Abstract
Pulmonary hypertension was once thought to be a rare condition and only managed in specialized centers. Now however, with the advent of echocardiography, it is found in many clinical scenarios, in the neonate with chronic lung disease, in the acute setting in the intensive care unit, in connective tissue disease and in cardiology pre- and postoperatively. We have a better understanding of the pathological process and have a range of medication which is starting to be able to palliate this previously fatal condition. This review describes the areas that are known in this condition and those that are less familiar. The basic physiology behind pulmonary hypertension and pulmonary vascular disease is explained. The histopathologic process and the various diagnostic tools are described and are followed by the current and future therapy at our disposal.
Introduction
Pulmonary arterial hypertension (PAH) can be implicated in the morbidity and mortality of many areas of cardiac and noncardiac pathology, and is an important complication in many patients with congenital heart disease.Citation1–Citation3 Traditionally, a diagnosis of PAH was accompanied by a bleak prognosis, with the survival with “primary” pulmonary hypertension being only 2.8 years. However, in recent years, increased awareness of this condition combined with more effective intervention at an early stage has reduced the number of patients with advanced pulmonary vascular disease (PVD), and improved survival rates.Citation1
Definition
Pulmonary arterial hypertension can be defined as an increase in pulmonary arterial pressure (PA pressure) in the pulmonary vascular bed,Citation1 and is defined as a mean PA pressure of more than 25 mmHg at rest or 30 mmHg with exercise.Citation2 However, in clinical practice, echocardiography is often used instead of cardiac catheterization, and thus pulmonary hypertension (PH) is more commonly considered to occur when systolic PA pressure > half systolic systemic pressure.Citation3 This allows for age-related changes since a pressure of 30 mmHg in a 300 g baby has different implications to that in a 70 kg adult. PA pressure is formed by a combination of pulmonary blood flow and pulmonary vascular resistance, known as a modification of Ohm’s law (V = I × R) or Darcy’s law.Citation4 In the fetus, the pulmonary vascular resistance (PVR) is high, which maintains a high mean PA pressure and so blood is preferentially shunted from the pulmonary artery to the systemic circulation via the arterial duct. Within a few days of birth, the PVR falls rapidly, leading to a consequent fall in PA pressure.Citation5 However, if there is a disease process which falls to allow the PVR to fall or if there is a pathological increase in either pulmonary blood flow or pulmonary vascular resistance, pulmonary hypertension will be maintained or will recur at a later age.
Causes of pulmonary hypertension
As the Venice classification states,Citation6 pulmonary hypertension can be divided into a number of causes, including pulmonary arterial hypertension, pulmonary venous hypertension, and pulmonary hypertension due to respiratory causes. This can be simplified for clinical practice in pediatrics (Table ).Citation3 One of the main distinctions is between primary or idiopathic pulmonary arterial hypertension (IPAH), which is extremely rare in pediatric patients, and secondary pulmonary hypertension, which constitutes the remainder of the causes. Of these secondary causes, congenital heart disease with a large left to right shunt secondary to a VSD or AVSD and chronic lung disease are the most common after the first few days of life.Citation3
Table 1 Causes of pulmonary hypertension in pediatrics
Pathogenesis
The histopathologic changes seen in PH are characterized by a process of vascular remodeling, which varies somewhat depending on the etiology. This involves proliferation of smooth muscle cells into peripheral (usually nonmuscular) arteries,Citation7 together with medial hypertrophy in normally muscular arteries. Impaired growth and loss of arterioles leads to a reduction in arterial density, and this is eventually accompanied by dilatation complexes, plexogenic lesions and fibroid necrosis.Citation8 In turn, this leads to a process of luminal obliteration.Citation9,Citation10 It is generally accepted that the early stages of this are reversible and the later stages are not.
Clinical features
Many of the symptoms of pediatric PH are nonspecific, and clinical features may be subtle even in advanced disease.Citation11 At birth, children commonly have cyanosis with hepatomegaly, an active right ventricle on palpation and a loud pulmonary second sound on auscultation.Citation3 This persistent pulmonary hypertension of the newborn (PPHN) may be difficult to treat but is well recognized in neonatal units and may improve with therapy to allow discharge home. If IPAH is left untreated, the most common presenting symptom is breathlessness, and children frequently present with poor appetite, faltering growth, lethargy, tachypnea, tachycardia, and irritability.Citation11,Citation12 Symptomatic severity has been connected to prognosis, reinforcing the need for early diagnosis and management.Citation13 The child with congenital heart disease and a post-tricuspid shunt is however completely different. They may present late (especially if they have upper airway obstruction as in Down’s syndrome) or may have inoperable or only partly palliated disease (such as pulmonary atresia with ventricular septal defect and aorto-pulmonary collateral arteries). Hence a systematic approach to investigation is required in order to identify those patients with PH.
Clinically, the severity of PH is assessed according to a modification of the New York Heart Association (NYHA)/World Health Organization (WHO) classification of functional capacity (Table ).Citation14
Table 2 Functional classification of pulmonary hypertension
Diagnosis and investigation
A summary of the investigations recommended in the current British Cardiac Society (BCS) GuidelinesCitation15 is shown in Table .
Table 3 Imaging investigations recommended for the assessment of PH
A chest radiograph may show pulmonary oligemia, with pruning of peripheral vesselsCitation16 there may be right ventricular hypertrophy (RVH) on electrocardiogram (ECG), and a low partial pressure of oxygen (PaO2) on arterial blood gas measurement. Echocardiography is a useful and sensitive investigation to identify possible secondary causes of PAH (such as ASD [rarely], VSD or cardiomyopathy), to provide a numerical assessment of the tricuspid regurgitation (TR) jet, and to assess left ventricular (LV) function. However, the results of derived formulae from echocardiography are to some extent operator-dependent. For older children, a six-minute walk test (6MWT) is a standardized method for assessing exercise tolerance in children,Citation17 and it correlates well with the WHO functional class system.
Cardiac catheterization remains the diagnostic gold standard for PH. Current UK guidelines state that right heart catheterization is essential in the investigation of new patients with suspected PH, and should be undertaken to assess specific and accurate measurements of PA pressure and PVR.Citation15 Vasodilator testing with inhaled nitric oxideCitation16 or prostacyclinCitation17 can also be carried out at the time of catheterization to assess the degree of reversibility of PH. In recent years, magnetic resonance imaging (MRI) has enabled detailed visualization of cardiac anatomy and pulmonary blood flow,Citation20 and may even be used to assess degree of vessel compliance.Citation21
Control of pulmonary vascular resistance
There are a number of different pathways involved in the control of PVR, many of which are therapeutic targets in the management of PAH, and are therefore essential to an understanding of the management of this condition (Table ).
Table 4 Summary of mechanisms in PHT
Patients with PAH have increased levels of circulating endothelin-1.Citation22,Citation23 As well as causing vasoconstriction, endothelin-1 leads to smooth muscle and fibroblast proliferation via endothelin A (ETA), and/or endothelin B (ETB) receptors.Citation24 Serotonin levels are also raised in PAH, which stimulates mitogenesis of vascular cells, and increased expression of the serotonin transporter is found in hypertensive arteries.Citation25 Patients with severe PAH have a relative deficiency of vasodilator pathways; they produce less endogenous prostacyclin, have reduced nitrogen oxide synthase (NOS) expression, and reduced vasoactive intestinal peptide (VIP) in the lungs.Citation26,Citation27
Prevention of pediatric pulmonary hypertension
Despite the advent of more efficacious therapeutics, prevention of pediatric pulmonary hypertension remains a priority. Patients with congenital heart defects secondary to a left to right shunt lesion should undergo early surgery to prevent development of PVD. This is particularly crucial in those patients with an AVSD or VSD.Citation28 Surgical and post-operative care is also crucial, and improvements in this area are likely to be a contributing factor in the declining incidence of postoperative pulmonary hypertension in children following cardiac surgery.Citation29 This involves adequate ventilation, chest physiotherapy, and, if necessary, antibiotics.
It is also essential to maintain good oxygenation, relatively low CO2 and pH towards the upper limit of normal, in order to reduce the PA pressure as much as possible. Consequently, the effectiveness of pulmonary vasodilatation will also be maximized. Use of sedation with fentanyl and clonidine should also be used as prophylaxis against pulmonary hypertensive crises.Citation30
Treatment of pediatric pulmonary hypertension
Current guidelines recommend that patients with PH should be managed by an experienced multiprofessional team at a specialist centre, with appropriate expertise and support for children and their families. Long-term community care involving clinical nurse specialists is also beneficial.Citation15
Response to treatment is less predictable in children, and therefore, close monitoring and rapid alteration of treatment as necessary is required in pediatric patients.Citation15 Research has suggested that prompt medical treatment of PAH, even in patients with established disease and Eisenmenger’s syndrome, may reduce the need for lung transplantation.Citation31 There are fewer therapeutic options currently available for patients with PH in comparison to those with PAH.
Initially, treatment of PH involves thorough investigation of potential underlying causes; treatment will thus be directed accordingly. For example, patients whose PAH is caused by upper airway obstruction might undergo adenotonsillectomy, and those with cystic fibrosis, asthma or bronchopulmonary dysplasia should be managed with the relevant therapeutics. Drugs with a propensity to cause vasoconstriction such as sympathomimmetic decongestants with α-adrenergic properties should be avoided in children with pulmonary hypertension.
It is important to note that UK guidelines differ from those in other parts of the world; the American College of Chest Physicians (ACCP) and the European Society of Cardiologists (ESC) suggest that those in NYHA class III or IV should be treated with a specific oral therapy.Citation32,Citation33
Recent guidelines have suggested a treatment algorithm for the management of pediatric PAHCitation15 (Figure ).
Figure 1 Current British Cardiovascular Society (BCS) algorithm for the management of PAH in children.
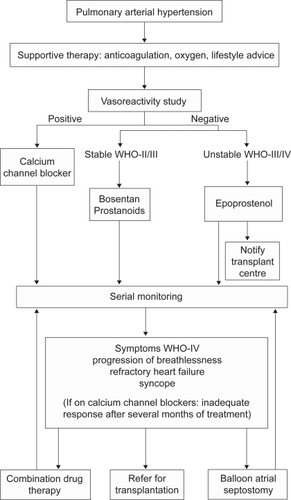
Treatment of acute disease
Children presenting with syncope, right heart failure or post-operative PAH must be diagnosed and treated promptly and safely.Citation34 This may be in an inhaled form (see Nitric oxide, below), orally with sildenafil, intravenously (see Epoprostenol, below), or with hemodynamic support (see Extracorporeal membranous oxygenation, below).Citation3
Oxygen
Good ventilation in the intensive care unit is essential (see Treatment of pediatric pulmonary hypertension, above), and oxygen therapy has been shown to be of benefit acutely for both hypoxic and nonhypoxic patients with PAH.Citation15 Additionally, some patients may benefit from the use of domiciliary nocturnal oxygen therapy, both acutely, and alongside maintenance therapy.Citation35
Nitric oxide
Nitric oxide (NO) inhalation is currently among the first line treatments for post-operative pediatric pulmonary hypertension or in the child with acute severe new presentation of PAH being managed on the intensive care unit,Citation36,Citation37 as it reduces PA pressure rapidly.Citation38 Inhaled NO therapy is also used acutely in persistent pulmonary hypertension of the newborn.Citation39 The mode of action of NO is via the stimulation of guanylyl cyclase and hence increased production of cyclic guanylate monophosphate (cGMP) in pulmonary smooth muscle cells; this causes uptake of calcium into the sarcoplasmic reticulum, which then leads to muscle relaxation, reduced PA pressure and PVR, and hence increased oxygenation. Any NO absorbed from the lungs into the systemic circulation is quickly deactivated by combination with hemoglobin, minimising any effect on the systemic circulation. Therefore, NO acts solely on the pulmonary vasculature, leaving systemic arterial pressure unaffected.Citation40,Citation41
Oxygenation index (OI) is a calculation combining fraction of inspired oxygen (FiO2), mean airway pressure and PaO2 as an indicator of degree of respiratory compromise;Citation42 OI of more than 25 is the usual indication for initiation of treatment with NO.Citation43 Acutely, doses start at 20 parts per million (ppm),Citation44 although post-operative PAH may be treated with doses as low as 3–10 ppm.Citation45 Sustained high doses of NO may lead to methemoglobinemia,Citation45,Citation46 and acute withdrawal of treatment may precipitate rebound PAH.Citation44 The latter may be avoided by the use of phosphodiesterase inhibitors (see below), prostacyclins (see below) or discontinuation of NO followed by resumption of treatment at half the dose.Citation47 Despite this, there is no clear evidence that short-term use of NO causes significant cardiac and respiratory complications.Citation48
NO administration has been shown to reduce the need for extracorporeal membrane oxygenation (ECMO) treatment in term or near term neonates with hypoxic respiratory failure.Citation42 However, a recent Cochrane review found that there was no significant difference in short-term post-operative mortality or mean PA pressure with administration of inhaled NO compared to placebo or conventional management (hyperventilation, use of sodium bicarbonate, intravenous inotropes and vasodilatory agents, and sedatives).Citation49 Current European guidance suggests that there is insufficient evidence at present to recommend the use of prophylactic post-operative inhaled NO in patients with congenital heart disease at risk of PAH.Citation50 However, there is sufficient evidence to support a trial of NO therapy (with a starting dose of 20 ppm, increasing to 40 ppm if there is no response) in patients with significant peri-operative PAH.Citation50 Treatment should be discontinued after 30 minutes if there has been no clinically significant response.
Extracorporeal membrane oxygenation (ECMO)
The use of extracorporeal membrane oxygenation (ECMO) has been associated with complications including interstitial and alveolar hemorrhage and secondary epithelial alterations.Citation51 For this reason, since the establishment of inhaled NO therapy as a treatment for pediatric PAH, the use of ECMO has significantly decreased.Citation52 Despite this, there are still some incidences in which ECMO therapy is necessary, such as those infants with severe respiratory or cardiac disease in addition to pulmonary arterial hypertension; one study of children with hypoxemic respiratory failure found that treatment with NO alone was successful in only 29% cases.Citation53
Maintenance therapies
Aims of maintenance therapies for pediatric pulmonary hypertension will generally be to lower PA pressure in order to reduce or reverse the rate of progression of PVD, and thus to obtain functional improvement in terms of increased activity levels.
Current guidelines state that the choice of therapy should be based on WHO functional class and response to vasodilator testing during cardiac catheterization.Citation15 The age of the child is also an important factor, as several drugs used to treat adults are unsuitable for use in young children; subcutaneous treprostinil frequently causes significant pain, and unwell children may be unable to tolerate inhaled doses of iloprost every 2–3 hours.Citation15
In the UK, current statistics suggest that only a minority of patients are responsive to vasoreactivity studies and treatment with calcium channel antagonists (see below).Citation15 Therefore, the majority of children are treated with specific disease-targeted therapies, such as intravenous epoprostenol (the only drug for children tested in a placebo-controlled trial),Citation54 bosentan,Citation55 and sildenafil. Choice of drug is dependent upon a number of factors, including side effect profile, route of administration, patient preferences, and physician experience.Citation33
Calcium channel antagonists
The calcium channel antagonist, nifedipine is one of the oldest agents used for PAH,Citation56,Citation57 and amlodipine has been suggested to be beneficial for IPAH.Citation58 An initial trial of these agents should be carried out, and calcium channel antagonists should only be continued in those patients who demonstrate an adequate vasodilator response (defined by a decrease in mean PA pressure and PVR by >20%).Citation59 These patients may need no further medication.Citation56,Citation58 However, calcium channel antagonists are only efficacious in 5%–10% of children and adults, whereas nonresponders demonstrate a continually rising PVR. Some patients may switch from being ‘responders’, demonstrating a stable response to calcium channel blockers for a period of several years, to ‘nonresponders,’ who suddenly require additional medication.
Phosphodiesterase inhibitors
Sildenafil, a phosphodiesterase inhibitor, given orally, has been shown to be a potent and selective pulmonary vasodilator.Citation60 Sildenafil works by selectively inhibiting phos-phodiesterase V (PDE V),Citation61 which is responsible for cGMP breakdown in lung tissue. The resultant increase in cGMP leads to calcium-mediated relaxation of vascular smooth muscle. These effects on pulmonary vasculature appear to occur independently of the cause of PAH, suggesting a role in the management of IPAH.Citation62
An initial small-scale pilot study suggested that sildenafil improves hemodynamics and exercise capacity in children with primary pulmonary hypertension and PAH secondary to congenital heart disease.Citation63 Since then, research has suggested that sildenafil works by improving endothelial-dependent vasodilatation, and by reducing plasma concentrations of endothelin-1 and von Willebrand factor, mediating a fall in PA pressure and PVR.Citation64 In IPAH and Eisenmenger’s syndrome, there are reduced levels of endothelial progenitor cells; sildenafil has been hypothesized to increase numbers of these cells in the long term, thereby reducing endothelial dysfunction.Citation65 Another study found that patients with PAH secondary to congenital heart defects treated with sildenafil had improved exercise capacity, WHO functional class and hemodynamics.Citation66 Similarly, Baharani and colleagues found that patients with CHD had significant improvements in six-minute walking distance (6-MWD) and systolic PA pressure when treated with sildenafil in comparison with placebo.Citation67 Sildenafil has been suggested to improve mean PA pressure, PVR, and exercise tolerance in PAH.Citation68 Research has suggested that there is a direct dose-response relationship between sildenafil and exercise capacity, measured by 6-MWD; one study observed a significant improvement in 6-MWD in patients taking 50 mg sildenafil three times daily (tds) compared to 25 mg tds or 12.5 mg tds.Citation69
Sildenafil has also been shown to work synergistically with NO, enhancing the efficacy of exogenous NO, which further increases vasodilatation.Citation70 This combined approach enables transfer of patients from intensive care, and facilitates weaning from high doses of nitric oxide, by reducing the rebound effect commonly seen with discontinuation of inhaled NO.Citation18,Citation71 In the long-term, sildenafil demonstrates marked down-regulatory responses which may limit its use,Citation67 although this may be lessened when administered alongside NO. Side-effects of sildenafil may include nausea, abdominal discomfort, headache, dizziness and flushingCitation72 and potentially, in the long-term, memory loss.Citation73
More recently, research has suggested that another PDE type 5 inhibitor, vardenafil, may be more effective than sildenafil in vitro.Citation74,Citation75 These studies have suggested that vardenafil acts directly to reduce calcium influx in the pulmonary artery, in addition to its vasodilatory effects via cGMP.
Endothelin receptor antagonists
Endothelin 1 (ET-1) acts on ETA and ETB receptors to promote mitosis of pulmonary artery smooth muscle cells; activation of ETA and ETB on smooth muscle cells causes vasoconstriction, whereas activation of ETB on the endothelial cell releasing NO causes vasodilatation. This is thought to contribute significantly to the imbalance between vasodilatation and vasoconstriction in pulmonary hypertensionCitation76 and high levels of ET-1 have been found in the lung and circulation of patients with PAH.Citation77,Citation78
Bosentan, a nonselective (endothelin receptor A and B) antagonist has been shown to reduce mean PA pressure and PVR, and increase quality of life in patients with IPAH.Citation17 One study found that in pediatric patients with congenital heart disease or connective tissue disease, treatment with bosentan for a median period of 14 months improved WHO functional class in 46%.Citation79 Bosentan has also been found to be effective in patients with Eisenmenger’s syndrome, reducing PA pressure and PVR and improving exercise capacity, without reducing oxygen saturations (the BREATHE-5 trial).Citation80,Citation81 However, bosentan has been shown to cause hepatic dysfunction in some patients; a recent study identified deranged liver function tests in 2.7% of children under 12 years of age on bosentan therapy, compared to 7.8% of those aged over 12 years.Citation82 There is some evidence to suggest that in the long-term, there is a progressive decline in the beneficial treatment effects of bosentan, particularly in children.Citation83
Sitaxsentan, a more ETA selective ERA has been investigated in recent trials. Research has suggested that that it may play an important role in the management of PAH associated with connective tissue diseases, and it may have a more prolonged action than bosentan in children with congenital heart disease.Citation84,Citation85 One study of 247 patients found that use of sitaxsentan 100 mg once daily improved exercise capacity and WHO functional class in adult patients with PAH, and it has reduced hepatic toxicity compared to bosentan.Citation86 As well as bosentan, sitaxsentan has also been shown to be effective in patients with Eisenmenger’s syndrome.Citation87 Data on the use of sitaxsentan is limited in children at the current time.
Ambrisentan, another selective ERA antagonist has recently been shown to improve exercise capacity in patients with PAH compared to placebo, and is well-tolerated.Citation88
Prostacyclin agonists
Prostacyclin (PGI2) is an endogenous vasodilatory mediator in the pulmonary vasculature; prostacyclin agonists act via cyclic AMP-dependent pathways in smooth muscle cells. Its effects include reducing PVR, inhibiting platelet aggregation, and reducing smooth muscle cell proliferation. In patients with pulmonary hypertension, pulmonary endothelial cells have decreased expression of prostacyclin synthaseCitation89 and urinalysis demonstrates a decrease in stable prostacyclin metabolites.Citation26
Preliminary results of treatment with prostacyclins have been promising. Barst and colleaguesCitation90 found a 90% survival rate at four years for children with severe IPAH treated with prostacyclin, and studies have suggested that prostacyclins improve hemodynamic function and quality of life.Citation91 Prostacyclins have been shown to work synergistically with NO, since they act through a different pathway and hence the therapeutic effects are additive. Therefore, these drugs may also be used when NO treatment has failed, when weaning from NO (as also occurs with sildenafil), or when therapeutic resistance has occurred.Citation47,Citation92 They may be administered intravenously (epoprostenol, treprostinil), by inhalation (iloprost) or orally (beraprost). However, the side-effects of prostacyclin agonists have limited their use to some extent; adverse events associated with epoprostenol include flushing, jaw pain, headaches, rashes and thrombocytopenia.Citation93 Epoprostenol requires a continuous infusion and has a short half-life (<6 minutes). Interruption of the infusion can lead to rapid increases in PVR, hemodynamic collapse and death.Citation93
Epoprostenol has been shown to improve the survival of patients with IPAH in the long-term,Citation94 and is the gold standard starting treatment for patients with severe IPAH. It also has a role acutely in neonates with persistent pulmonary hypertension of the newborn (PPHN), post-operatively in children with heart disease, and in patients with IPAH, to reduce PVR prior to cardiac catheterization. However, epoprostenol has dose-limiting effects, and may cause severe rebound pulmonary arterial hypertension.Citation95
Treprostinil was originally used as a subcutaneous infusion, which has a longer half-life and increased stability, so does not have to be kept in cooled storage.Citation96 However, this route of administration is extremely painful, which has limited its use in the pediatric population. In recent research, intravenous preparations of treprostinil have been investigated, which have been suggested to cause fewer adverse effects than epoprostenol. However, whilst successful transfer rates have been achieved, high central-line infection rates were seen.Citation97
Beraprost, an oral preparation, is well-tolerated and widely used in parts of Europe and in Japan, but research has suggested it is less efficacious than intravenous preparations.Citation98,Citation99
The inhaled form, iloprost, is easy to administer, has fewer side-effects as it acts directly on the lungsCitation100 and it avoids the potential risks of infection seen with indwelling central lines.Citation95 Iloprost must be administered approximately every two hours, and therefore has a greater role in the acute setting. It has been shown to be effective in improving hemodynamic parameters, exercise tolerance and quality of life in patients with PAH.Citation101,Citation102
Novel therapies
Rho-kinase inhibitors
Developments in recent years have suggested that a new class of drug, rho kinase inhibitors, may be beneficial in the treatment of pediatric PAH.Citation103,Citation104 Rho kinase causes vasoconstriction of vascular smooth muscle through phosphorylation and consequent inhibition of myosin phosphatase.Citation105 It has also been shown that rho kinase acts by activation of the enzyme myosin light chain kinase (MLCK), which causes phosphorylation of the hyper-constrictive segments of arteries in vitro.Citation106 Animal models have suggested that activation of rho kinase is associated with pulmonary vasoconstriction and proliferation, impaired endothelial vasodilatation and pulmonary remodelling, and that administration of its antagonists reverse these processes.Citation107,Citation108 Preliminary results have suggested that administration of intravenous fasudil, a selective rho-kinase inhibitor, may cause acute pulmonary vasodilatation and reduction in PA pressure in patients with severe PAH refractory to other therapies.Citation103,Citation109
Vasoactive intestinal peptide
Recent research investigating the role of vasoactive intestinal peptide (VIP) in rats has produced promising results.Citation110 This study showed that ETA antagonists, ETB antagonists, and VIP all prevented the pulmonary vasoconstriction caused by ET1. Unlike ETA and ETB antagonists, VIP did not induce an increase in airway resistance and thus this peptide may exert protective effects against both the vascular and bronchial adverse effects of ETI. This may therefore have a role in patients with PAH and co-existing chronic lung disease. Clearly therefore, in vivo experimentation is now needed.
Estradiol derivatives
There has been limited investigation into the use of 2-ethoxyestradiol, a nonestrogenic metabolite of estradiol in the treatment of PAH.Citation111 This study found that 2-ethoxyestradiol lowered right ventricle (RV) peak systolic pressure and consequently led to reduced vascular remodelling and mortality in rats with PAH. This suggests that in vitro anti-proliferative agents including synthetic analogues of estradiol metabolites may be protective against development of PVD.
Apoptosis and gene therapy
Limited research has suggested that modulation of the process of apoptosis (programmed cell death) involved in the process of vascular remodelling in PAH could be one possible therapeutic opportunity.Citation112,Citation113 For example, survivin (also known as Birc5), is one of a family of genes known to inhibit apoptosis; the use of molecular antagonists of survivin to increase cell death and prevent vascular remodelling may, in the future, hold therapeutic potential.Citation114 Similarly, preliminary research has suggested that the use of the 3-hydroxy-3-methyl-glutaryl-CoA (HMG CoA) reductase inhibitor, pravastatin, and the Cox-2 inhibitor, celecoxib, prevent the development of monocrotaline-induced PAH in rats.Citation115,Citation116 Simvastatin has been found to be ineffective in this respect in vitro.Citation117
Serotonin pathways
Research has found that patients with PAH have increased plasma serotonin levels,Citation118 and that over-expression of the serotonin transporter gene (SERT) increases PA pressure.Citation119 Hypoxia and monocrotaline-induced PAH in animals has been shown to be inhibited by the selective serotonin reuptake inhibitor (SSRI), fluoxetine.Citation120,Citation121 So far however, studies in humans have not produced statistically significant results.Citation122
l-Arginine
An alternative approach to inhaled NO therapy is to increase endogenous NO synthesis. Plasma l-arginine is a substrate for endothelial nitrogen oxide synthase,Citation123 and experimentalCitation124 and clinicalCitation125 data have suggested that patients with a left-to-right shunt lesion may be deficient in l-arginine, both pre- and post-operatively. However, research has suggested that low levels of plasma arginine do not correlate with the degree of post-operative PAHCitation126. Further trials are needed to ascertain whether peri-operative therapy with l-arginine may be beneficial.
Anticoagulation
Whilst most children with PAH will not be treated with anticoagulation, it is recommended that those at high risk of thromboembolism (such as patients with IPAH and reduced cardiac output, indwelling venoatrial shunt or severe poly-cythemia), should be prescribed warfarin.Citation127
Other therapies
Although rare, children with systemic sclerosis and coexisting pulmonary hypertension should be treated with corticosteroids, which appear to slow disease progression in this group.Citation128
Combination treatment
Currently, there is some debate as to the optimal management of patients who demonstrate clinical deterioration despite maximal targeted therapy with one agent. The use of combination therapies has been widely adopted across the US and Europe, but currently, there is little clear consensus as to the most effective and safe combinations.Citation129 In Switzerland and other countries, combination therapies involving two or three of bosentan, iloprost, and sildenafil are commonly used but their relative merits are not yet clarified.Citation130
Combination of sildenafil with an endothelin receptor antagonist (notably bosentan) has been recently validated in several trials, improving hemodynamic variables, exercise capacity, and quality of life in patients with IPAH or PAH unresponsive to monotherapy.Citation131–Citation133 Limited trials of treatment with bosentan and a prostacyclin agonist such as epoprostenol or iloprost have suggested this may be an effective combination.Citation134,Citation135 Inhaled prostacyclin and milrinone have also been postulated to work synergistically to reduce PVR.Citation136
However, the pharmacokinetics involved are complex; at steady state, when used in combination, bosentan reduces the maximum plasma concentration of sildenafil, and sildenafil increases the concentration of bosentan.Citation137 Further research is therefore needed to establish the clinical applications for these complex drug interactions. It is also important to note that dual treatments combine the adverse effects of both drugs, and thus the incidence of side-effects may be increased.Citation138
Management of refractory pulmonary hypertension
Those patients who remain symptomatic despite medical therapies may require more invasive forms of management.
Atrial septostomy
Children with PAH and without adequate right to left shunting across the atria commonly develop recurrent syncopal episodes.Citation12 Atrial septostomy, either by cardiac catheterization or surgically, has been shown to be beneficial in patients experiencing recurrent syncope, by creating a left to right shunt and consequently maintaining cardiac output.Citation139 In turn, this procedure has been suggested to reduce the signs and symptoms of right heart failure.Citation141–Citation144 Improved survival rates have also been demonstrated following atrial septostomy; Kerstein and colleaguesCitation139 demonstrated increased survival at one and two years, 87% and 76%, respectively.
Lung transplantation
Lung transplantation is currently only considered as a last resort,Citation145 in part due to concerns regarding long-term survival rates; one study found that survival rates were 77% at one year, 62% at two years, and 55% after five years.Citation146 Since 1986, over 1055 children have undergone transplantation worldwide.Citation147 Between January 1990 and June 2006, there were 977 pediatric lung transplantation; of these, the primary indication in 104 patients (10.6%) was IPAH, 34 (3.5%) due to CHD, 18 for PVD (1.8%), and 17 (1.7%) for Eisenmenger’s syndrome.Citation147 However, the use of lung transplantations is restricted by waiting times, risks of surgery, and the problems accompanying transplant rejection, and thus cannot be considered as a viable treatment option earlier in the course of disease.
Treatment outcomes and prognosis
Currently, there is no PAH-specific quality of life measure, although recent studies of outcomes in pediatric patients suggest that although physical activity is limited to approximately 50% of normal, psychological scores are 80%–90% of normal.Citation147,Citation148 These scores do not correlate with age, time since diagnosis, PVR or cause of PAH.
Treatment with disease-targeted therapies for secondary and idiopathic PAH has been shown to improve survival in a number of studies;Citation15,Citation129 a number of recent trials have investigated the relative efficacy of management options in pulmonary arterial hypertension (Table ).Citation149
Table 5 Key trials of pharmacological agents for the management of pediatric PAH
A recent meta-analysis of trials of modern treatment modalities including phosphodiesterase inhibitors, endothelin receptor antagonists and prostacyclin agonists found that overall, active treatment resulted in reduction in mortality of 43% (risk ratio 0.57, 95% confidence interval [CI] 0.35–0.92; p = 0.023) (Figure ).Citation149
Figure 2 Meta-analysis of active treatment versus placebo in trials of current management strategies in pulmonary arterial hypertension.
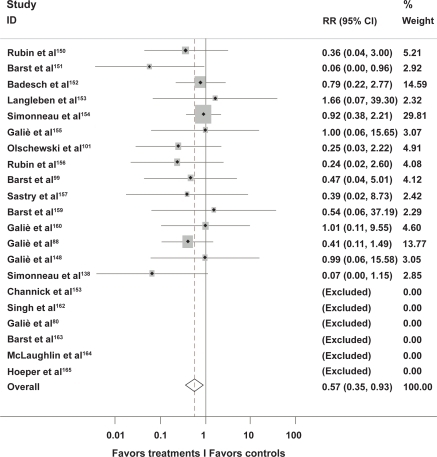
Survival rates are typically higher in those with secondary PAH compared to IPAH, and for those with IPAH, combination therapy is more effective than monotherapy.Citation141,Citation166 Children with postoperative PAH have previously had particularly poor outcomes, and therefore early detection in these patients is crucial.Citation15 There is currently limited evidence regarding the optimal management of patients with pulmonary venous hypertension, and for patients with other forms of pulmonary hypertension.
Conclusion
Advances in available therapeutics for pulmonary hypertension have improved survival in pediatric patients. General supportive care, early diagnosis and prompt surgery for patients with PAH secondary to CHD is crucial. An individualized approach must be taken in deciding treatment options, depending on the symptoms, clinical signs, and hemodynamic status of each patient.
Following this, treatment options are complex and current BCS guidelines suggest that treatment should universally depend upon the NYHA functional class and response to vasodilator therapy.Citation15 Acutely, treatment options include oxygen therapy, NO and ECMO. Current maintenance therapies include calcium channel antagonists, phosphodiesterase inhibitors, endothelin receptor antagonists and prostacyclin agonists, with or without anticoagulation. For patients who remain symptomatic or whom demonstrate equivocal hemodynamic response to therapy with a single agent, combination therapy may be considered. Atrial septostomy and lung transplantation are generally considered as the next stage in patients who have been unresponsive to medical therapy. A recent systematic review of oral treatment methods in PAH highlighted the problems of small sample size and limited follow-up period in many of the available studies this is particularly problematic in pediatric populations.Citation167 More in vivo research is needed to assess the efficacy of the increasing number of novel therapies.
Disclosure
Dr. Hawkins reports no conflict of interest in this work. Dr. Tulloh has received lecture fees and honoria from Actelion, Pfizer and GlaxoSmithkline.
References
- AndrewsRPulmonary hypertension in pediatricsCurr Opin Ped200214603605
- GibbsJRecommendations on the management of pulmonary hypertension in clinical practiceHeart200186Suppl 111311410544
- TullohRCongenital heart disease in relation to pulmonary hypertension in paediatric practicePaediatr Respir Rev2005617418016153566
- FreedMDInvasive diagnostic and therapeutic techniques Part 1: cardiac catheterisationAdamsFHHeart Disease in Infants, Children and AdolescentsLondonWilliams and Wilkins2000130146
- AndrewsRAtrial septal defect with failure to thrive in infancy: hidden pulmonary vascular disease?Pediatr Cardiol20022352853012211202
- SimonneauGClinical classification of pulmonary hypertensionJ Am Coll Cardiol20044312 Suppl S5S12S15194173
- RabinovichMPathobiology of pulmonary hypertensionAnnu Rev Pathol2007236939918039104
- RabinovitchMVascular structure in lung tissue obtained at biopsy correlated with pulmonary hemodynamic findings after repair of congenital heart defectsCirculation1984696556676697454
- PietraGGHistopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension RegistryCirculation198980119812062805258
- JefferyTKMolecular and cellular basis of pulmonary vascular remodelling in pulmonary hypertensionProg Cardiovasc Dis20024517320212525995
- RichSPrimary pulmonary hypertension. A national prospective studyAnn Intern Med19871072162233605900
- WidlitzAPulmonary arterial hypertension in childrenEur Respir J20032115517612570125
- D’AlonzoGESurvival in patients with primary pulmonary hypertension. Results from a national prospective registryAnn Intern Med19911153433491863023
- RichSClinical insights into the pathogenesis of primary pulmonary hypertensionChest19981143 Suppl237S241S9741575
- National Pulmonary Hypertension Centres of the UK and IrelandConsensus statement on the management of pulmonary hypertension in clinical practice in the UK and IrelandThorax200863ii1ii4118308974
- ManesAPulmonary hypertension: classification and diagnostic algorithmItal Heart J2005683483916270476
- MaiyaSResponse to bosentan in children with pulmonary hypertensionHeart20069266467016216850
- AtzAMCombined effects of nitric oxide and oxygen during acute pulmonary vasodilator testingJ Am Coll Cardiol19993381381910080486
- MikhailGAn evaluation of nebulized prostacyclin in patients with primary and secondary pulmonary hypertensionEur Heart J1997189149915049458458
- MuthuranguVNovel method of quantifying pulmonary vascular resistance by use of simultaneous invasive pressure monitoring and phase-contrast magnetic resonance flowCirculation2004110782683415302793
- MuthuranguVMeasurement of total pulmonary arterial compliance using invasive pressure monitoring and MR flow quantification during MR-guided cardiac catheterizationAm J Physiol Heart Circ Physiol20052893H1301H130615879483
- HoeperMMGoal-oriented treatment and combination therapy for pulmonary arterial hypertensionEur Respir J20052685886316264047
- LanglebenDEndothelin-1 in acute lung injury and the adult respiratory distress syndromeAm Rev Respir Dis1993148(6 Pt 1):164616508256914
- DavieNET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cellsAm J Respir Crit Care Med2002165339840511818328
- HervéPIncreased plasma serotonin in primary pulmonary hypertensionAm J Med19959932492547653484
- ChristmanBWAn imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertensionN Engl J Med1992327270751603138
- PetkovVVasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertensionJ Clin Invest200311191339134612727925
- BandoKPulmonary hypertension after operations for congenital heart disease: analysis of risk factors and managementJ Thorac Cardiovasc Surg1996112616001607discussion 160716098975852
- LindbergLHow common is severe pulmonary hypertension after pediatric cardiac surgery?J Thorac Cardiovasc Surg200212361155116312063463
- OhsumiHEffects of fentanyl on carotid sinus baroreflex control of circulation in rabbitsAm J Physiol1989256(3 Pt 2):R625R6312493749
- AdriaenssensTAdvanced therapy may delay the need for transplantation in patients with the Eisenmenger syndromeEur Heart J200627121472147716707548
- BadeschDBMedical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelinesChest200713161917192817565025
- GalièNGuidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of CardiologyEur Heart J200425242243227815589643
- TaylorCJRisk of cardiac catheterization under anaesthesia in children with pulmonary hypertensionBr J Anaesth200798565766117401143
- SandovalJNocturnal oxygen therapy in patients with the Eisenmenger syndromeAm J Respir Crit Care Med200116491682168711719310
- SteudelWInhaled nitric oxide: basic biology and clinical applicationsAnesthesiology19999141090112110519513
- AdatiaIEffect of intracardiac repair on biosynthesis of thromboxane A2 and prostacyclin in children with a left to right shuntBr Heart J19947254524567818962
- BeghettiMContinuous low dose inhaled nitric oxide for treatment of severe pulmonary hypertension after cardiac surgery in paediatric patientsBr Heart J199573165687888265
- DavidsonDInhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: a randomized, double-masked, placebo-controlled, dose-response, multicenter study. The I-NO/PPHN Study GroupPediatrics1998101(3 Pt 1):3253349480993
- ThébaudBInhaled and exhaled nitric oxideCell Mol Life Sci1999558911031112
- LowsonSMAlternatives to nitric oxideBr Med Bull20047011913115531733
- OrtitzRMExtracorporeal membrane oxygenation in pediatric respiratory failurePediatr Clin North Am198734139463808772
- The Neonatal Inhaled Nitric Oxide Study GroupInhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failureNew Eng J Med19973365976049036320
- KinsellaJPInhaled nitric oxide therapy in childrenPaediatr Respir Rev20056319019816153568
- AbmanSHNeonatal pulmonary hypertension: a physiologic approach to treatmentPediatr Pulmonol Suppl20042612712815029626
- LevinDLPersistent pulmonary hypertension of the newborn infantJ Pediatr1976894626630784932
- HermonMIntravenous prostacyclin mitigates inhaled nitric oxide rebound effect: A case control studyArtif Organs1999231197597810564300
- GöthbergSResidual pulmonary hypertension in children after treatment with inhaled nitric oxide: a follow-up study regarding cardiopulmonary and neurological symptomsActa Paediatr200089121414141911195228
- BizzarroMInhaled nitric oxide for the postoperative management of pulmonary hypertension in infants and children with congenital heart diseaseCochrane Database Syst Rev20094CD005055
- MacraeDJInhaled nitric oxide therapy in neonates and children: reaching a European consensusIntensive Care Med200430337238014722629
- ChouPPulmonary changes following extracorporeal membrane oxygenation: autopsy study of 23 casesHum Pathol19932444054128491481
- ClarkRHLow-dose nitric oxide therapy for persistent pulmonary hypertension: 1 year follow-upJ Perinatol200323430030312774137
- FakiogluHHypoxic respiratory failure in term newborns: clinical indicators for inhaled nitric oxide and extracorporeal membrane oxygenation therapyJ Crit Care200520328829316253800
- LammersAEEpoprostenol treatment in children with severe pulmonary hypertensionHeart2007673974317065181
- MaiyaSResponse to bosentan in children with pulmonary hypertensionHeart200692566467016216850
- WimmerMExperience with long-term nifedipine therapy in paediatric cardiological patientsPadiatr Padol19902531811932116616
- HoudeCProfile of paediatric patients with pulmonary hypertension judged by responsiveness to vasodilatorsBr Heart J19937054614688260279
- MawatariEAmlodipine prevents monocrotaline-induced pulmonary arterial hypertension and prolongs survival in rats independent of blood pressure loweringClin Exp Pharmacol Physiol200734759460017581214
- AdatiaIInhaled nitric oxide and hemodynamic evaluation of patients with pulmonary hypertension before transplantationJ Am Coll Cardiol1995257165616647759720
- WeimannJSildenafil is a pulmonary vasodilator in awake lambs with acute pulmonary hypertensionAnesthesiology20009261702171210839922
- ReffelmannTTherapeutic potential of phosphodiesterase 5 inhibition for cardiovascular diseaseCirculation2003108223924412860892
- LeibovitchLTherapeutic applications of sildenafil citrate in the management of paediatric pulmonary hypertensionDrugs2007671577317209664
- HumplTBeneficial effect of oral sildenafil therapy on childhood pulmonary arterial hypertension: twelve-month clinical trial of a single-drug, open-label, pilot studyCirculation2005111243274328015956137
- RossiRSildenafil improves endothelial function in patients with pulmonary hypertensionPulm Pharmacol Ther200821117217717428713
- DillerGPCirculating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertensionCirculation2008117233020303018519847
- BarnettCFSildenafil in the treatment of pulmonary hypertensionVasc Health Risk Manag20062441142217323595
- SpringRMSildenafil for pulmonary hypertension: dose-dependent improvement in exercise performancePulm Pharmacol Ther200821351652118249145
- GalièNSildenafil citrate therapy for pulmonary arterial hypertensionN Engl J Med20053532148215716291984
- BharaniAThe efficacy and tolerability of sildenafil in patients with moderate-to-severe pulmonary hypertensionIndian Heart J200355555912760589
- AtzAMSildenafil augments the effect of inhaled nitric oxide for postoperative pulmonary hypertensive crisesJ Thorac Cardiovasc Surg2002124362862912202882
- KothariSSChronic oral sildenafil therapy in severe pulmonary artery hypertensionIndian Heart J200254440440912462669
- LeeSHCurrent treatment strategies for pulmonary arterial hypertensionJ Intern Med2005258319921516115293
- Della TorreFSildenafil in pulmonary hypertensionSarcoidosis Vasc Diffuse Lung Dis2005221787915881285
- TeixeiraCEDifferential effects of the phosphodiesterase type 5 inhibitors sildenafil, vardenafil, and tadalafil in rat aortaJ Pharmacol Exp Ther2006316265466116204472
- ToqueHAVardenafil, but not sildenafil or tadalafil, has calcium-channel blocking activity in rabbit isolated pulmonary artery and human washed plateletsBr J Pharmacol2008154478779618536732
- LangIMPaediatric pulmonary vascular diseasePaediatr Respir Rev20045323824815276136
- GiaidAExpression of endothelin-1 in the lungs of patients with pulmonary hypertensionN Engl J Med199332824173217398497283
- CacoubPEndothelin-1 in pulmonary hypertensionN Engl J Med199332926196719688247067
- RosenzweigEBEffects of long-term bosentan in children with pulmonary arterial hypertensionJ Am Coll Cardiol200546469770416098438
- GalièNBosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled studyCirculation20061141485416801459
- GiannakoulasGBosentan in mild pulmonary hypertensionLancet2008372965117301731author reply 1731.
- BeghettiMSafety experience with bosentan in 146 children 2–11years old with pulmonary arterial hypertension: results from the European Postmarketing Surveillance programPediatr Res200864220020418414142
- Van LoonRLELong-term effect of bosentan in adults versus children with pulmonary arterial hypertension associated with systemic-to-pulmonary shunt: does the beneficial effect persist?Am Heart J200715477678217893008
- ApostolopoulouSCSitaxsentan in pulmonary arterial hypertensionChest200312351772; author reply 1772–177312740304
- BarstRJClinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: open-label pilot studyChest200212161860186812065350
- BenzaRLSitaxsentan for the treatment of pulmonary arterial hypertension: a 1-year, prospective, open-label observation of outcome and survivalChest2008134477578218625676
- MehtaPKEndothelin receptor antagonists improve exercise tolerance and oxygen saturations in patients with Eisenmenger syndrome and congenital heart defectsTex Heart Inst J200835325626118941642
- GalièNAmbrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2Circulation2008117233010301918506008
- TudorAObservations on peripheral microcirculation in young hypertensive patientsRom J Intern Med200543127378
- BarstRJVasodilator therapy for primary pulmonary hypertension in childrenCirculation19999991197120810069788
- RosenzweigEBLong-term prostacyclin for pulmonary hypertension with associated congenital heart defectsCirculation199999141858186510199883
- RosenzweigEBPulmonary arterial hypertension in childrenPediatr Pulmonol200438122215170869
- TakaokaSCurrent therapies for pulmonary arterial hypertensionSemin Cardiothorac Vasc Anesth20071113714817536117
- BarstRJDiagnosis and treatment of pulmonary artery hypertensionCurr Opin Pediatr1996855125198946133
- BadeschDBProstanoid therapy for pulmonary arterial hypertensionJ Am Coll Cardiol20044312 Suppl S56S61S15194179
- SimonneauGContinuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trialAm J Respir Crit Care Med2002165680080411897647
- IvyDDTransition of stable pediatric patients with pulmonary arterial hypertension from intravenous epoprostenol to intravenous treprostinilAm J Cardiol200799569669817317374
- VizzaCDLong term treatment of pulmonary arterial hypertension with beraprost, an oral prostacyclin analogueHeart200186666166511711462
- BarstRJBeraprost therapy for pulmonary arterial hypertensionJ Am Coll Cardiol200341122119212512821234
- BakerSEInhaled iloprost in pulmonary arterial hypertensionAnn Pharmacother2005397–81265127415976392
- OlschewskiHInhaled iloprost for severe pulmonary hypertensionN Engl J Med2002347532232912151469
- GesslerTInhaled prostanoids in the therapy of pulmonary hypertensionJ Aerosol Med Pulm Drug Deliv200821111218518827
- IshikuraKBeneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertensionCirc J200670217417816434811
- AlapatiVRMechanisms of U46619- and 5-HT-induced contraction of bovine pulmonary arteries: role of chloride ionsBr J Pharmacol200715181224123417592513
- SomlyoAPSignal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin IIJ Physiol2000522Pt 2:17718510639096
- ShimokawaHRho-kinase is an important therapeutic target in cardiovascular medicineArterioscler Thromb Vasc Biol20052591767177516002741
- AbeKLong-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in ratsCirc Res200494338539314670839
- FukumotoYRecent progress in the treatment of pulmonary arterial hypertension: expectation for rho-kinase inhibitorsTohoku J Exp Med2007211430932017409670
- FukumotoYAcute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertensionHeart200591339139215710736
- JanosiTDifferential roles of endothelin-1 ETA and ETB receptors and vasoactive intestinal polypeptide in regulation of the airways and the pulmonary vasculature in isolated rat lungExp Physiol200893111210121918567602
- TofovicSP2-Ethoxyestradiol is antimitogenic and attenuates monocrotaline-induced pulmonary hypertension and vascular remodelingVascul Pharmacol20084846174183
- GurbanovEThe key role of apoptosis in the pathogenesis and treatment of pulmonary hypertensionEur J Cardiothorac Surg200630349950716870458
- MichelakisEDEmerging concepts and translational priorities in pulmonary arterial hypertensionCirculation20081181486149518824655
- AltieriDCSurvivin and apoptosis controlAdv Cancer Res200388315212665052
- GuerardPThe HMG-CoA reductase inhibitor, pravastatin, prevents the development of monocrotaline-induced pulmonary hypertension in the rat through reduction of endothelial cell apoptosis and overexpression of eNOSNaunyn Schmiedebergs Arch Pharmacol2006373640141416896805
- RakotoniainaZCelecoxib but not the combination of celecoxib +atorvastatin prevents the development of monocrotaline-induced pulmonary hypertension in the ratNaunyn Schmiedebergs Arch Pharmacol2008378324125118542928
- McMurtryMSStatin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: the rapamcyin-atorvastatin-simvastatin studyAm J Physiol Lung Cell Mol Physiol20072934L933L94017675370
- HervéPIncreased plasma serotonin in primary pulmonary hypertensionAm J Med19959932492547653484
- MacLeanMROverexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertensionCirculation2004109172150215515078799
- MarcosESerotonin transporter inhibitors protect against hypoxic pulmonary hypertensionAm J Respir Crit Care Med2003168448749312773327
- GuignabertCSerotonin transporter inhibition prevents and reverses monocrotaline-induced pulmonary hypertension in ratsCirculation2005111212812281915927991
- KawutSMSelective serotonin reuptake inhibitor use and outcomes in pulmonary arterial hypertensionPulm Pharmacol Ther200619537037416483811
- ArimaMClinical features of acute pulmonary thromboembolism in younger patientsCirc J200367433033312655164
- CastilloLWhole body nitric oxide synthesis in healthy men determined from [15N] arginine-to-[15N] citrulline labelingProc Natl Acad Sci U S A19969311460114658876157
- McMullanDMAlterations in endogenous nitric oxide production after cardiopulmonary bypass in lambs with normal and increased pulmonary blood flowCirculation2000102supplIII172III17811082382
- BarrFEEffect of cardiopulmonary bypass on urea cycle intermediates and nitric oxide levels after congenital heart surgeryJ Pediatr2003142263012520250
- GorenfloMPlasma L-arginine and metabolites of nitric oxide synthase in patients with left-to-right shunt after intracardiac repairChest200512741184118915821193
- TanakaEPulmonary hypertension in systemic lupus erythematosus: evaluation of clinical characteristics and response to immunosuppressive treatmentJ Rheumatol200229228228711838845
- HoeperMMDrug treatment of pulmonary arterial hypertension: current and future agentsDrugs200565101337135415977967
- FasnachtMSSwiss Society for Pulmonary Arterial HypertensionThe Swiss registry for pulmonary arterial hypertension: the paediatric experienceSwiss Med Wkly20071373536510513
- MogollónMVCombination therapy with sildenafil and bosentan reverts severe pulmonary hypertension and allows heart transplantation: case reportTransplant Proc20063882522252317097987
- LiuCEndothelin receptor antagonists for pulmonary arterial hypertensionCochrane Database Syst Rev20063CD00443416856046
- DriscollJAMedical therapy for pulmonary arterial hypertensionExpert Opin Pharmacother200891658118076339
- HumbertMTreatment of pulmonary arterial hypertensionN Engl J Med2004351141425143615459304
- McLaughlinVVRandomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertensionAm J Respir Crit Care Med2006174111257126316946127
- HaraldssonAInhaled prostacyclin for treatment of pulmonary hypertension after cardiac surgery or heart transplantation: a pharmacodynamic studyJ Cardiothorac Vasc Anesth19961078648688969392
- BurgessGMutual phar-macokinetic interactions between steady-state bosentan and sildenafilEur J Clin Pharmacol2008641435018040672
- SimonneauGAddition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trialAnn Intern Med2008149852153018936500
- KersteinDBlade balloon atrial septostomy in patients with severe primary pulmonary hypertensionCirculation1995917202820357534664
- RichSAtrial septostomy as palliative therapy for refractory primary pulmonary hypertensionAm J Cardiol1983519156015616189385
- LawMAAtrial septostomy improves survival in select patients with pulmonary hypertensionAm Heart J2007153577978417452153
- HausknechtMJSuccessful palliation of primary pulmonary hypertension by atrial septostomyAm J Cardiol19906515104510461691586
- SandovalJGraded balloon dilation atrial septostomy in severe primary pulmonary hypertension. A therapeutic alternative for patients nonresponsive to vasodilator treatmentJ Am Coll Cardiol199822973049708453
- CheeverKHAn overview of pulmonary arterial hypertension: risks, pathogenesis, clinical manifestations, and managementJ Cardiovasc Nurs2005202108116quiz 117–118.15855858
- HuddlestonCBLung transplantation in childrenAnn Surg2002236327027612192313
- AuroraPRegistry of the International Society for Heart and Lung Transplantation: eleventh official pediatric lung and heart/lung transplantation report – 2008J Heart Lung Transplant200827997898318765189
- HaworthSGRole of the endothelium in pulmonary arterial hypertensionVascul Pharmacol200645531732517005453
- Galie’NTreatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomized controlled trialLancet20083712093210018572079
- GalieNA meta-analysis of randomized controlled trials in pulmonary arterial hypertensionEur Heart J20093039440319155250
- RubinLJTreatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trialAnn Intern Med19901124854912107780
- BarstRJA comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study GroupN Engl J Med19963342963028532025
- BadeschDBContinuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trialAnn Intern Med200013242543410733441
- ChannickRNEffects of the dual endothelin receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled studyLancet20013581119112311597664
- LanglebenDEffects of the thromboxane synthetase inhibitor and receptor antagonist terbogrel in patients with primary pulmonary hypertensionAm Heart J2002143E412040360
- GalieNEffects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomised, double-blind placebo-controlled trialJ Am Coll Cardiol2002391496150211985913
- RubinLJBosentan therapy for pulmonary arterial hypertensionN Engl J Med200234689690311907289
- SastryBKSClinical efficacy of sildenafil in primary pulmonary hypertension. 1: A randomized, placebo-controlled, doubleblind, crossover studyJ Am Coll Cardiol2004431149115315063421
- HumbertMCombination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2Eur Respir J20042435335915358690
- BarstRJSitaxsentan therapy for pulmonary arterial hypertensionAm J Respir Crit Care Med2004169444144714630619
- GalieNThe Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group. Sildenafil citrate therapy for pulmonary arterial hypertensionN Engl J Med20053532148215716291984
- WilkinsMRSildenafil versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) studyAm J Respir Crit Care Med20051711292129715750042
- SinghTA randomized, placebo controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertensionAm Heart J2006151851.e1851.e5
- BarstRJTreatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentanJ Am Coll Cardiol200647102049205616697324
- McLaughlinVVRandomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertensionAm J Respir Crit Care Med20061741257126316946127
- HoeperMCombining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertensionEur Resp J20064691694
- HislopADevelopmental biology of the pulmonary circulationPaediatr Respir Rev200561354315698814
- TorresFSystematic review of randomised, double-blind clinical trials of oral agents conducted in patients with pulmonary arterial hypertensionInt J Clin Pract200761101756176517877662