Abstract
Obstructive sleep apnea (OSA) is associated with high cardiovascular morbidity and mortality. Recent studies have shown that it is associated with atherosclerosis and left ventricular dysfunction markers. The aim of this study was to assess the cardiovascular effects of OSA depending on its severity, in patients without clinically diagnosed cardiovascular disease. One hundred thirty newly diagnosed, nondiabetic OSA patients (mean age 49 ± 10 years), without vasoactive treatment were included. They underwent clinical and ambulatory blood pressure measurements, echocardiography, carotid ultrasound examination, and a carotid–femoral pulse wave velocity (PWV) measurement. Seventy-five percent of the subjects were hypertensive according to the clinical or ambulatory measurement. More patients with the most severe forms (respiratory disturbance index >37/hour) had a nondipper profile (52% vs 34%; P = 0.025) and their left ventricular mass was higher (40 ± 7 vs 36 ± 8 g/m, p = 0.014). This last parameter was independently and inversely associated with mean nocturnal oxygen saturation (P = 0.004). PWV and carotid intima-media thickness did not differ between one OSA severity group to another, but the prevalence of carotid hypertrophy was higher when mean SaO2 was below 93.5% (29.5 vs 16%; P = 0.05). Our study shows that in OSA patients without clinically diagnosed cardiovascular disease, there is a significant left ventricular and arterial effect, which is even more marked when OSA is severe.
Introduction
Obstructive sleep apnea (OSA) is a common but underdiagnosed diseaseCitation1 that causes increased cardiovascular morbidity and mortality, including arterial hypertension (HT), coronary heart disease, heart rhythm and conduction disorders, heart failure, and stroke.Citation2–Citation12
Although OSA is often associated with certain cardiovascular risk factors such as HT, obesity, diabetes, and dyslipidemia, it is legitimate to mention the direct role that OSA plays in the development of atherosclerosis. In fact, repeated episodes of hypoxia, hypercapnia, microarousals, and changes in intrathoracic pressure trigger pathophysiological mechanisms such as sympathetic hyperactivity,Citation13–Citation15 oxidative stress,Citation16 systemic inflammation,Citation17 hypercoagulability,Citation18 and even endothelial dysfunction.Citation19 And all these abnormalities combine chronically to bring about the development of vascular lesions. Ultrasonography – a noninvasive, quick and reproducible technique – can be used to evaluate the atherosclerotic process at an early stage.Citation20 It analyzes vascular remodeling, measures the parietal thickness (intima-media thickness [IMT]) and detects atheromatous plaques. Like IMT and carotid plaques, carotid–femoral pulse wave velocity (PWV) is an early and independent marker of cardiovascular morbidity and mortality, notably coronary and cerebral morbidity and mortality.Citation21–Citation23 PWV is a noninvasive way of assessing aortic stiffness. The large arteries play a crucial role in cardiac structure so increased arterial stiffness contributes independently to arterial pressure and to an increase in left ventricular afterload, thereby promoting left ventricular hypertrophy (LVH).Citation24
A few studies have focused on structural and functional modifications to the large arteries in OSA, with the majority of them reporting increases in carotid IMT and PWV.Citation25–Citation34 Similarly, some studies have found a link between OSA and LVH.Citation35–Citation38 The link that connects OSA to these cardiovascular abnormalities remains ambiguous due to the frequent presence of confounding factors in the studies.
We performed our study on a large sample of patients with newly diagnosed and untreated OSA, without any known cardiovascular disease or vasoactive treatment. The main aim was to assess arterial structural and functional modifications, as well as remodeling of the left ventricle (LV), in this population. To the best of our knowledge, no study has simultaneously analyzed these three cardiovascular abnormalities in OSA patients without clinically diagnosed cardiovascular disease.
Materials and methods
Study population
The patients included in the study were referred to the Sleep Laboratory at the University Hospital of Grenoble (France) for symptoms indicating OSA between November 2001 and July 2007. The diagnosis of OSA was confirmed using polysomnography or ventilation polygraphy. We did not include patients if they presented a known cardiovascular disease including HT, any pathology affecting arterial blood pressure (BP) regulation (such as heart failure, Parkinson’s disease and heart or kidney transplantation), atrial fibrillation or frequent extrasystole (>10/min), chronic respiratory insufficiency or previous treatment of OSA by means of nasal continuous positive airway pressure (nCPAP), maxillofacial surgery or a mandibular advancement prosthesis (oral appliance). We excluded diabetic patients (fasting glycemia ≥7.0 mmol/L or antidiabetic treatment), patients for whom the 24-hour ambulatory BP monitoring (ABPM) was invalid and patients who were on vasoactive drugs. Ethical approval was obtained from the local ethics committee and all of the participants gave their informed consent. The registration number for this study is NCT00764218.
Blood pressure and heart rate measurements
Clinical BP was measured by mercury sphygmomanometer on three occasions in line with European Society of Hypertension–European Society of Cardiology guidelines.Citation39 Systolic BP (SBP) and diastolic BP (DBP) were assessed. Pulse pressure (PP) was calculated using the following formula: PP = SBP minus DBP. Clinical HT was defined as a clinical SBP ≥140 mmHg and/or a clinical DBP ≥ 90 mmHg.Citation39 Increased clinical PP was defined as PP ≥ 65 mmHg.Citation40 Clinical heart rate (HR) was measured by pulse palpation (30 seconds) after the third measurement of BP in a supine position.
ABPM was carried out with a Spacelabs 90207® device (Spacelabs International, Redmond, WA, USA). Measurements were taken every 15 minutes over 24 hours. Daytime (07:00 to 22:00) HT was defined as daytime SBP ≥ 135 mmHg and/or daytime DBP ≥ 85 mmHg, and nocturnal (22:00 to 07:00) HT as nocturnal SBP ≥ 120 mmHg and/or nocturnal DBP ≥ 70 mmHg.Citation39 Nondipping status was defined as a nocturnal BP reduction of <10%.
Respiratory measurements
Full polysomnography was performed in 94 of the 130 patients (72%). Continuous recordings were taken with electrode positions C3/A2-C4/A1-Cz/01 of the international 10–20 Electrode Placement System, eye movements, chin electromyogram and ECG modified V2 lead. Sleep was scored manually according to standard criteria.Citation41 Airflow was measured using nasal pressure associated with the sum of buccal and nasal thermistor signals. Respiratory efforts were monitored with abdominal and thoracic bands. An additional respiratory effort signal (ie, pulse transit time) was recorded concurrently. Pulse transit time allowed us to identify “autonomic activations”, and as a consequence, microarousals.Citation42 We were thus able to use the same rules and definition for hypopnea whatever the diagnostic method used for the diagnosis of sleep apnea. Oxygen saturation (SaO2) was measured using a pulse oximeter (Biox-Ohmeda 3700®; Ohmeda, Liberty Corner, NJ, USA). The same variables were measured in the remaining 36 patients, except for sleep variables which were not recorded. Apnea was defined as a complete cessation of airflow for ≥10 seconds, and hypopnea as a ≥50% reduction in the nasal pressure signal or a 30%–50% decrease associated with either oxygen desaturation of ≥3% or an arousal (defined according to the Chicago report or by autonomic activations on pulse transit time), both lasting for ≥10 seconds.Citation42,Citation43 Apnea was classified as obstructive, central or mixed according to the presence or absence of respiratory efforts. The classification of hypopnea as obstructive or central was based on the pulse transit time signal and the shape of the inspiratory part of nasal pressure (flow limited aspect or not). The respiratory disturbance index (RDI) was calculated and defined as the number of episodes of apnea and hypopnea per hour of sleep (full polysomnography) or per hour of recording (polygraphy without electroencephalogram recording). In our study, a diagnosis of OSA was retained if RDI was ≥15 per hour. Subjects were split into two groups depending on the severity of their OSA, using the median RDI: group A (RDI < 37/hour, N = 65) and group B (RDI > 37/hour, N = 65). In an additional analysis, they were then split according to the median of the mean nocturnal SaO2: group 1 (SaO2 < 93.5%, N = 65) and group 2 (SaO2 > 93.5%, N = 65).
Echocardiography
The echocardiogram was carried out using an HP Sonos 2500® (Hewlett Packard, Santa Clara, CA, USA) machine equipped with a 2.5 MHz probe. The examination was performed in M-mode with 2D guidance in the long axis of the left parasternal view. LV internal end-diastolic (LVD) and end-systolic diameters, as well as interventricular septum and posterior wall (LVPW) thicknesses, were measured over five consecutive cycles. Systolic function was assessed by the LV ejection fraction (LVEF) according to the Teicholz formula. LV mass (LVM) was measured according to the Penn convention using the Devereux formula and was normalized for body surface area and height2.7 to derive the LV mass index (LVMI and LVMI-height2.7).Citation44,Citation45 LVH was defined as an LVMI of ≥ 111 g/m2 or ≥ 50 g/m2.7 in men and of ≥106 g/m2 or ≥47 g/m2.7 in women.Citation46,Citation47 LV geometry was analysed according to the presence or absence of LVH and the calculated relative parietal thickness at the end of ventricular diastole (RWT = 2 × LVPW/LVD). All echocardiograms were recorded by the same experienced operator. We were able to perform a complete LV geometry analysis in 116 of the 130 patients (89%).
Carotid ultrasonography
B-mode ultrasonography was performed using an HP Sonos 2500® (Hewlett Packard) machine using a sectorial 7.5 MHz probe. The method used to determine the mean common carotid IMT and luminal diameter has been previously described.Citation48 Both common carotid arteries were studied consecutively in the long axis with a probe incidence allowing good quality images. The IMT was defined as the distance separating the most internal parts of these lines and the luminal diameter by the distance between the blood–intima interfaces on the anterior and posterior walls. The images were recorded in end-diastole and then analyzed by specific validated software (TIMC laboratory, CHU Grenoble, France). IMT and diameter measurements were carried out on areas free of atheroma and then averaged. The IMT and luminal diameter values for any given subject were the mean values for the two common carotid arteries. Carotid wall hypertrophy was defined as a common carotid IMT above 0.8 mm.Citation49 A plaque was defined as an echogenic structure encroaching into the vessel lumen with a distinct area and with an IMT more than 50% greater than those of the neighboring sites. Carotid ultrasonography was performed by two operators who were blinded to the other study data. The analysis of carotid parameters using the specific software was performed by the same operator throughout the entire study.
Aortic pulse wave velocity
To determine the carotid-femoral PWV, two pulse transducers were fixed on the skin over the right common carotid and femoral arteries. The time delay was measured with a Complior® device (Artech Medical, Pantin, France), between the feet of simultaneously recorded pulse waves and averaged over 10 consecutive cycles. The carotid-femoral PWV was calculated as the distance between the arterial sites divided by the time delay. Increased PWV was defined as PWV > 12 m/second.Citation39
Biological parameters
All of the subjects had plasma assays of total cholesterol (enzymatic colorimetry, normal: 4.62–7.04 mmol/L), triglycerides (enzymatic colorimetry, normal: 0.63–2.58 mmol/L), high-density lipoprotein (HDL) cholesterol (enzymatic colorimetry, normal: 1.0–1.62 mmol/L), low-density lipoprotein (LDL) cholesterol (Friedwald formula, normal: 2.60–4.67 mmol/L), glucose (enzymatic method, normal: 3.8–5.8 mmol/L) and creatinine (enzymatic colorimetry, normal: 62–106 μmol/L).
Statistical analysis
Statistical analyses were performed using SPSS software (SPSS Inc, Chicago, IL, USA). We assessed the normality of data distribution. Continuous data were expressed as mean ± standard deviation (SD). Relationships between continuous variables were evaluated using Pearson’s correlation analysis when data were normally distributed and using Spearman’s correlation analysis when they were not normally distributed. Noncontinuous variables, expressed as percentages, were compared using a Chi-squared test. Comparisons between groups for continuous variables were made using a Student’s t-test (or a Mann–Whitney U test when the data were not normally distributed). We performed multivariate analysis using stepwise regression. Variables included in our analysis were all the variables significantly (P < 0.05) associated with the explained variable using univariate analysis. We considered values of P < 0.05 significant for all analyses. We have chosen to include all P values under 0.20 in the tables.
Results
We included 130 patients (109 men, 84%), with a mean age of 49 ± 10 years. The general characteristics of the population are presented in . Most of the patients exhibited moderate to severe OSA (63 of the 130 subjects (48%) had a RDI of 30–50 per hour, and 31 (24%) had a RDI upper than 50 per hour). Body mass index (BMI) was 26.5 ± 3 kg/m2, 73 patients (56%) were overweight and 15 (11.5%) were obese. Groups A and B were comparable in terms of age, sex, smoking status and all biological parameters. Patients with RDI over 37 per hour (ie, Group B) had a significantly higher BMI.
Table 1 General, hemodynamic, biological, and respiratory characteristics of the global population and according to the severity of OSA (RDI)
Clinical BP and ABPM parameters, as well as HR, did not vary from group to group (). Forty-five patients (35%) had clinical HT, 66 (51%) had diurnal HT, and 89 (68%) had nocturnal HT. In total, 98 patients (75%) were hypertensive measured clinically or with ABPM. Out of the 45 patients presenting clinical HT, grades were as follows: 28 HT grade 1 (62%), 13 HT grade 2 (29%) and four HT grade 3 (9%). Eight subjects (18%) had isolated systolic HT, 15 (33%) had isolated diastolic HT, and 22 (49%) had systolo-diastolic HT. Fifty-five patients (42%) had a nondipper profile for either SBP or DBP. There was a trend for a higher prevalence of clinical HT in group B (41% vs 28%, respectively; P = 0.07) than in group A, and a nondipper profile was more common in the most severe cases of OSA ().
Table 2 Cardiovascular parameters in the global population and according to the severity of OSA (RDI)
Arterial parameters are presented in . There are no significant differences between groups A and B in any of these parameters. Carotid IMT and PWV were positively correlated with age (P < 0.0001), clinical SBP, DBP, and PP (P < 0.0001 for all but the correlation between IMT and DBP of P < 0.01), as well as with all BP parameters measured using ABPM (P < 0.05). Carotid IMT was correlated with mean SaO2 (r = −0.21, P = 0.017; ) but PWV and PP were not correlated with any respiratory parameters. Clinical PP was positively correlated with age (r = 0.19, P = 0.03) and carotid IMT, PWV and clinical PP correlated with each other. In a multivariate analysis, IMT and PWV were independently associated with age and with clinical SBP (P < 0.0001). Clinical PP was independently associated with PWV (P < 0.0001) and IMT (P = 0.005).
Figure 1 Relationship between carotid IMT and mean nocturnal SaO2 (r = −0.21, P = 0.017).
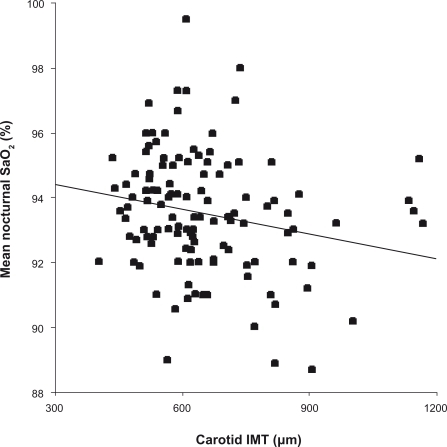
Patients presenting carotid parietal hypertrophy were older, had a higher clinical SBP and presented more severe OSA (). Subjects with carotid atheromatous plaques were older (P < 0.0001), presented higher clinical SBP and DBP (P < 0.001), and higher total (P = 0.018) and LDL (P = 0.028) cholesterol.
Table 3 Factors related to carotid hypertrophy
All of the echocardiographic parameters are summarized in . LVEF did not change from group to group depending on the severity of the OSA. LVH was present in 5% to 9.5% of the patients depending on the criteria chosen (LVMI or LVMI-height2.7), with no significant difference between the two groups. Patients with the most severe OSA had significantly higher LV wall thickness, LVM (before and after indexation) and RWT. LVMI-height2.7 was significantly correlated with age (r = 0.19, P = 0.04), clinical SBP (r = 0.25, P = 0.006), and DBP (r = 0.22, P = 0.02), diurnal and nocturnal SBP and PP (P < 0.05), mean nocturnal SaO2 (r = −0.27, P = 0.003), minimal SaO2 (r = −0.19, P = 0.036) and SaO2 < 90% (r = 0.23, P = 0.011), but not with RDI. LVMI-height2.7 was also correlated with IMT (r = 0.19, P = 0.038) and PWV (r = 0.18, P = 0.05). In a multivariate analysis, LVMI-height2.7 was independently correlated with clinical SBP (β = −0.25, P = 0.01) and mean nocturnal SaO2 (β = 0.23, P = 0.004).
Patients with mean SaO2 < 93.5% (Group 1) were older (P = 0.001) and had a higher BMI (P = 0.003). They also tended to have worse HT as evaluated both clinically and measured using ABPM () and presented significantly higher LVMI-height2.7 (P = 0.003) (), as well as a greater prevalence of carotid parietal hypertrophy (29.5% vs 16%, P = 0.05).
Figure 2 LVMI-height2.7 according to the mean nocturnal SaO2 group (median) (P = 0.003 between groups 1 and 2).
Abbreviations: LVMI, left ventricular mass index; SaO2, oxygen saturation.
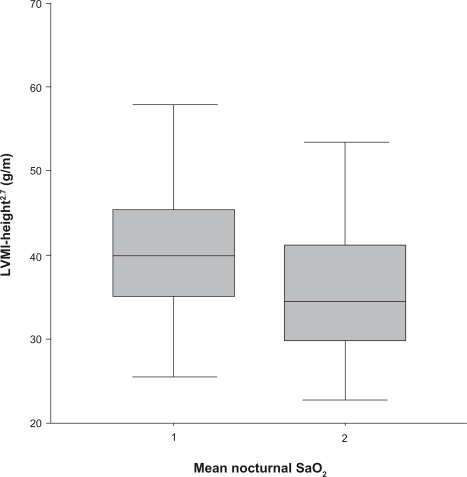
Table 4 Cardiovascular anomalies according to the severity of OSA (mean nocturnal SaO2)
Discussion
HT: a common disease in OSA
The prevalence of HT in OSA patients is estimated to be nearly 50%.Citation50 Although the link between the two pathologies was long disputed due to the numerous confounding factors, OSA is now recognized as one of the causes of secondary HT.Citation39,Citation51 In our study, 75% of patients were hypertensive, although at inclusion, none of them were known to be hypertensive. In the general population, the prevalence of undiagnosed HT is around 25%,Citation52 which is three times lower than in our population. Our results show the extent to which HT is under-diagnosed in OSA patients. They also serve as a reminder of the utility of ABPM, which is a recommended technique in this pathology.Citation39 In our study, this OSA-associated HT showed the following characteristics: predominantly nocturnal HT associated with a high prevalence of nondipper status and primarily diastolic HT. This is in line with the data found in the literature. We did not find any difference in BP values according to the severity of the OSA. This is certainly linked to the fact that, despite high prevalence of HT, the population studied was very moderately hypertensive (62% grade 1 HT). In fact, we were studying an OSA population without clinically diagnosed cardiovascular disease, which was consequently not greatly affected by severe HT.
From a pathophysiological point of view, episodes of apnea and hypopnea are responsible for changes in BP. The occurrence of a cortical microarousal leads to a hypertensive peak.Citation53,Citation54 Moreover, repeated episodes of hypoxia and hypercapnia trigger pathophysiological mechanisms including sympathetic hyperactivity.Citation13,Citation55 and the secretion of vasoactive substances.Citation56,Citation57 Finally, each apnea is accompanied by a cycle of desaturation and reoxygenation which leads to the production of free radicals,Citation58 systemic inflammation,Citation17 and coagulation abnormalities.Citation18 These biochemical and cellular modifications contribute to endothelial dysfunctionCitation19,Citation59 and atherosclerotic degeneration. It has been hypothesized that chronic hypoxia has an effect on vascular remodeling on the basis of results from animal and tissue culture experiments.Citation60,Citation61
OSA, carotid remodeling, and arterial stiffness
Carotid ultrasonography and evaluation of arterial stiffness are useful for the early detection of atherosclerosis at an asymptomatic stage.Citation62,Citation63 Increased carotid IMT is a marker of structural vascular damages whereas arterial stiffness, as estimated by PP and PWV, reflects functional modifications on the large arteries. In our study, carotid IMT, PP and PWV values were not very high compared to previous studies conducted on apnea patients. This partly explains the absence of a significant difference between our two OSA severity groups for these arterial parameters. These “normal” values are probably due to the relatively low number of major vascular risk factors in our population.
Almost a quarter of patients presented carotid hypertrophy and 19% had carotid plaques. These values are particularly high for a population potentially considered at low cardiovascular risk, according to the literature.Citation64 Furthermore, carotid hypertrophy was more common in patients with the most severe OSA (mean SaO2 < 93.5%) and it was associated with worse respiratory parameters. In line with other studies,Citation25–Citation30,Citation33 our results suggest that OSA contributes to the genesis of atherosclerosis. However, it remains to be formally demonstrated that the association between OSA and carotid hypertrophy is independent, as the confounding factors are numerous. A few studies state that OSA has a direct effect on IMT.Citation25,Citation28,Citation30 However, most of these studies have significant limitations as they cover a large number of major vascular risk factors and subject numbers are low, eg, Silvestrini and colleagues studied a sample of 23 obese patients.Citation25 Similarly, in the study by Schulz and colleagues, 20% of patients were diabetic, 58% were dyslipidemic, and 65% were hypertensive.Citation30 In contrast, in our study, there were no diabetic patients, only 11.5% were obese, and only 7.5% were dyslipidemic. We recently published a study in 83 OSA patients after exclusion of the majority of confounding factors and found an independent association between IMT and SaO2, but only in normotensive subjects.Citation28 In addition to the relatively low number of major vascular risk factors in our study, the relatively young age of the population and the recent diagnosis of the OSA potentially contributed to the exclusion of patients with more advanced atherosclerosis. However, our population must not be considered at low cardiovascular risk. This is well demonstrated by the high prevalence of carotid wall abnormalities.
We did not find a significant link between the severity of OSA and aortic stiffness. This is inconsistent with the findings of a number of other studies and can be explained first and foremost by the characteristics of the patients included.Citation26,Citation28,Citation31,Citation32,Citation34 In fact, whilst in our study OSA was at a mild stage in most subjects and whilst there were few major vascular risk factors, the majority of other studies focusing on PWV looked at very severe OSA.Citation31,Citation32,Citation34 Although carotid IMT appears to increase early on in the disease, the increase in arterial stiffness could be the consequence of prolonged exposure to OSA.
OSA and cardiac hypertrophy
Our results, like those of other studies, show a link between LVM and the severity of OSA.Citation35–Citation38,Citation65–Citation67 However, the prevalence of LVH was moderate in our study (9.5%), with no significant difference between the groups of differing OSA severity. In Noda and colleagues’ study, prevalence was nearly 40% but the patient population had been suffering from OSA for a longer period of time, was more obese and had worse HT.Citation67 We can question the independence of the link between OSA and LVM, because OSA patients are more often hypertensive, obese or diabetic, and these are all well-characterized risk factors for LVH. Hedner and colleagues were the first to consider this link, showing that LVM was around 15% higher in normotensive OSA patients than in normotensive control subjects.Citation65 Similarly, in the study by Cloward and colleagues, HT prevalence was 52% and that of LVH was 88%, suggesting that LVH cannot be explained by HT alone.Citation37 However, the results of these studies must be analyzed with care because both included obese patients in whom it is well known that it is difficult to measure LVM by ultrasound. Furthermore, neither used ABPM, although 24-hour BP monitoring is more closely associated with LVM. Conversely, a more recent study conducted by Niroumand and colleagues in 533 OSA patients, in whom confounding factors were rigorously controlled for, showed that OSA was not directly involved in the LVM increase.Citation36 In our study, the link between LVM and mean nocturnal SaO2 was independent of other factors, suggesting that OSA is directly involved in the occurrence of LVH.
From a pathophysiological point of view, the cardiac changes observed during OSA are linked to an increase in LV afterload, due to a number of different mechanisms. Thus, repeated episodes of nocturnal hypoxia and microarousals – as a result of the sympathetic hyperactivity that they cause – contribute to increase BP.Citation15 On the other hand, the degree of negative intrathoracic pressure generated by inspiratory effort during OSA has the direct effect of raising transmural pressure and LV afterload, independently of BP.Citation68–Citation70 Finally, our study, like several others, suggests that arterial stiffness is a possible left ventricular remodeling mechanism during OSA.Citation71,Citation72
Study limitations
The main limitation of our study is the absence of a control group. However, several studies have already shown that OSA patients present greater atherosclerotic and cardiac effects than healthy subjects. The aim of our prospective study performed on a large sample of newly diagnosed OSA patients was to determine whether the severity of OSA was associated with the cardiovascular effects independently of traditional major cardiovascular risk factors. As far as we know, this is the first study to have simultaneously evaluated structural and functional cardiovascular parameters in newly diagnosed OSA subjects.
It is difficult to assert that OSA is at an early stage. However, absence of known cardiovascular disease, particularly known hypertension, and absence of former OSA treatment is in favor of a recent disease.
We have used simplified techniques for assessing OSA diagnosis and severity in a subgroup of the included patients. However, in our study, LVM and carotid hypertrophy were mainly correlated with SaO2 parameters which are properly assessed by simplified sleep studies.
Conclusion
Our population of newly diagnosed OSA patients without clinically diagnosed cardiovascular diseases presented early signs of atherosclerosis and nascent cardiac damage. The severity of the OSA appeared to play more of a role in LV remodeling than in arterial modification. A more systematic study of functional and structural cardiac and arterial modifications in OSA patients could improve the stratification of their cardiovascular risk and help identify candidates for earlier and more aggressive OSA therapy.
Disclosures
The authors report no conflicts of interest in this work.
References
- KapurVStrohlKPRedlineSIberCO’ConnorGNietoJUnderdiagnosis of sleep apnea syndrome in U.S. communitiesSleep Breath20026495412075479
- MillerWPCardiac arrhythmias and conduction disturbances in the sleep apnea syndrome. Prevalence and significanceAm J Med1982733173217124758
- YoungTPeppardPPaltaMPopulation-based study of sleep-disordered breathing as a risk factor for hypertensionArch Intern Med1997157174617529250236
- NietoFJYoungTBLindBKAssociation of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health StudyJAMA20002831829183610770144
- MooeTFranklinKAHolmströmKRabbenTWiklundUSleep-disordered breathing and coronary artery disease: long-term prognosisAm J Respir Crit Care Med20011641910191311734445
- WolkRKaraTSomersVKSleep-disordered breathing and cardiovascular diseaseCirculation200310891212847053
- Ancoli-IsraelSDuhamelERStepnowskyCEnglerRCohen-ZionMMarlerMThe relationship between congestive heart failure, sleep apnea, and mortality in older menChest20031241400140514555572
- KanagalaRMuraliNSFriedmanPAObstructive sleep apnea and the recurrence of atrial fibrillationCirculation20031072589259412743002
- LaviePLavieLHererPAll-cause mortality in males with sleep apnoea syndrome: declining mortality rates with ageEur Respir J20052551452015738297
- YaggiHKConcatoJKernanWNLichtmanJHBrassLMMohseninVObstructive sleep apnea as a risk factor for stroke and deathN Engl J Med20053532034204116282178
- BaguetJPNarkiewiczKMallionJMUpdate on hypertension management: obstructive sleep apnea and hypertensionJ Hypertens20062420520816331122
- BassettiCLMilanovaMGuggerMSleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcomeStroke20063796797216543515
- LeuenbergerUJacobESweerLWaravdekarNZwillichCSinowayLSurges of muscle sympathetic nerve activity during obstructive apnea are linked to hypoxemiaJ Appl Physiol1995795815887592221
- SomersVKDykenMEClaryMPAbboudFMSympathetic neural mechanisms in obstructive sleep apneaJ Clin Invest199596189719047560081
- NarkiewiczKSomersVKThe sympathetic nervous system and obstructive sleep apnea: implications for hypertensionJ Hypertens199715161316199488212
- SvatikovaAWolkRLermanLOOxidative stress in obstructive sleep apnoeaEur Heart J2005262435243916105851
- ShamsuzzamanASWinnickiMLanfranchiPElevated C-reactive protein in patients with obstructive sleep apneaCirculation20021052462246412034649
- von KänelRLoredoJSAncoli-IsraelSMillsPJNatarajanLDimsdaleJEAssociation between polysomnographic measures of disrupted sleep and prothrombotic factorsChest200713173373917356087
- NietoFJHerringtonDMRedlineSBenjaminEJRobbinsJASleep apnea and markers of vascular endothelial function in a large community sample of older adultsAm J Respir Crit Care Med200416935436014551166
- PignoliPTremoliEPoliAIntimal plus medial thickness of the arterial wall: a direct measurement with ultrasound imagingCirculation198674139914063536154
- BlacherJAsmarRDjaneSLondonGMSafarMEAortic pulse wave velocity as a marker of cardiovascular risk in hypertensive patientsHypertension1999331111111710334796
- LaurentSBoutouyriePAsmarRAortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patientsHypertension2001371236124111358934
- Willum-HansenTStaessenJATorp-PedersenCPrognostic value of aortic pulse wave velocity as index of arterial stiffness in the general populationCirculation200611366467016461839
- RomanMJGanauASabaPSImpact of arterial stiffening on left ventricular structureHypertension20003648949411040224
- SilvestriniMRizzatoBPlacidiFBaruffaldiRBianconiADiomediMCarotid artery wall thickness in patients with obstructive sleep apnea syndromeStroke2002331782178512105352
- KaynakDGöksanBKaynakHDegirmenciNDagliogluSIs there a link between the severity of sleep-disordered breathing and atherosclerotic disease of the carotid arteriesEur J Neurol20031048749312940827
- SuzukiTNakanoHMaekawaJObstructive sleep apnea and carotid-artery intima-media thicknessSleep20042712913314998249
- BaguetJPHammerLLévyPThe severity of oxygen desaturation is predictive of carotid wall thickening and plaque occurrenceChest20051283407341216304292
- MinoguchiKYokoeTTazakiTIncreased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apneaAm J Respir Crit Care Med200517262563016120716
- SchulzRSeegerWFegbeutelCChanges in extracranial arteries in obstructive sleep apnoeaEur Respir J200525697415640325
- DragerLFBortolottoLALorenziMCFigueiredoACKriegerEMLorenzi-FilhoGEarly signs of atherosclerosis in obstructive sleep apneaAm J Respir Crit Care Med200517261361815901608
- TsioufisCThomopoulosKDimitriadisKThe incremental effect of obstructive sleep apnoea syndrome on arterial stiffness in newly diagnosed essential hypertensive subjectsJ Hypertens20072514114617143185
- WattanakitKBolandLPunjabiNMShaharERelation of sleep-disordered breathing to carotid plaque and intima-media thicknessAtherosclerosis200819712513117433330
- ProtogerouADLaabanJPCzernichowSStructural and functional arterial properties in patients with obstructive sleep apnoea syndrome and cardiovascular comorbiditiesJ Hum Hypertens20082241542218075519
- KraicziHPekerYCaidahlKSamuelssonAHednerJBlood pressure, cardiac structure and severity of obstructive sleep apnea in a sleep clinic populationJ Hypertens2001192071207811677374
- NiroumandMKupersteinRSassonZHanlyPJImpact of obstructive sleep apnea on left ventricular mass and diastolic functionAm J Respir Crit Care Med20011631632163611401886
- ClowardTVWalkerJMFarneyRJAndersonJLLeft ventricular hypertrophy is a common echocardiographic abnormality in severe obstructive sleep apnea and reverses with nasal continuous positive airway pressureChest200312459460112907548
- DursunogluDDursunogluNEvrengülHImpact of obstructive sleep apnoea on left ventricular mass and global functionEur Respir J20052628328816055877
- 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC)J Hypertens2007251105118717563527
- AsmarRVolSBrisacAMTichetJTopouchianJReference values for clinic pulse pressure in a nonselected populationAm J Hypertens20011441541811368460
- RechtschaffenAKalesAA manual of standardized terminology, techniques and scoring system for sleep stages of human subjectsWashington DCNational Institutes of Health1968
- ArgodJPépinJLSmithRPLévyPComparison of esophageal pressure with pulse transit time as a measure of respiratory effort for scoring obstructive nonapneic respiratory eventsAm J Respir Crit Care Med2000162879310903225
- The Report of an American Academy of Sleep Medicine Task Force. Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical researchSleep19992266768910450601
- DevereuxRBReichekNEchocardiographic determination of left ventricular mass in manCirculation197755613618138494
- DevereuxRBAlonsoDRLutasEMEchocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findingsAm J Cardiol1986574504582936235
- GanauADevereuxRBRomanMJPatterns of left ventricular hypertrophy and geometric remodeling in essential hypertensionJ Am Coll Cardiol199219155015581534335
- De SimoneGDevereuxRBRomanMJAldermanMHLaraghJHRelation of obesity and gender to left ventricular hypertrophy in normotensive and hypertensive adultsHypertension1994236006068175168
- BaguetJPMallionJMMoreau-GaudryANoirclercMPéoc’hMSichéJPRelationships between cardiovascular remodeling and the pulse pressure in never treated hypertensionJ Hum Hypertens200014233010673727
- Bonithon-KoppCDucimetièrePTouboulPJPlasma angiotensin-converting enzyme activity and carotid wall thickeningCirculation1994899529548124834
- HlaKMYoungTBBidwellTPaltaMSkatrudJBDempseyJSleep apnea and hypertension. A population-based studyAnn Intern Med19941203823888304655
- The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 reportJAMA20032892560257212748199
- SegaRTrocinoGLanzarottiAAlterations of cardiac structure in patients with isolated office, ambulatory, or home hypertension: Data from the general population (Pressione Arteriose Monitorate E Loro Associazioni [PAMELA] Study)Circulation20011041385139211560854
- WeissJWRemsburgSGarpestadERinglerJSparrowDParkerJAHemodynamic consequences of obstructive sleep apneaSleep1996193883978843530
- MorganBJDempseyJAPegelowDFBlood pressure perturbations caused by subclinical sleep-disordered breathingSleep19982173774611286350
- LesskeJFletcherECBaoGUngerTHypertension caused by chronic intermittent hypoxia – influence of chemoreceptors and sympathetic nervous systemJ Hypertens199715159316039488210
- KanagyNLWalkerBRNelinLDRole of endothelin in intermittent hypoxia-induced hypertensionHypertension20013751151511230327
- MøllerDSLindPStrungeBPedersenEBAbnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apneaAm J Hypertens20031627428012670743
- WilcoxCSReactive oxygen species: roles in blood pressure and kidney functionCurr Hypertens Rep2002416016611884272
- KatoMRoberts-ThomsonPPhillipsBGImpairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apneaCirculation20001022607261011085964
- OkamotoRHataniMTsukitaniMThe effect of oxygen on the development of atherosclerosis in WHHL rabbitsAtherosclerosis19834747536870989
- KourembanasSMoritaTChristouHHypoxic responses of vascular cellsChest1998114Suppl 125S28S9676610
- GrobbeeDEBotsMLCarotid artery intima-media thickness as an indicator of generalized atherosclerosisJ Intern Med19942365675737964435
- HerringtonDMBrownWVMoscaLRelationship between arterial stiffness and subclinical aortic atherosclerosisCirculation200411043243715262851
- SassCHerbethBChapetOSiestGVisvikisSZannadFIntima-media thickness and diameter of carotid and femoral arteries in children, adolescents and adults from the Stanislas cohort: effect of age, sex, anthropometry and blood pressureJ Hypertens199816159316029856359
- HednerJEjnellHCaidahlKLeft ventricular hypertrophy independent of hypertension in patients with obstructive sleep apnoeaJ Hypertens199089419462174947
- DaviesRJCrosbyJProtheroAStradlingJRAmbulatory blood pressure and left ventricular hypertrophy in subjects with untreated obstructive sleep apnoea and snoring, compared with matched control subjects, and their response to treatmentClin Sci (Lond)1994864174248168336
- NodaAOkadaTYasumaFNakashimaNYokotaMCardiac hypertrophy in obstructive sleep apnea syndromeChest1995107153815447781343
- BudaAJPinskyMRIngelsNBJrDaughtersGT2ndStinsonEBAldermanELEffect of intrathoracic pressure on left ventricular performanceN Engl J Med1979301453459460363
- VirolainenJVentiläMTurtoHKupariMEffect of negative intrathoracic pressure on left ventricular pressure dynamics and relaxationJ Appl Physiol1995794554607592202
- BradleyTDHallMJAndoSFlorasJSHemodynamic effects of simulated obstructive apneas in humans with and without heart failureChest20011191827183511399711
- TanriverdiHEvrengulHKaftanAEffect of obstructive sleep apnea on aortic elastic parameters: relationship to left ventricular mass and functionCirc J20067073774316723796
- TavilYKanbayASenNThe relationship between aortic stiffness and cardiac function in patients with obstructive sleep apnea, independently from systemic hypertensionJ Am Soc Echocardiogr20072036637217400115