDL metabolism and reverse cholesterol transportCirc Res20059612213215976321
- LiXChyuKYNetoJRDifferential effects of apolipoprotein A-I-mimetic peptide on evolving and established atherosclerosis in apolipoprotein E-null miceCirculation20041101701515353488
- LichtensteinAHAusmanLMJalbertSMEffects of different forms of dietary hydrogenated fats on serum lipoprotein cholesterol levelsN Engl J Med199934019334010379016
- LiuPYLiYHWuHLPlatelet-activating factor-acetylhydrolase A379V (exon 11) gene polymorphism is an independent and functional risk factor for premature myocardial infarctionJ Thromb Haemost200641023816689754
- Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study GroupPrevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levelsN Engl J Med19983391349579841303
- LundEGPetersonLBAdamsADDifferent roles of liver X receptor alpha and beta in lipid metabolism: effects of an alpha-selective and a dual agonist in mice deficient in each subtypeBiochem Pharmacol2006714536316325781
- MacknessMIDurringtonPMacknessBParaoxonase 1 activity, concentration and genotype in cardiovascular diseaseCurr Opin Lipidol20041539940415243212
- MacknessMIDurringtonPNMacknessBHow high-density lipoprotein protects against the effects of lipid peroxidationCurr Opin Lipidol200011383810945720
- MaedaKNoguchiYFukuiTThe effects of cessation from cigarette smoking on the lipid and lipoprotein profiles: a meta-analysisPrev Med2003372839014507483
- ManninenVTenkanenLKoskinenPJoint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatmentCirculation19928537451728471
- ManttariMHuttunenJKKoskinenPLipoproteins and coronary heart disease in the Helsinki Heart StudyEur Heart J199011Suppl H26312073911
- MarcilMYuLKrimbouLCellular cholesterol transport and efflux in fibroblasts are abnormal in subjects with familial HDL deficiencyArterioscler Thromb Vasc Biol199919159699888879
- MarchesiMBoothEADavisTApolipoprotein A-IMilano and 1-palmitoyl-2-oleoyl phosphatidylcholine complex (ETC-216) protects the in vivo rabbit heart from regional ischemia-reperfusion injuryJ Pharmacol Exp Ther200431110233115375174
- MartinGDuezHBlanquartCStatin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-IJ Clin Invest200110714233211390424
- MeksawanKPendergastDRLeddyJJEffect of low and high fat diets on nutrient intakes and selected cardiovascular risk factors in sedentary men and womenJ Am Coll Nutr2004231314015047679
- MeroNVan TolAScheekLMDecreased postprandial high density lipoprotein cholesterol and apolipoproteins A-I and E in nor-molipidemic smoking men: relations with lipid transfer proteins and LCAT activitiesJ Lipid Res19983914935029684753
- MezdourHJonesRDengremontCHepatic lipase deficiency increases plasma cholesterol but reduces susceptibility to atherosclerosis in apolipoprotein E-deficient miceJ Biol Chem19972721357059153204
- McDonaldMCDhadlyPCockerillGWReconstituted high-density lipoprotein attenuates organ injury and adhesion molecule expression in a rodent model of endotoxic shockShock200320551714625480
- McNeishJAielloRJGuyotDHigh density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATP-binding cassette transporter-1Proc Nat Acad Sci USA20009742455010760292
- MiaoBZondloSGibbsSRaising HDL cholesterol without inducing hepatic steatosis and hypertriglyceridemia by a selective LXR modulatorJ Lipid Res2004451410715145986
- MillerMKwiterovichPOIsolated low HDL-cholesterol as an important risk factor for coronary heart diseaseEur Heart J199011Suppl H9142073916
- MillerNEThelleDSFordeOHThe Tromso heart-study. High-density lipoprotein and coronary heart-disease: a prospective case-control studyLancet19771965867464
- MineoCDeguchiHGriffinJHEndothelial and antithrombotic actions of HDLCirc Res20069813526416763172
- MoestrupSKKozyrakiRCubilin, a high-density lipoprotein receptorCurr Opin Lipidol2000111334010787174
- MoffattRJEffects of cessation of smoking on serum lipids and high density lipoprotein-cholesterolAtherosclerosis1988748593214483
- MortonREGreeneDJSuppression of lipid transfer inhibitor protein activity by oleate. A novel mechanism of cholesteryl ester transfer protein regulation by plasma free fatty acidsArterioscler Thromb Vasc Biol199717304189409291
- MotojimaKPetersJMGonzalezFJPPAR alpha mediates peroxisome proliferator-induced transcriptional repression of nonperoxisomal gene expression in mouseBiochem Biophys Res Commun199723015589020034
- MRC/BHFHeart Protection Study of cholesterol-lowering therapy and of antioxidant vitamin supplementation in a wide range of patients at increased risk of coronary heart disease death: early safety and efficacy experienceEur Heart J19962072541
- Multiple Risk Factor Intervention TrialRisk factor changes and mortality results. Multiple Risk Factor Intervention Trial Research GroupJAMA19822481465777050440
- NaikSUWangXDa SilvaJSPharmacological activation of liver X receptors promotes reverse cholesterol transport in vivoCirculation200611390716365197
- NanjeeMNCookeCJGarvinRIntravenous apoA-I/lecithin discs increase pre-beta-HDL concentration in tissue fluid and stimulate reverse cholesterol transport in humansJ Lipid Res20014215869311590214
- NaqviTZShahPKIveyPAEvidence that high-density lipoprotein cholesterol is an independent predictor of acute platelet-dependent thrombus formationAm J Cardiol199991011710569655
- NavabMAnantharamaiahGMReddySTApolipoprotein A-I mimetic peptides and their role in atherosclerosis preventionNat Clin Pract Cardiovasc Med20063540716990839
- NavabMAnantharamaiahGMReddySTHuman apolipoprotein A-I and A-I mimetic peptides: potential for atherosclerosis reversalCurr Opin Lipidol200415645915529023
- NavabMBerlinerJAWatsonADThe Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial LectureArterioscler Thromb Vasc Biol199616831428673557
- NissenSETardifJCNichollsSJEffect of torcetrapib on the progression of coronary atherosclerosisN Engl J Med200735613041617387129
- NissenSETsunodaTTuzcuEMEffect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trialJAMA2003290229230014600188
- NobecourtEDaviesMJBrownBEThe impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin: cholesterol acyltransferaseDiabetologia2007506435317216278
- NoferJRLevkauBWolinskaISuppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipidsJ Biol Chem200127634480511432865
- NorataGDCallegariEMarchesiMHigh-density lipoproteins induce transforming growth factor-beta2 expression in endothelial cellsCirculation200511128051115911702
- O’ConnellBJDenisMGenestJHigh-density lipoproteins and endothelial functionCirculation200110419788311602504
- OliverWRShenkJLSnaithMRA selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transportProc Natl Acad Sci20019853061111309497
- OramJFLawnRMABCA1: The gatekeeper for eliminating excess tissue cholesterolJ Lipid Res2001421173911483617
- OutRHoekstraMHildebrandRBMacrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor-deficient miceArterioscler Thromb Vasc Biol200626229530016857950
- PapadakisJAGanotakisESJagroopIAStatin + fibrate combination therapy fluvastatin with bezafibrate or ciprofibrate in high risk patients with vascular diseaseInt J Cardiol199932374410402106
- PauciulloPBorgninoCPaolettiREfficacy and safety of a combination of fluvastatin and bezafibrate in patients with mixed hyperlipidaemia (FACT study)Atherosclerosis20001504293610856536
- PloschTKokTBloksVWIncreased hepatobiliary and fecal cholesterol excretion upon activation of the liver X receptor is independent of ABCA1J Biol Chem200227733870712105210
- PoonMZhangXDunskyKApolipoprotein(a) induces monocyte chemotactic activity in human vascular endothelial cellsCirculation199796251499355887
- QureshiAIGilesWHCroftJBApolipoproteins A-1 and B and the likelihood of non-fatal stroke and myocardial infarction – data from The Third National Health and Nutrition Examination SurveyMed Sci Monit20028CR311612011770
- RimmEBWilliamsPFosherKModerate alcohol intake and lower risk of coronary heart disease: meta-analysis of effects on lipids and haemostatic factorsBMJ19993191523810591709
- RittershausCWMillerDPThomasLJVaccine-induced antibodies inhibit CETP activity in vivo and reduce aortic lesions in a rabbit model of atherosclerosisArterioscler Thromb Vasc Biol20002021061210978256
- RossnerSBjorvellHEarly and late effects of weight loss on lipoprotein metabolism in severe obesityAtherosclerosis198764125303606709
- RubenstrunkAHanfRHumDWSafety issues and prospects for future generations of PPAR modulatorsBiochim Biophys Acta2007 Epub ahead of print
- RubinEMKraussRMSpanglerEAInhibition of early atherogenesis in transgenic mice by human apolipoprotein AINature199135326571910153
- RubinsHBDavenportJBabikianVReduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol. The Veterans Affairs HDL Intervention Trial (VA-HIT)Circulation200110328283311401940
- SacksFMPfefferMAMoyeLAThe effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigatorsN Engl J Med1996335100198801446
- SakaiNMatsuzawaYHiranoKDetection of two species of low density lipoprotein particles in cholesteryl ester transfer protein deficiencyArterioscler Thromb1991117191988006
- SakuKAhmadMGlas-GreenwaltPActivation of fibrinolysis by apolipoproteins of high density lipoproteins in manThromb Res198539184035643
- SavagePDBrochuMAdesPAGender alters the high-density lipoprotein cholesterol response to cardiac rehabilitationJ Cardiopulm Rehabil2004242485415286530
- Scandinavian Simvastatin Survival Study GroupRandomized trial of cholesterol lowering in 4,444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S)Lancet1994344138397968073
- SchaeferJRSchweerHIkewakiKMetabolic basis of high density lipoproteins and apolipoprotein A-I increase by HMG-CoA reductase inhibition in healthy subjects and a patient with coronary artery diseaseAtherosclerosis19991441778410381291
- SchmitzGDrobnikWATP-binding cassette transporters in macrophages: promising drug targets for treatment of cardiovascular diseaseCurr Opin Investig Drugs200238538
- SchmitzGLangmannTHigh-density lipoproteins and ATP-binding cassette transporters as targets for cardiovascular drug therapyCurr Opin Investig Drugs2005690719
- SegrestJPLiLAnantharamaiahGMStructure and function of apolipoprotein A-I and high-density lipoproteinCurr Opin Lipidol2000111051510787171
- ShahPKEmerging HDL-based therapies for atherothrombotic vascular diseaseCurr Treat Options Cardiovasc Med20079607017378977
- ShahPKLow-density lipoprotein lowering and atherosclerosis progression: does more mean less?Circulation200210620394012379568
- ShahPKAminJLow high density lipoprotein level is associated with increased restenosis rate after coronary angioplastyCirculation1992851279851555271
- ShahPKKaulSNilssonJExploiting the vascular protective effects of high-density lipoprotein and its apolipoproteins: an idea whose time for testing is coming, part IICirculation2001104249850211705831
- ShahPKNilssonJKaulSEffects of recombinant apolipoprotein A-I(Milano) on aortic atherosclerosis in apolipoprotein E-deficient miceCirculation19989778059498542
- ShihDMGuLXiaYRMice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosisNature199839428479685159
- SoloffLAIntermediate lipoproteins, atherosclerosis, and GofmanCirculation1998977089495311
- StaelsBDallongevilleJAuwerxJMechanism of action of fibrates on lipid and lipoprotein metabolismCirculation1998982088939808609
- StaelsBVu-DacNKosykhVAFibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibratesJ Clin Invest199595705127860752
- StefanickMLMackeySSheehanMEffects of diet and exercise in men and postmenopausal women with low levels of HDL cholesterol and high levels of LDL cholesterolN Engl J Med199833912209647874
- SuganoMMakinoNSawadaSEffect of antisense oligonucleotides against cholesteryl ester transfer protein on the development of atherosclerosis in cholesterol-fed rabbitsJ Biol Chem1998273503369478952
- SuganoMTsuchidaKMakinoNHigh-density lipoproteins protect endothelial cells from tumor necrosis factor-alpha-induced apoptosisBiochem Biophys Res Commun2000272872610860844
- SugataniJMiwaMKomiyamaYHigh-density lipoprotein inhibits the synthesis of platelet-activating factor in human vascular endothelial cellsJ Lipid Mediat Cell Signal19961373888998599
- SviridovDKingwellBHoangASingle session exercise stimulates formation of pre beta 1-HDL in leg muscleJ Lipid Res200344522612562839
- SviridovDMiyazakiOTheodoreKDelineation of the role of pre-beta 1-HDL in cholesterol efflux using isolated pre-beta 1-HDLArterioscler Thromb Vasc Biol2002221482812231570
- SzaparyPORaderDJThe triglyceride-high-density lipoprotein axis: an important target of therapy?Am Heart J20041482112115308990
- TallARYvan-CharvetLWangNThe failure of torcetrapib: was it the molecule or the mechanism?Arterioscler Thromb Vasc Biol2007272576017229967
- TardifJCEffect of reconstituted high density lipoprotein infusions on coroanry atherosclerosis2007Paper presented at Annual Meeting of the American College of CardiologyMarch 24–27New Orleans, LA
- TaylorAJSullenbergerLELeeHJArterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statinsCirculation20041103512715537681
- TerasakNHiroshimaAKoieyamaTT-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient miceFEBS Lett200353661112586329
- ThorntonJSymesCHeatonKModerate alcohol intake reduces bile cholesterol saturation and raises HDL cholesterolLancet19832819226137650
- Tilly-KiesiMTikkanenMJEffects of lovastatin and gemfibrozil on high-density lipoprotein subfraction density and composition in patients with familial hypercholesterolemiaJ Lab Clin Med19921201103101613316
- TothPPHigh-density lipoprotein as a therapeutic target: clinical evidence and treatment strategiesAm J Cardiol20059650K58K discussion 34K–35K
- US Department of Health and Human ServicesPhysical activity and health: A report of the Surgeon General: Adolescents and young adults fact sheet1999 http://www.cdc.gov/nccdphp/sgr/adoles.htm
- VanceDEVan den BoschHCholesterol in the year 2000Biochim Biophys Acta200015291811111073
- WilsonPWAbbottRDCastelliWPHigh density lipoprotein cholesterol and mortality. The Framingham Heart StudyArteriosclerosis19888737413196218
- Van der GaagMSvan TolAVermuntSHAlcohol consumption stimulates early steps in reverse cholesterol transportJ Lipid Res20014220778311734581
- VaughanAMOramJFABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteinsJ Biol Chem200528030150715994327
- VegaGLMaPTCaterNBEffects of adding fenofibrate (200 mg/day) to simvastatin (10 mg/day) in patients with combined hyperlipidemia and metabolic syndromeAm J Cardiol2003919566012686335
- VenkateswaranALaffitteBAJosephSBControl of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alphaProc Natl Acad Sci2000971209710211035776
- WangMBriggsMRHDL: the metabolism, function, and therapeutic importanceChem Rev200410411193714719972
- WatsonADBerlinerJAHamaSYProtective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoproteinJ Clin Invest1995962882918675659
- West of Scotland Coronary Prevention GroupWest of Scotland Coronary Prevention Study: identification of high-risk groups and comparison with other cardiovascular intervention trialsLancet1996348133913428918276
- WestmanJRoobolCCarlsonLAIn vitro reverse cholesterol transport from THP-1-derived macrophage-like cells with synthetic HDL particles consisting of proapolipoprotein A1 or apolipoprotein A1 and phosphatidylcholineScand J Clin Lab Invest19955523337624733
- WilliamsPTInteractive effects of exercise, alcohol, and vegetarian diet on coronary artery disease risk factors in 9242 runners: the National Runners’ Health StudyAm J Clin Nutr19976611972069356539
- WilliamsPTThe relationships of vigorous exercise, alcohol, and adiposity to low and high high-density lipoprotein-cholesterol levelsMetabolism200453700915164315
- WoodPDStefanickMLDreonDMChanges in plasma lipids and lipoproteins in overweight men during weight loss through dieting as compared with exerciseN Engl J Med1988319117393173455
- YamashitaSMatsuzawaYOkazakiMSmall polydisperse low density lipoproteins in familial hyperalphalipoproteinemia with complete deficiency of cholesteryl ester transfer activityAtherosclerosis1988707123355618
- YuhannaISZhuYCoxBEHigh-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthaseNat Med20017853711433352
- ZeiherAMSchachlingerBHohnloserSHCoronary atherosclerotic wall thickening and vascular reactivity in humans. Elevated high-density lipoprotein levels ameliorate abnormal vasoconstriction in early atherosclerosisCirculation1994892525328205660
- ZhangYZanottiIReillyMPOverexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivoCirculation2003108661312900335