
Abstract
Aim: A confirmed wild-type RAS tumor status is commonly required for prescribing anti-EGFR treatment for metastatic colorectal cancer. This noninterventional, observational research project estimated RAS mutation prevalence from real-world sources. Materials & methods: Aggregate RAS mutation data were collected from 12 sources in three regions. Each source was analyzed separately; pooled prevalence estimates were then derived from meta-analyses. Results: The pooled RAS mutation prevalence from 4431 tumor samples tested for RAS mutation status was estimated to be 43.6% (95% CI: 38.8–48.5%); ranging from 33.7% (95% CI: 28.4–39.3%) to 54.1% (95% CI: 51.7–56.5%) between sources. Conclusion: The RAS mutation prevalence estimates varied among sources. The reasons for this are not clear and highlight the need for further research.
Keywords: :
Colorectal cancer (CRC) was the third most common malignancy among men and the second most common malignancy among women globally in 2012 [Citation1]. Approximately, 60% of CRC cases occur in developed regions and incidence rates can vary up to tenfold around the world, with the lowest rates observed in South Central Asia and Africa (excluding Southern Africa) [Citation2]. A quarter of patients with CRC already have metastatic disease at the time of their initial presentation and diagnosis, and almost 50% will subsequently develop metastases [Citation3]. Once metastatic disease has emerged, the 5-year survival rate is approximately 5–15%, with a median overall survival of 9–30 months [Citation4].
In the past decade, new treatment options have become available to patients with metastatic CRC (mCRC) that include the use of irinotecan- and oxaliplatin-containing chemotherapies, the use of antiangiogenic drugs, such as bevacizumab, and the introduction of monoclonal antibodies (mAbs), such as cetuximab and panitumumab, which target the EGFR [Citation5]. A number of studies have shown that RAS biomarker status (exons 2, 3 and 4 of the KRAS and NRAS oncogenes) is predictive of patients’ treatment response to anti-EGFR mAbs specifically, and patients who have RAS wild-type tumors are significantly more likely to benefit from anti-EGFR treatment [Citation6–10]. As a result, treatment guidelines for anti-EGFR therapies recommend that patients with mCRC should have confirmed wild-type RAS tumor status prior to the initiation of treatment [Citation11]. There have been a limited number of studies that have attempted to use real-world data to estimate the prevalence of RAS mutations among patients with mCRC. The majority of data relating to RAS mutation prevalence in mCRC, which have been published previously, are reported from clinical trials.
The primary objective of this research project was to estimate the prevalence of RAS mutations in patients with mCRC using different real-world data sources. These estimates include an overall estimate and estimates calculated according to patients’ demographic and clinical characteristics. Additional objectives included estimating the prevalence of non-KRAS exon 2 mutations (KRAS exons 3, 4 and NRAS exons 2, 3, 4) in patients with a confirmed wild-type KRAS exon 2 status and estimating the prevalence of BRAF mutations among patients with mCRC.
Materials & methods
Data sources
This noninterventional, observational research project used aggregate patient data, rather than individual patient data, from multiple real-world data sources to estimate RAS mutation prevalence. Data were collected from 12 data sources (three local mCRC registries, two industry-sponsored studies [Amgen] and seven pathology centers) across 12 countries (Algeria, Argentina, Bahrain, Czech Republic, Egypt, Greece, Hungary, Kuwait, Lebanon, Poland, Saudi Arabia and the United Arab Emirates) (). Recruitment periods for the observational studies and registries providing the aggregate data spanned a range of different time periods (from 2012 to 2015). The study period for the majority of the data sources was between January and December 2014.
Table 1. Data sources included in the analysis.
The data were collected using a standardized online data collection form for all samples according to both their RAS mutation status and a number of demographic and clinical variables. In addition, the turnaround times for RAS testing of the samples were also requested. The online data collection form was distributed to each data source, with data entry organized locally. A pilot trial of the online form was conducted to ensure any issues with the electronic data collection were addressed before full implementation of the form. Data checks were subsequently carried out after receiving data from each source to determine if there were any errors or missing data that required correction or clarification. Turnaround time was defined as the number of days between the laboratory receiving the request for a RAS test to be performed and the result being reported to the requesting oncologist. Only aggregate data were requested with no possible link to individual patient information.
Patient eligibility criteria
To be eligible for the study, patients were required to have a recorded diagnosis of mCRC and a tumor sample that had been tested for RAS mutation status. Some samples were initially tested for KRAS exon 2 status only, with testing for the remaining RAS exons (KRAS exons 3, 4 and NRAS exons 2, 3, 4) conducted at a later time point. In practice, samples were likely to have been collected at different time points (e.g., time of surgery or immediately prior to initiation of anti-EGFR therapy), including some which may have been collected prior to metastatic diagnosis (most likely stage III CRC).
Statistical analysis
The prevalence of RAS and BRAF mutations (defined generically herein as ‘biomarker’ [bm]) was estimated as follows:
The prevalence of RAS and BRAF mutations was estimated from all samples taken from patients with mCRC. The prevalence of non-KRAS exon 2 mutations (i.e., mutations of KRAS exons 3, 4 and NRAS exons 2, 3, 4) was also estimated for all samples from patients with mCRC and a confirmed wild-type KRAS exon 2 status. The prevalence estimates are reported with their corresponding 95% CIs. The Clopper–Pearson (exact) method was used for CI calculation. For each data source, prevalence was also estimated according to a number of covariate patient characteristics, including patient age, gender, the site the tumor sample was taken from, the clinical indication for sampling, presence or absence of metastases, tumor staging and line of treatment at the time of testing. Comparison between the prevalence estimates by different variables was carried out based on 95% CIs and p-values (using Pearson χ2 testing).
The meta-analysis was performed using a random effects model to consolidate the results of the primary outcome analysis, and account for the heterogeneity of the data sources, particularly with respect to variables such as testing methods, time periods and patient characteristics. The Freeman–Tukey double arcsine transformation method was used to stabilize the variance. The Hodges–Lehman estimator – a robust and nonparametric estimator of a population's location parameter – was used for the overall point estimate of prevalence. The meta-analysis was carried out using the ‘meta’ command in Stata (StataCorp. 2013. Stata Statistical Software: Release 2013, StataCorp LP, TX, USA).
Results
Aggregate data from 4431 tumor samples from 12 data sources were included in the RAS mutation prevalence analysis. Aggregate data from 2561 tumor samples from six data sources were included in the BRAF analysis. Aggregate data from 2606 tumor samples from 11 data sources were included in the analysis of non-KRAS exon 2 (KRAS exons 3, 4 and NRAS exons 2, 3, 4) samples with KRAS exon 2 wild-type samples. The size of these data sources varied from 100 tumor samples (Czech Republic pathology centers) to 1664 tumor samples (Polish pathology center) ().
RAS mutation prevalence
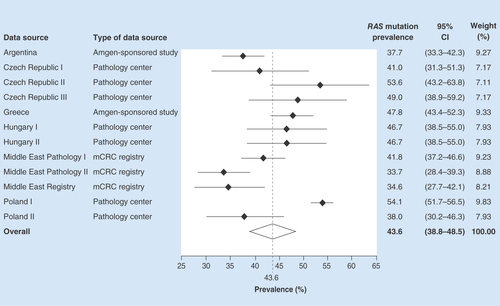
The overall pooled RAS mutation prevalence across the 12 data sources included in the analysis was estimated to be 43.6% (95% CI: 38.8–48.5%). RAS mutation prevalence varied between data sources, ranging from 33.7% (95% CI: 28.4–39.3%) in the Middle Eastern pathology dataset and 34.6% (95% CI: 27.7–42.1%) in the Middle Eastern mCRC registry to 53.6% (95% CI: 43.2–63.8%) and 54.1% (95% CI: 51.7–56.5%) in two European pathology centers, in the Czech Republic and Poland, respectively ().
Forest plot showing biomarker prevalence by data source.
mCRC: Metastatic colorectal cancer.
RAS prevalence by patient characteristics
Pooled RAS mutation prevalence estimates varied according to certain demographics and clinical variables. Although none of the differences were statistically significant, numerically greater RAS mutation prevalence estimates were observed
in: patients who were aged ≥70 years compared with younger patients; tumor samples isolated from metastatic rather than primary tumor sites; and tumor samples with a ratio of ≥20% neoplastic cells to normal cells in the tumor sample versus <20% ().
Table 2. Pooled RAS prevalence estimates by patient characteristics.
There were no differences in prevalence estimates for any of the other variables assessed including: patient gender; the tissue from which the sample DNA was isolated (left colon, right colon or rectum); whether or not there was evidence of liver metastases at the time the sample was taken; if there were synchronous or metachronous metastases; or tumor stage ().
BRAF mutation prevalence
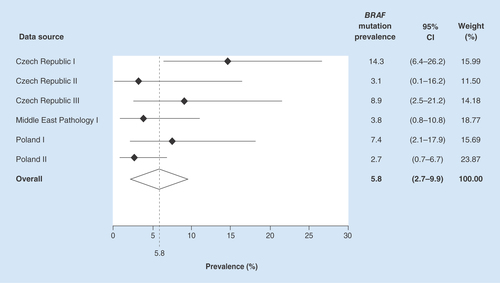
The overall pooled BRAF mutation prevalence was estimated as 5.8% (95% CI: 2.7–9.9%) across the six data sources. The BRAF mutation prevalence estimates varied from 2.7% (95% CI: 0.7–6.7%) in one of the Poland pathology datasets to 14.3% (95% CI: 6.4–26.2%) in one of the Czech Republic pathology datasets ().
Non-KRAS exon 2 mutation prevalence in KRAS exon 2 wild-type samples
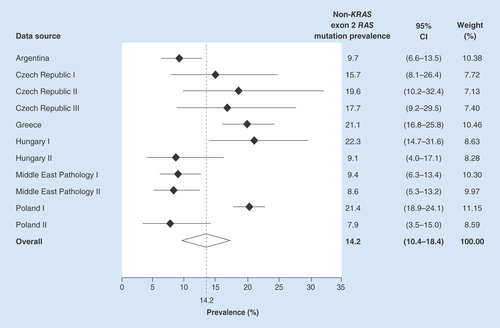
The overall pooled non-KRAS exon 2 mutation prevalence, in KRAS exon 2 wild-type samples, was estimated as 14.2% (95% CI: 10.4–18.4%). The prevalence estimates varied from 8.6% (95% CI: 5.3–13.2%) in one of the Middle Eastern mCRC pathology datasets to 22.3% (95% CI: 14.7–31.6%) in a Hungarian pathology center ().
RAS testing turnaround time
Eleven data sources reported RAS testing turnaround times for 3832 samples. Nearly all (94.9%) of the RAS mutation test results were reported to the requesting physician within 10 working days of the test being requested, with 57.0% being reported in 1–5 days, 38.0% in 6–10 days and only 5.1% in >10 days ().
Table 3. RAS mutation testing turnaround time by data source.
Discussion
To our knowledge, this is the largest research project to date to carry out a pooled analysis of RAS mutation prevalence specifically using real-world data. The overall pooled RAS mutation prevalence across all data sources included in the study was estimated to be 43.6% (95% CI: 38.8–48.5%). However, the RAS mutation prevalence estimates for individual data sources included in this analysis varied considerably. Elucidating the reasons for this variability was beyond the scope of the current study but may relate to the ethnic diversity of the patient cohorts in the various countries/regions from which the data were derived. Numerous epidemiological data show major geographical variation with significantly higher risk of CRC in affluent societies and prospective cohort data have linked dietary habits and lifestyle factors to CRC, which may contribute to RAS mutation heterogeneity [Citation12]. The largely lower rate of CRC seen in developing countries of Asia and Africa compared with western countries may also relate to the fact that studies on the prevalence and raw data are very blurred and not definite [Citation13]. The current study highlights the need for further research regarding the reasons for geographical variation in RAS mutation prevalence.
The overall prevalence estimate from this study is somewhat different to estimates reported from past clinical trials. A retrospective analysis of pooled data from five randomized controlled studies (RCTs) of panitumumab estimated the overall unadjusted RAS mutation prevalence in patients with mCRC as 55.9% (95% CI: 53.9–57.9%); however, the prevalence estimate presented here is more consistent with the results of another recent real-world study, which found the overall RAS mutation prevalence reported by European pathology laboratories to be 48.5% (95% CI: 46.4–50.6%) [Citation11,Citation12]. This suggests that the actual prevalence in real-world clinical practice may be lower, meaning that more patients are potential candidates for anti-EGFR treatment than might have been previously thought. Potential reasons for this apparent imbalance may relate to the necessary application of restrictive RCT selection criteria to determine which patients are eligible to participate in controlled trials and which may result in study cohorts not representing the wider patient population observed in routine clinical practice. The wider availability of next-generation sequencing for patient selection or characterization in an RCT setting may also have driven the apparent bias toward an overestimate of the prevalence of RAS mutation among patients with mCRC. This approach may have a lower detection limit than standard laboratory tests for the detection of low copy number mutant alleles [Citation13]. Alternatively, standard laboratory tests may be less sensitive than those used during RCTs.
The overall pooled BRAF mutation prevalence of 5.8% was similar to findings from previous RCTs, where BRAF mutation prevalence was estimated as 8.1% (95% CI: 6.7–9.6%) [Citation11]. Similarly, the prevalence of other RAS mutations (KRAS exons 3, 4 and NRAS exons 2, 3, 4) in samples with a confirmed KRAS exon 2 wild-type status was numerically lower than the estimate reported by a recent meta-analysis of both panitumumab and cetuximab RCTs, which found that 19.9% (95% CI: 16.7–23.4%) of samples had a non-KRAS exon 2 mutation. However, the results are within the margin of error of each other [Citation14].
The RAS testing turnaround time was no greater than 10 working days for nearly all of the samples (94.9%) included in the different data sources. This is marginally higher than reported in a survey of pathology centers in Europe, carried out at the end of 2014, which estimated that for 90.8% of the laboratories the turnaround time was less than 10 working days [Citation12]. However, this estimate only includes the time between a laboratory receiving the sample and reporting back to the physician. Hence, it is likely that the time from a physician making the request to receiving the sample result is actually longer.
Data collection methodology among pathology laboratories has previously been shown to be highly variable, with a number of different DNA extraction and mutation testing methods being used [Citation15]. This is important to note given the recognized differences in sensitivity among different genotype-ascertainment methodologies [Citation16,Citation17]. In addition, potential quality issues of sample preparation could explain some of the variability. One of the limitations of this research project is that it did not collect data relating to the quality of the DNA samples tested or ascertain if the pathology laboratories involved in the testing were participating in an external quality assurance scheme. This highlights the importance of external quality assurance schemes, particularly given the number of RAS testing methods that have not yet been validated and the lack of standardization among pathology laboratories [Citation16,Citation18,Citation19]. Blood-based RAS mutation analysis is now available in many countries and may address some of these concerns if used in future clinical trials [Citation20]. Although institutes may or may not be accredited by an independent body, it is a requirement of the European Medicines Authority that RAS mutational status should be determined by an experienced laboratory using validated test methods.
Additionally, the prevalence estimates reported here should be interpreted with caution as the data sources are highly heterogeneous. The heterogeneity of the data could be explained by differences in data collection methodology and the original purposes of the data collection. Differences between eligibility criteria, patient characteristics and study periods may have contributed to RAS prevalence differences. Some of the included samples may have been initially tested for KRAS exon 2 status with testing for the remaining RAS exons (KRAS exons 3, 4 and NRAS exons 2, 3, 4) conducted at a later time point. In practice, samples were likely to have been collected at different time points (e.g., time of surgery), including some which may have been collected prior to metastatic diagnosis (most likely stage III CRC).
Conclusion
This pooled analysis of RAS mutation prevalence estimates from real-world data sources found the overall RAS mutation prevalence among patients with mCRC to be somewhat lower than reported in clinical trials.
Future perspective
New knowledge of biomarkers involved in mCRC will impact the future screening, diagnosis and treatment of the disease. This may lead to new anticancer agents that are selective for certain subgroups of patients and to more precise use of the currently available agents. This will increase treatment options available to clinicians and may lead to tailored individualistic approaches, decreasing treatment cost and the adverse effects related to mAb therapy.
RAS mutation status is an important predictive biomarker of successful outcome for patients with metastatic colorectal cancer (mCRC) treated with anti-EGFR therapies such as cetuximab and panitumumab.
Therefore, in many countries, a confirmed wild-type RAS (exons 2, 3, 4 of KRAS and NRAS) tumor status is a requirement for prescribing anti-EGFR treatment to patients with mCRC.
Our currently limited understanding of RAS mutation prevalence in mCRC is largely derived from clinical trials; this research project aims to estimate the prevalence on RAS mutations based on real-world data sources.
Aggregate patient data were collected from a range of geographically diverse real-world sources, by means of an online standardized form, and included in an overall meta-analysis.
The overall pooled RAS mutation prevalence for 4431 tumor samples across 12 data sources was estimated to be 43.6%.
RAS mutation prevalence varied between data sources, ranging from low estimates of 33.7% in a Middle Eastern pathology dataset and 34.6% in a Middle Eastern mCRC registry to the highest estimates of 53.6% and 54.1% in two European pathology centers, in the Czech Republic and Poland, respectively.
The overall pooled RAS mutation prevalence was lower than previously reported in a pooled analysis of randomized controlled studies of anti-EGFR treatment; however, it was more consistent with the results of a similar, albeit smaller, real-world study.
Therefore, the actual real-world prevalence may be lower than reported in randomized controlled studies, meaning that more patients are potential candidates for anti-EGFR treatment than might have been previously thought.
Authors’ contributions
All authors were involved in, and contributed to, the drafting and critical review of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Acknowledgements
The authors would like to thank and acknowledge the following people for their contributions to this article: A Bray (Amgen Ltd) for study management support; S Jani and the Adelphi Research team for data management; E Benes-Javor (Adelphi Research) for providing statistical input; D Spanopoulos (Amgen Ltd) for data management support; and Adelphi Communications for editorial assistance and support (funded by Amgen Ltd).
Financial & competing interests disclosure
This research project was funded by Amgen Ltd. Amgen was also involved in the study design, data collection and analysis, decision to publish and preparation of the manuscript. Individual conflict of interest disclosures are given below. G Kafatos is a compensated employee of Amgen Ltd as an Observational Research Senior Manager and a stockholder in Amgen Ltd. D Niepel is a compensated employee of Amgen GmbH as a Medical Director and a stockholder in Amgen GmbH. K Lowe is a compensated employee of Amgen Inc. as an Epidemiology Senior Manager and a stockholder in Amgen Inc. T Garawin is a compensated employee of Amgen Inc. as the Head of Oncology (Middle East) and a stockholder in Amgen Inc. Z Traugottová is a compensated employee of Amgen s.r.o. as a Senior Medical Advisor and a stockholder in Amgen Ltd. A Bilalis is a compensated employee of Amgen Hellas EPE as a Senior Medical Advisor. E Molnar is a compensated employee of Amgen Kft as a Senior Medical Advisor. J Timar has received honoraria from Amgen, Boehringer Ingelheim, Eli Lilly, Merck Serono, Pfizer and Roche. P Fabian has received consultation fees from Amgen and Pfizer, and honoraria from Amgen, AstraZeneca, Merck Serono, Pfizer and Roche. J Han van Krieken has received research funding from Amgen and Merck Serono, speaker fees from Amgen, Merck Serono, Agilent (DAKO) and Roche and consulting fees from Milestone Pharmaceuticals and Sakura. J Trojan has received speaker fees from Amgen and Bayer, and consulting fees from Bayer, Eli Lilly and Merck Serono. S Murray has received consulting fees from Amgen. P Wójcik is a Co-owner and Board member of Oncogene Diagnostics sp. z o.o. S Jenkins-Anderson, H Westhead, E Toth, N Gouvas, G Papaxoinis, N Mokhtar, H Vosmikova, A Skalova, A Tysarowski and M Barugel have no competing interests to declare. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Amgen Ltd. provided funding for medical writing assistance and journal publication fees.
Additional information
Funding
References
- Torre LA , SiegelRL, WardEM, JemalA. Global cancer incidence and mortality rates and trends-an update. Cancer Epidemiol. Biomarkers Prev.25, 16–27 (2016).
- Ferlay J , SoerjomataramI, DikshitRet al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer136, E359–E386 (2015).
- van Cutsem E , CervantesA, NordlingerB, ArnoldD. ESMO Guidelines Working Group. Collaborators (20). Ann. Oncol.25(Suppl. 3), iii1–iii9 (2014).
- Tay RY , WongR, HawkesEA. Treatment of metastatic colorectal cancer: focus on panitumumab. Cancer Manag. Res.7, 189–198 (2015).
- Moorcraft SY , SmythEC, CunninghamD. The role of personalized medicine in metastatic colorectal cancer: an evolving landscape. Therap. Adv. Gastroenterol.6, 381–395 (2013).
- Douillard JY , OlinerKS, SienaSet al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med.369, 1023–1034 (2013).
- Schwartzberg LS , RiveraF, KarthausMet al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J. Clin. Oncol.32, 2240–2247 (2014).
- Heinemann V , von WeikersthalLF, DeckerTet al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol.15, 1065–1075 (2014).
- Seymour MT , BrownSR, MiddletonGet al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol.14, 749–759 (2013).
- Peeters M , DouillardJY, van CutsemEet al. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin. Oncol.31, 759–765 (2013).
- Peeters M , KafatosG, TaylorAet al. Prevalence of RAS mutations and individual variation patterns among patients with metastatic colorectal cancer: a pooled analysis of randomised controlled trials. Eur. J. Cancer51, 1704–1713 (2015).
- Boleij A , TackV, TaylorAet al. RAS testing practices and RAS mutation prevalence amongst mCRC patients: results from a Europe-wide survey of pathology centres. BMC Cancer16, 825 (2016) ( In Press ).
- Peeters M , OlinerKS, ParkerAet al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin. Cancer Res.19, 1902–1912 (2013).
- Sorich MJ , WieseMD, RowlandAet al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann. Oncol.26, 13–21 (2015).
- Boleij A , TopsBB, RomboutPDet al. RAS testing in metastatic colorectal cancer: excellent reproducibility amongst 17 Dutch pathology centers. Oncotarget6, 15681–15689 (2015).
- Wong NA , GonzalezD, Salto-TellezMet al. RAS testing of colorectal carcinoma – a guidance document from the Association of Clinical Pathologists Molecular Pathology and Diagnostics Group. J. Clin. Pathol.67, 751–757 (2014).
- Malapelle U , CarlomagnoC, de LucaCet al. KRAS testing in metastatic colorectal carcinoma: challenges, controversies, breakthroughs and beyond. J. Clin. Pathol.67, 1–9 (2014).
- van Krieken JH , JungA, KirchnerTet al. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch.453, 417–431 (2008).
- Tack V , LigtenbergMJ, TembuyserLet al. External quality assessment unravels interlaboratory differences in quality of RAS testing for anti-EGFR therapy in colorectal cancer. Oncologist20, 257–262 (2015).
- Li Y , FuXH, YuanJQet al. Colorectal cancer: using blood samples and tumor tissue to detect K-ras mutations. Expert Rev. Anticancer Ther.15, 715–725 (2015).