
Abstract
In psychopathology research, endophenotypes are a subset of biomarkers that indicate genetic vulnerability independent of clinical state. To date, an explicit expectation is that endophenotypes be specific to single disorders. We evaluate this expectation considering recent advances in psychiatric genetics, recognition that transdiagnostic vulnerability traits are often more useful than clinical diagnoses in psychiatric genetics, and appreciation for etiological complexity across genetic, neural, hormonal and environmental levels of analysis. We suggest that the disorder-specificity requirement of endophenotypes be relaxed, that neural functions are preferable to behaviors as starting points in searches for endophenotypes, and that future research should focus on interactive effects of multiple endophenotypes on complex psychiatric disorders, some of which are ‘phenocopies’ with distinct etiologies.
Nearly a decade ago, I (TP Beauchaine) wrote an editorial for this journal in which I summarized differences between endophenotypes and other biomarkers [Citation1]. It is an honor to write a sequel to that editorial following Irving Gottesman's recent death. Gottesman brought the endophenotype concept to psychiatry from evolutionary biology, where John and Lewis [Citation2] discovered that grasshopper adaptation to local environments could not be explained by exophenotypic variation, but could be explained by endophenotypic variation – genetically induced individual differences expressed at the microscopic rather than macroscopic level of analysis. Although this insight ran counter to Darwin's account of evolution by natural selection of exophenotypes, it is now clear that endophenotypes of all kinds confer selective advantages in local environments. The sickle-cell trait protecting against malaria infection provides one example.
Ten years ago, there was widespread optimism in the psychopathology research community that identifying endophenotypes could lead to major breakthroughs in psychiatric genetics. Such optimism was spawned in large part by Gottesman and Gould's [Citation3] now famous paper on the promise of endophenotypes. This paper, which has been cited thousands of times, defined the endophenotype concept, explained how it could be applied in schizophrenia research to pinpoint molecular genetic targets, and identified eye-tracking dysfunction and sensorimotor gating as likely endophenotypes [Citation1,Citation3]. In a follow-up article, Gould and Gottesman [Citation4] restricted the definition of endophenotype to make the concept more useful for psychiatric genetics. Our objectives in writing this essay are to revisit the definition of endophenotype, and to contextualize its current scientific utility in behavioral neuroscience given claims that core assumptions made by those who conduct endophenotype research are mistaken [Citation5], lingering confusion about important distinctions between endophenotypes and other types of biomarkers, major advances in psychiatric genetics, widespread recognition – as exemplified in the Research Domain Criteria initiative – that vulnerability traits are often more useful than clinical diagnoses as dependent variables in psychiatric genetics research [Citation6], and greater appreciation for etiological complexity across genetic, neural, hormonal and environmental levels of analysis. Discussing these points illustrates why the endophenotype concept is still promising, even though endophenotypes for specific psychiatric disorders remain elusive [Citation7]. Failed/incomplete attempts to identify endophenotypes could lead the field to prematurely abandon the concept. However, few studies conducted to date have evaluated endophenotypes of heritable vulnerability traits (e.g., impulsivity, irritability, anhedonia) that cut across traditional diagnostic boundaries. Refocusing endophenotype research on neurobiological mechanisms of such traits rather than traditional diagnoses may hold promise moving forward, as discussed below [Citation8]. Throughout this article, we maintain a clear distinction between vulnerability traits as latent (i.e., not overtly observable) liabilities to psychopathology, and symptoms as manifest (i.e., directly observable) signs of latent vulnerability. This is an important distinction to maintain when discussing endophenotypes, which by definition are not captured by overt symptoms, but are closer to pathophysiology.
Pathognomonic signs versus transdiagnostic vulnerabilities
Throughout the 20th century and up until quite recently, psychiatry and related disciplines were focused intensely on discovering pathognomonic signs of mental illness [Citation9]. Pathognomonic signs indicate unequivocally that a person has a specific disorder. In medicine, these signs are fundamental to effective diagnosis and treatment. For example, malaria parasite antigens in the bloodstream confirm that a patient is either infected, or suffered an infection in the past. No other disease process and no other etiology is possible. If antigens are identified, antimalarial treatment can be initiated to prevent primary or relapse infections. This simple example illustrates why pathognomonic signs are so valuable; they are sensitive, specific, and denote a fundamental understanding of disease process, facilitating prevention and more effective treatment.
An (often implicit) assumption of the search for pathognomonic signs in psychiatry is that different mental disorders arise from consistent, predictable, and largely independent pathophysiologies [Citation9,Citation10]. As elaborated below, this assumption undergirds the logic of the endophenotype concept, and must be met – at least to some degree – for endophenotypes to be discovered (see ‘The endophenotype concept defined’ section). Yet research conducted over the past decade suggests that very few psychiatric disorders arise from single disease processes, and that occurrences of highly similar neuropsychiatric conditions represent phenocopies with distinct pathogenic mechanisms (e.g., the intellectual disabilities and ‘autisms’). Moreover, many psychiatric conditions represent extreme behavioral expressions of multifactorially inherited traits for which no specific gene – and perhaps no particular combination of genes – is necessary or sufficient for psychiatric morbidity. In rare cases, the same disorders may be influenced strongly by highly deleterious genetic variants (e.g., 22q11.2 deletion in schizophrenia), yet that same variant may confer liability not only to schizophrenia but also to a range of phenotypes spanning cognitive disability to affective disorder to epilepsy. Furthermore, genetic and neural underpinnings of what have traditionally been conceptualized as distinct disorders are known to overlap [Citation11]. These genetic and neural processes give rise to transdiagnostic vulnerabilities that interact complexly with other genetic liabilities and other vulnerability traits, and are therefore not specific to current diagnostic classes [Citation9,Citation12–13]. Viewed in this context, and by way of example, the fact that dexamethasone nonsuppression characterizes some but not all forms of major depression warrants continual revisitation (rather than abandonment) of the test's usefulness as a biomarker of important etiological subtypes of depression with very different long-term courses [Citation14,Citation15]. More generally, such findings suggest that searches for new endophenotypes will be unproductive as long as we expect them to fulfill the criterion of being pathognomonic for disease entities that arise from heterogeneous causes and constitute, in essence, phenocopies of disparate biological processes.
The endophenotype concept defined
Gottesman and Gould described endophenotypes as ‘measurable components unseen by the unaided eye along the pathway between disease and distal genotype’ (p. 636) [Citation3]. They were agnostic regarding preferred levels of analysis, stating that endophenotypes could be represented in neurophysiology, biochemistry, neurocognition, self-report, or assorted other outcomes. Regardless of the level of analysis, to be useful endophenotypes must mark genetic vulnerability, and be amenable to more precise measurement than overt phenotypes. Gottesman and Gould recognized that measurement error is introduced at every level of analysis between genotypes and phenotypes, and that many if not most phenotypes are therefore imprecise (see ).
Relations among genotypes, endophenotypes and phenotypes (A) and putative differences in measurement precision (B). Endophenotypes, which can be specified at many levels of analysis including neurobiological and behavioral, fall along pathways between (i.e., ‘bridge’) genotypes and phenotypes. A traditional assumption is that measurement precision of endophenotypes exceeds that of phenotypes (see text). The leftmost graph of (B) depicts perfect measurement precision of a dichotomous genetic vulnerability, distributed in equal proportions (50:50) in a sample of n = 1000. This level of precision is associated with genotyping a monogenic trait. The middle graph depicts measurement of an endophenotype of large effect size. The right graph depicts a phenotype measured with 50% error – a situation common to ratings of observed behavior [Citation16]. As envisioned by Gottesman and Gould [Citation3], better measurement precision of endophenotypes facilitates identification of genotypes.
![Figure 1. Levels of analysis and measurement precision of genotypes, endophenotypes, and phenotypes.Relations among genotypes, endophenotypes and phenotypes (A) and putative differences in measurement precision (B). Endophenotypes, which can be specified at many levels of analysis including neurobiological and behavioral, fall along pathways between (i.e., ‘bridge’) genotypes and phenotypes. A traditional assumption is that measurement precision of endophenotypes exceeds that of phenotypes (see text). The leftmost graph of (B) depicts perfect measurement precision of a dichotomous genetic vulnerability, distributed in equal proportions (50:50) in a sample of n = 1000. This level of precision is associated with genotyping a monogenic trait. The middle graph depicts measurement of an endophenotype of large effect size. The right graph depicts a phenotype measured with 50% error – a situation common to ratings of observed behavior [Citation16]. As envisioned by Gottesman and Gould [Citation3], better measurement precision of endophenotypes facilitates identification of genotypes.](/cms/asset/9abc0dba-5429-4ca5-b908-1d2ccf14fee2/ibmm_a_12321058_f0001.jpg)
Following from this underlying philosophy, Gould and Gottesman [Citation4] specified the following six criteria for endophenotypes:
An endophenotype must segregate with illness in the general population;
An endophenotype must be heritable;
An endophenotype must be state independent, manifesting whether illness is present or in remission;
An endophenotype must cosegregate with the disorder within families;
An endophenotype must be present at a higher rate within affected families than in the general population;
An endophenotype should be a characteristic that can be measured reliably, and specific to the illness of interest.
These criteria specify how endophenotypes differ from other biomarkers (e.g., state-dependent biochemical features, surrogate end points, etc.) [Citation1]. By definition, endophenotypes mark genetic vulnerability independent of clinical state, so biomarkers that change with illness episodes do not qualify. For example, even though low levels of brain-derived neurotrophic factor (BDNF) are observed among individuals with bipolar disorder, such findings are restricted to acute manic episodes, and therefore change with clinical state [Citation17]. Thus, low BDNF does not qualify as an endophenotype and cannot be used to infer genetic liability during depressive or euthymic states. As psychiatry progresses from exclusively syndromal toward pathophysiological diagnosis, findings such as these will have clinical as well as research implications. Continuing with this example, if normal BDNF levels are used to incorrectly rule out bipolar disorder in a currently depressed individual who has yet to experience his/her first manic episode, antidepressant treatment for unipolar depression would ensue, with possible iatrogenic effects including increased suicide risk [Citation18]. This example and others illustrate why endophenotypes should not be equated with state-dependent biomarkers. One hope is that endophenotypes will eventually be used as prospective markers of vulnerability to psychopathology, facilitating prevention [Citation1,Citation3].
Complications for the endophenotype concept?
The endophenotype concept has been immensely successful in affecting how psychiatrists and other psychopathologists think about gene-behavior relations, and in stimulating new research. In 2003, when Gottesman and Gould published their now famous article [Citation3], 38 papers referenced endophenotypes. As of 4 August, 2017, 42,985 papers have done so. Although we cannot possibly summarize this literature, several themes present real or apparent problems for the endophenotype concept as envisioned by Gould and Gottesman. Some of these problems emerged soon after their second paper was published, when Flint and Munafò [Citation5] conducted a large meta-analysis to evaluate effect sizes for putative endophenotypes of several psychiatric disorders. The authors found no evidence to support the hypothesis that effect sizes for endophenotypes exceed those for exophenotypes (although this has traditionally been an assumption of endophenotypes, it need not be expected when endophenotypic liabilities confer additive, incremental and/or interactive vulnerability to disease [discussed later in this Perspective]).
More recently, a special issue of Psychophysiology was devoted to evaluating 17 putative electrophysiological endophenotypes of schizophrenia liability among nearly 5000 twins and parents in the Minnesota Twin Family Study, using genome-wide scans [Citation19]. Like the schizophrenia phenotype itself, electrophysiological markers were heritable, yet common molecular genetic variants (ascertained via genome-wide association) singly accounted for small proportions of their heritable variance [Citation20]. Similar findings for exophenotypes have plagued psychiatric genetics [Citation21], and suggest that the molecular genetic bases of schizophrenia are inordinately complex [Citation22] – a state of affairs that applies to most psychiatric disorders. Although some have suggested that such results undermine the endophenotype concept altogether, any such conclusion is premature given several considerations outlined below. Even for schizophrenia, as sample sizes have grown for molecular genetic analysis (some on the order five- to six-times the size of the Minnesota study), it has become tenable statistically to ascertain joint influences of numerous common variants, to identify allelic combinations that signal vulnerability to disease, and to explore associations between candidate endophenotypes and specific profiles of polygenic risk. In addition, preliminary studies using small samples suggest that genetic vulnerability may be manifested in certain neural functions [Citation23]. However, state dependence of these functions remains to be determined.
Loose definitions of endophenotypes
Gottesman and Gould [Citation3] were painstaking in their selection of putative endophenotypes, taking great care to ensure that their six criteria (see ‘The endophenotype concept defined’ section) were met. Irregularities in smooth pursuit eye tracking, for example, which are measured by sophisticated eye-tracking equipment, are observed in about 80% of those with schizophrenia, about 45% of their first-degree relatives, and about 10% of community controls [Citation3]. This 10% figure is compatible with the estimated base rate of schizophrenia liability in the general population (genetic liability is not fully penetrant, so only a fraction of the 10% develop the disorder) [Citation24]. These and other findings indicate that eye-tracking dysfunction segregates with illness in the population, is heritable, is state-independent, cosegregates with illness in pedigrees, is more common in pedigrees than in the general population, and can be measured precisely and is specific to schizophrenia liability [Citation1,Citation3]. Thus, eye-tracking dysfunction meets all six of Gould and Gottesman's criteria. It is also worth noting that in choosing eye-tracking dysfunction and sensorimotor gating as endophenotypes, Gottesman and Gould passed over many other biomarkers of schizophrenia that although useful in certain contexts, did not meet their criteria.
In contrast to such precaution, many papers in the psychopathology literature treat the terms endophenotype and biomarker interchangeably. In fact, many who study biological correlates of mental disorders refer to any and all such correlates as endophenotypes, without considering Gould and Gottesman's criteria [Citation25]. Furthermore, for most disorders, systematic evaluations of putative endophenotypes using the Gould and Gottesman's criteria have not been conducted. In the remainder of this article, we offer suggestions for revising the endophenotype concept, and for conducting more systematic evaluations given advances in our understanding of psychopathology in the past decade, particularly in the area of psychiatric genetics.
Problems with choosing endophenotypes based on current diagnostic categories: the case of ADHD
Several problems arise from the practice of considering all neurobiological correlates of psychopathology as endophenotypes [Citation25]. Some of these problems are illustrated in research on ADHD, for which several putative endophenotypes have been suggested [Citation26]. Genetic association studies consistently yield significant odds ratios for those with ADHD versus controls on several variants that affect dopamine (DA) neurotransmission [Citation27]. Some of these genes are expressed heavily in the striatum [Citation28], which is downregulated and therefore under-responsive in anticipation of incentives among those with ADHD [Citation28]. These deficits in striatal responding are a core neural substrate of impulsive decision-making, a hallmark of the ADHD phenotype [Citation29]. Collectively, such findings imply that striatal responding to incentives might be an endophenotype of ADHD [Citation26], particularly given a plausible causal pathway from genetic variation → neural responding → behavior. However, when applying Gould and Gottesman's criteria, it is readily apparent that striatal responding to incentives does not qualify as an endophenotype. Although some aspects of striatal DA function are heritable [Citation30], striatal under-responding to reward is also observed among those with other disruptive behavior disorders, including conduct disorder, antisocial personality disorder and substance-use disorders [Citation12,Citation31]. This lack of specificity might be explained away by high co-morbidities of ADHD with other externalizing disorders, yet additional findings present more intractable problems.
Blunted striatal responding to incentives is also observed among depressed children, adolescents, and adults [Citation32,Citation33], and among those who self-injure [Citation34]. As a result, it has been advanced as an endophenotype of depression by research groups who study mood disorders (in parallel to those who study ADHD) [Citation35]. Currently, although low striatal responding appears to mark premorbid risk for depression [Citation36], which suggests state independence, it is not specific to any syndrome (e.g., major depression) or class of disorders (e.g., disruptive behavior disorders). Although one course of action is to reject striatal responding as an endophenotype – another is to relax disorder specificity as a criterion for endophenotypes, and to include state-independent biomarkers that confer vulnerability to multiple disease states for which at least some individuals share pathophysiology.
Diagnostic categories versus transdiagnostic vulnerabilities
An emerging consensus in the psychopathology research community is that complex functional interactions among a limited number of neural and hormonal systems – far fewer in quantity than syndromes defined in the psychiatric nomenclature – give rise to many if not most mental health conditions [Citation6,Citation12]. From this perspective, endophenotypes might be more effectively reconstrued as markers of genetic liability to transdiagnostic vulnerability traits (e.g., impulsivity, irritability, anhedonia). As Skuse noted over 15 years ago, ‘…a focus on traits, rather than syndromes, is appropriate and could in due course contribute to the redefinition of traditional psychiatric syndromes’ (p. 395) [Citation37]. When reframed in this way, common neural correlates of psychopathology among what have traditionally been considered as distinct disorders are no longer a nuisance in our quest for greater specificity, but are instead opportunities to better understand common etiologies. This reformulated approach to understanding psychopathology undergirds the Research Domain Criteria initiative [Citation6], but has not filtered down fully into research on endophenotypes.
Building again on the example of blunted striatal DA function, this suggests several logical next steps, including (a) describe/demarcate boundaries of the vulnerability trait it confers; assess its heritability, as opposed to heritabilities of specific disorders; and evaluate patterns of shared molecular genetic risk among traditionally defined disorders encompassed by the trait. This approach represents a major shift in philosophy over disorder-specific searches for endophenotypes. Instead of traditionally defined disorders (phenotypes) serving as starting points from which we work backward to infer potential endophenotypes (then evaluate their associations with genetic liability), an alternative is to begin with neural function (a putative endophenotype) or a ‘gene-first’ approach as a starting point, working forward and backward to: specify common behavioral traits (phenotypes) that such neural functions imbue, and ascertain genetic liability.
Returning to the example of midbrain DA function, a now sizable body of literature indicates that low tonic striatal DA activity and blunted striatal DA reactivity during reward anticipation imbue an anhedonic, irritable mood state that characterizes ADHD, other externalizing disorders, unipolar depression, and intentional self-injury [Citation11]. Moreover, comorbidity rates between ADHD and unipolar depression are high [Citation38], the disorders share molecular genetic liability [Citation38,Citation39], and both are characterized by present bias in decision-making (preference for immediate overdelayed rewards) [Citation40]. Although the heritability of anhedonia has not been evaluated in large twin studies, irritability is moderately heritable [Citation41].
Collectively, these findings suggest that phenotypically dissimilar disorders share a common neural vulnerability – expressed psychologically as trait anhedonia and irritability. This state of affairs is depicted in . Low midbrain DA activity and reactivity to incentives – neural vulnerabilities to both ADHD and unipolar depression [Citation11] – emerge from interactions among numerous etiological inputs including normal allelic variation [Citation42–44], heritable CNVs [Citation45,Citation46], de novo mutations [Citation47], epigenetic changes in receptor function [Citation48], neurotoxic environmental risk exposures [Citation48] and neurohormonal influences [Citation49,Citation50], among other factors [Citation12]. These influences aggregate and interact to induce an anhedonic and irritable mood state that confers vulnerability to both externalizing disorders and unipolar depression. Blunted striatal responding to incentives is therefore not an endophenotype of any specific disorder. Rather, it is an etiologically complex transdiagnostic neural vulnerability. This raises the obvious question of why two affected individuals can display such different behavioral outcomes.
Blunted striatal responding to incentives (left panel, top) arises from aggregation of and complex interactions among normal allelic variation, heritable CNVs, de novo mutations, epigenetic factors, environmental risk exposures and neurohormonal influences. Very few of these are necessary or sufficient to produce the resulting subcortical endophenotype. Low striatal responding predisposes to trait anhedonia/irritability (a psychological state), producing a behavioral bias to reward seeking, which can temporarily upregulate aversive mood. Importantly, this behavioral bias can be moderated by other subcortical systems, including the amygdala and its interconnections (left panel, bottom). Strong amygdalar response to threat (a moderating subcortical endophenotype) predisposes to trait anxiety (a largely independent psychological state), which moderates trait anhedonia to produce a behavioral bias to immobilization. In most cases, these behavioral biases are insufficient to eventuate in psychopathology in the absence of prefrontal cortex dysfunction (a potentiating cortical endophenotype). Poor prefrontal regulation of subcortically generated emotions is observed in almost all forms of psychopathology (see text). For the sake of clarity, specific frontal structures that regulate subcortical systems (dorsolateral prefrontal cortex, ventromedial prefrontal cortex, orbitofrontal cortex, anterior cingulate cortex), and interconnections among subcortical systems (extended amygdala, bed nucleus of the stria terminalis) are omitted.
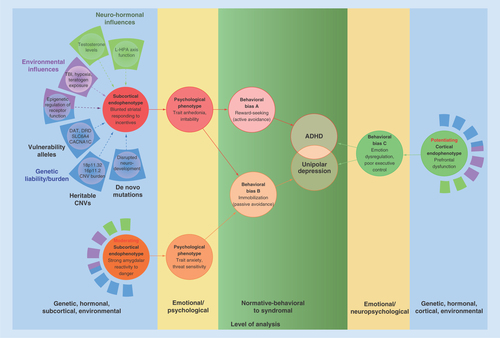
Etiological complexity I: moderating effects of other subcortical systems
Externalizing disorders are distinguished from unipolar depression by moderating effects of other subcortical and cortical networks on the midbrain DA system. These moderating influences, along with nonshared environmental influences, can shape trait vulnerability into different behavioral manifestations [Citation12,Citation51]. Among the most important of these moderators is amygdalar function. Unipolar depression is characterized by volumetric abnormalities in the amygdala [Citation52], and by amygdalar hyper-reactivity to threat [Citation53]. In contrast, externalizing disorders are characterized by amygdalar hyporeactivity to similar stimuli [Citation54]. As illustrated in , individual differences in amygdalar responding (and in its functional neuroanatomical connections) interact with individual differences in striatal function [Citation55] to moderate behavioral expression of anhedonia/irritability. Thus, anhedonia and irritability motivate reward-seeking behavior primarily among those with low amygdalar responding to threat because they lack anxiety and fear [Citation56]. For these individuals, appetitive behaviors upregulate midbrain DA levels, providing temporary relief from their chronically aversive mood state [Citation11]. In contrast, those who are hyper-responsive to threat engage primarily in passive avoidance rather than reward-seeking because they experience anxiety and fear, which inhibit appetitive behaviors. Like striatal DA responding, amygdalar reactivity to threat is heritable, and has its own set of intricate and interactive causal influences (see ), which cannot be reviewed here given space constraints. Thus, two subcortically founded, heritable traits (anhedonia/irritability vs threat sensitivity), imbued by different neurobiological mechanisms, interact to affect psychological adjustment and behavioral expressions of vulnerability.
Several important points follow from this discussion. First, heritable individual differences in neurobiological functions interact with one another, which can ‘shift’ behavioral distributions of each trait, thereby obscuring any 1:1 correspondence between genes and behavior [Citation13]. Thus, we should be circumspect about using behaviors as starting points to infer endophenotypes (discussed earlier in this Perspective). Second, etiology is inordinately complex [Citation12,Citation31,Citation57]. Etiological complexities that were either unrecognized or only nascently recognized when Gould and Gottesman defined endophenotypes are now known to operate in many forms of psychopathology. As noted above, for example, much greater appreciation for multifactorial inheritance – whereby few if any genes are necessary or sufficient for psychiatric morbidity – has emerged. Furthermore, rare copy number variants, many of which are not inherited, play a significant role in almost all major classes of psychopathology [Citation58], contributing further to genetic complexity. Third, epigenetic changes in gene expression are common, and can affect neural functions that subserve motivation, mood regulation, and social affiliation [Citation59]. These neural systems are interactive and interdependent, which further obfuscates any linear trajectories across genetic → neural → behavioral levels of analysis. Finally, nonepigenetic environmentally induced influences on neural responding can affect behavior, producing ‘phenotypes’ that are indistinguishable from those that are genetically mediated. Teratogen exposure, hypoxic events, and use of strong stimulants all downregulate striatal DA responding, with behavioral consequences that are indistinguishable from the heritable ADHD phenotype [Citation48].
Etiological complexity II: potentiating effects of cortical systems & general liability to psychopathology
Given the above discussion, it is important to note that subcortical neural function rarely determines psychopathology, except in extreme cases such as focal lesions [Citation60]. Many individuals who suffer from no psychiatric condition exhibit below normal striatal responding to incentives, and ordinary variation in such responding gives rise to individual differences in personality [Citation28]. Many people who are high on trait anxiety also function well in their day-to-day lives. Thus, subcortical neural systems are insufficient to explain the functional impairment that characterizes most forms of psychopathology [Citation9,Citation61]. Rather, psychopathology is most likely when strong, subcortically generated emotions are not regulated effectively by functional subdivisions of the prefrontal cortex [Citation51].
Effortful regulation of appetitive emotions, such as wanting, is exerted through dorsolateral prefrontal and orbitofrontal inhibition of striatal activity and reactivity, whereas effortful regulation of aversive emotions, such as anxiety and fear, is exerted through lateral prefrontal inhibition of amygdalar activity and reactivity [Citation62,Citation63]. Those with externalizing disorders exhibit diminished functional connectivity between striatal and frontal structures [Citation64], whereas depressed and anxious individuals exhibit altered functional connectivity between the amygdala and the ventrolateral PFC, the ventromedial PFC, and the anterior cingulate [Citation65]. In addition, poor functional connectivity between the amygdala and the orbitofrontal cortex, and poor top-down control of the amygdala by the medial PFC, are implicated in emotional lability [Citation66]. Thus, failures in prefrontal regulation of subcortical structures involved in approach- and avoidance-related affect are observed in both externalizing and internalizing disorders.
The two right panels of depict the potentiating role of poor emotion regulation and executive function on psychopathological outcomes. When the subcortical substrates of reward-seeking and immobilization are met with poor top-down cortical regulation, psychopathology becomes likely. Thus, gradations from normal personality variation to psychopathology, depicted in green, are determined by interactions among subcortical structures that generate emotion, and cortical structures that regulate emotion [Citation67].
Etiological complexity III: underexplored dimensions of general psychopathology
Historically, intellectual disabilities, autism and temperamental liabilities to personality disorder have been split off from many studies of psychopathology on the basis of preconceived notions that their origins are distinct from those that confer liability to common (formerly Axis I) DSM-defined disorders. The fact that comorbidities between the former and latter types of disorder are the rule rather than the exception, and that intellectual disability, autism, and personality disorder represent pathological tails along continuous distributions of trait vulnerabilities (i.e., general cognition, reciprocal social behavior, cooperativeness, respectively) in the population highlights the importance of accounting for effects of variation in these traits in any study relating phenotype → endophenotype → neural mechanism → gene in human populations. These ‘modifiers’ are rarely considered and often excluded in highly selective research designs. The example of autism is instructive because it reflects effects of an endophenotypic influence that bears a formidable 1:1 relationship to a highly heterogeneous disease state (autism spectrum disorder; ASD) [Citation68], and simultaneously, when present, accentuates severity of any co-morbid psychopathologies [Citation69].
When standardized methods for quantitative assessment of ASD symptoms and traits are applied to the general population, the unequivocal result from a host of studies, implementing numerous measurement instruments, is that characteristic traits and features are distributed continuously – not bimodally – in nature [Citation70]. Remarkably, these traits (deficits in reciprocal social behavior, impairment in social communication, repetitive behavior, restriction in range of interests) are as highly correlated in the general population as they are (by definition) among individuals with clinical ASD syndromes; thus exhibiting a unitary factor structure in the general population. Furthermore, when measurements of subclinical autistic traits are implemented in studies of families affected by ASD, they aggregate among first-degree relatives with a frequency and order of magnitude higher than observed in the general population [Citation71,Citation72]. This is not the case in families affected by other psychiatric conditions, however, when present, subclinical autistic traits exacerbate the severity of nearly any psychiatric condition with which they co-occur [Citation68]. Finally, in very large genetic–epidemiologic studies, genetic susceptibilities to these subclinical syndromes exhibit near-complete overlap with genetic underpinnings of the clinical-level syndromes [Citation71], suggesting strongly that continuous distributions observed in nature relate to quantitative accumulation of causal liability. Collectively, such findings fulfill criteria to establish that this trait cluster an autism endophenotype.
It is important to note that in the same way that height influences weight, neurodevelopmental characteristics of variation in general cognition, reciprocal social behavior and temperament can influence behavior and psychopathology such that specification of the role of any single endophenotype within an individual will ultimately require established maps of expectable relations between variables (analogous to the height vs weight norms for males and females used in pediatric practice) to ascertain relative contributions of multiple causal factors to a given neuropsychiatric syndrome [Citation73]. Moreover, when considering the role of inherited variation in neurodevelopmental traits on psychopathology, it is important to note that the recent explosion in genetic studies of neurodevelopmental disorders (made possible by steady reductions in costs of genotyping) have greatly expanded our understanding of the diversity of genetic influences on development and behavior. As noted above, causal heritable influences on neurodevelopmental outcomes can be common or rare, inherited or de novo (germline), specific to a disease (Rett Syndrome) or largely nonspecific (many chromosomal rearrangements). It is likely that this full diversity of types of genetic influence is operative in the development of psychopathology, and that diversity constitutes a confound when biomarker research is either not genetically informative or predicated on only a singular model of inheritance (e.g., additive genetic influence in twin studies).
Conclusion
Etiological complexity is the rule rather than the exception for major psychiatric disorders. Overt behaviors (i.e., phenotypes), including psychopathology, are affected by myriad interactive genetic factors (both heritable and nonheritable), interdependent neural systems that alter one another's functional outputs, and environmental insults that affect neural function. These influences bear complex relations to measured genetic liability, formidable as the sum total of heritable influences may be. Thus, behaviors may only occasionally be the best starting points for identifying endophenotypes. In fact, using the word phenotype to refer to behavioral syndromes can be misleading because it promotes anachronistic assumptions about 1:1 gene-behavior relations that do not exist for multifactorially inherited traits.
Future perspective: implications for the endophenotype concept
Most psychiatric disorders do not represent single disease entities, and single disease processes cut across psychiatric boundaries. This does not mean we should retire the endophenotype concept. Rather, endophenotypes may be useful in future research, provided we recognize the transdiagnostic nature of genetic and neural vulnerabilities, and that we evaluate interactive roles of genetic liabilities and neural systems in affecting behavior. Main effects models of psychopathology have probably run their course [Citation12], at least until such time as more biologically tractable parcellations of major psychopathologies are established. Endophenotypes should be sought that map onto genetically influenced neural functions rather than specific psychiatric disorders, and interactions among multiple endophenotypes and functional behavioral outcomes should be evaluated. To accomplish these objectives, effective collaborations among researchers who study different disorders, such as ADHD and unipolar depression (see ‘Discussion’ section earlier), will be required. At present, these literatures are largely uninformed by one another [Citation11]. Finally, the expectation that any single etiological model should explain a preponderance of variance in a neuropsychiatric outcome should neither dominate the discovery process nor result in dismissal of important leads that relate to subsets of individuals affected by what are now construed as single conditions. Endophenotypes that mark heritable vulnerability may only capture a subset of those who exhibit similarly disordered behavior. This does not represent failure; rather, it indicates that more work lies ahead in our quest to uncover the daunting complexity of psychopathology.
Endophenotypes are a subset of biomarkers that, by definition, lie closer to genetic liability than exophenotypes
According to their current definition, endophenotypes mark genetic liability to specific psychiatric disorders, independent of clinical state. Endophenotypes must
Segregate with illness in the general population.
Be heritable.
Be state independent, manifesting whether illness is present or in remission.
Cosegregate with the disorder within families.
Be present at higher rates within affected families than in the general population.
Be characteristics that can be measured reliably, and specific to the illness of interest.
Since these criteria were published, problems have emerged for the disorder-specificity criterion
Most psychiatric disorders are etiologically complex, including influences of normal allelic variation, heritable CNVs, de novo mutations, epigenetic changes in receptor function and neurohormonal influences, among others.
Few of these are necessary or sufficient to eventuate in psychopathology.
Transdiagnostic vulnerability traits are often more useful than clinical diagnoses as dependent variables in psychiatric genetics research.
Vulnerabilities to psychopathology interact with one another and with other behavioral traits (e.g., temperament, personality) to obscure 1:1 correspondences between genetic liability and behavior.
Many apparently similar cases of psychopathology represent ‘phenocopies’ with distinct etiologies.
Several recommendations would preserve the utility of the endophenotype concept moving forward
The disorder-specificity requirement should in most cases be relaxed.
Endophenotypes that mark vulnerability to multiple disorders can still be useful.
Neural functions are preferable to behaviors as starting points in searches for endophenotypes.
Future research should focus on interactive effects of multiple endophenotypes on complex psychiatric disorders.
Financial & competing interests disclosure
Preparation of this article was supported by grants UH2DE025980 (National Institute of Dental and Craniofacial Research, TP Beauchaine, PI) and U54 HD087011 (National Institute of Child Health and Human Development, Intellectual and Developmental Disabilities Research Center at Washington University, JN Constantino, PI). TP Beauchaine receives speaking honoraria and book royalties for related work, and JN Constantino receives royalties for commercial distribution of the Social Responsiveness Scale, a quantitative measure of autistic traits, use of which contributed to scientific advances described in this article. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was used in production of this manuscript.
Additional information
Funding
References
- Beauchaine TP . The role of biomarkers and endophenotypes in prevention and treatment of psychopathological disorders. Biomark. Med.3, 1–3 (2009).
- John B , LewisKR. Chromosome variability and geographical distribution in insects. Chromosome rather than gene variations provide the key to differences among populations. Science152, 711–721 (1966).
- Gottesman II , GouldTD. The endophenotype concept in psychiatry. Etymology and strategic intentions. Am. J. Psychiatry160, 636–645 (2003).
- Gould TD , GottesmanII. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav.5, 113–119 (2006).
- Flint J , MunafòMR. The endophenotype concept in psychiatric genetics. Psychol. Med.37, 163–180 (2007).
- Cuthbert BN , InselTR. Toward the future of psychiatric diagnosis. The seven pillars of RDoC. BMC Med.11, 126 (2013).
- Iacono WG , VaidyanathanU, VriezeSI, MaloneSM. Knowns and unknowns for psychophysiological endophenotypes. Integration and response to commentaries. Psychophysiology51, 1339–1347 (2014).
- Miller GA , ClaysonPE, YeeCM. Hunting genes, hunting endophenotypes. Psychophysiology51, 1329–1330 (2014).
- Beauchaine TP , ThayerJF. Heart rate variability as a transdiagnostic biomarker of psychopathology. Int. J. Psychophysiol.98, 338–350 (2015).
- van Praag HM . No functional psychopharmacology without functional psychopathology. Acta Psychiatr. Scand.122, 438–439 (2010).
- Zisner A , BeauchaineTP. Neural substrates of trait impulsivity, anhedonia, and irritability. Mechanisms of heterotypic comorbidity between externalizing disorders and unipolar depression. Dev. Psychopathol.28, 1179–1210 (2016).
- Beauchaine TP , ZisnerA, SauderCL. Trait impulsivity and the externalizing spectrum. Annu. Rev. Clin. Psychol.13, 343–368 (2017).
- Finucane B , ChallmanTD, MartinCL, LedbetterDH. Shift happens: family background influences clinical variability in genetic neurodevelopmental disorders. Genet. Med.18, 302–304 (2016).
- Sher L , OquendoMA, BurkeAK, CooperTB, MannJJ. Combined dexamethasone suppression-corticotrophin-releasing hormone stimulation test in medication-free major depression and healthy volunteers. J. Affect. Disord.151, 1108–1112 (2013).
- Coryell W , SchlesserM. The dexamethasone suppression test and suicide prediction. Am. J. Psychiatry158, 748–753 (2001).
- Hoyt WT , KernsMD. Magnitude and moderators of bias in observer ratings: a meta-analysis. Psychol. Methods4, 403–424 (1999).
- Li P-Y . State-dependent decrease in levels of brain-derived neurotrophic factor in bipolar disorder. A meta-analytic study. Neurosci. Lett.466, 139–143 (2009).
- Pacchiarotti I , BondDJ, BaldessariniRJet al. International Society for Bipolar Disorders Task Force report on antidepressant use in bipolar disorders. Am. J. Psychiatry170, 1249–1262 (2013).
- Iacono WG . Genome-wide scans of genetic variants for psychophysiological endophenotypes: introduction to this special issue of psychophysiology. Psychophysiology51, 1201–1202 (2014).
- Munafò MR , FlintJ. The genetic architecture of psychophysiological phenotypes. Psychophysiology51, 1331–1332 (2014).
- Plomin R . Missing heritability, polygenic scores, and gene–environment correlation. J. Child Psychol. Psychiatry54, 1147–1149 (2013).
- Genovese G , FromerM, StahlEAet al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci.19, 1433–1441 (2016).
- Windemuth A , CalhounVD, PearlsonGD, KocherlaM, JagannathanK, RuanoG. Physiogenomic analysis of localized fMRI brain activity in schizophrenia. Ann. Biomed. Eng.36, 877–888 (2008).
- Korfine L , LenzenwegerMF. The taxonicity of schizotypy: a replication. J. Abnorm. Psychol.104, 26–31 (1995).
- Lenzenweger MF . Thinking clearly about endophenotype-intermediate phenotype–biomarker distinctions in developmental psychopathology research. Dev. Psychopathol.25, 1347–1357 (2013).
- Castellanos FX , TannockR. Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat. Rev. Neurosci.3, 617–628 (2002).
- Li D , ShamPC, OwenMJ, HeL. Meta-analysis shows significant association between dopamine system genes and attention deficit hyperactivity disorder (ADHD). Hum. Mol. Genet.15, 2276–2284 (2006).
- Plichta MM , ScheresA. Ventral–striatal responsiveness during reward anticipation in ADHD and its relation to trait impulsivity in the healthy population: a meta-analytic review of the fMRI literature. Neurosci. Biobehav. Rev.38, 125–134 (2014).
- McClure SM , LaibsonDI, LoewensteinG, CohenJD. Separate neural systems value immediate and delayed monetary rewards. Science306, 503–507 (2004).
- Stokes PRA , ShotboltP, MehtaMAet al. Nature or nurture? Determining the heritability of human striatal dopamine function. An [18F]-DOPA PET study. Neuropsychopharmacology38, 485–491 (2013).
- Beauchaine TP , McNultyT. Comorbidities and continuities as ontogenic processes: toward a developmental spectrum model of externalizing behavior. Dev. Psychopathol.25, 1505–1528 (2013).
- Forbes EE , DahlRE. Altered reward function in adolescent depression. What, when, and how?J. Child Psychol. Psychiatry53, 3–15 (2012).
- Nestler EJ , CarlezonWA. The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry59, 1151–1159 (2006).
- Sauder CL , DerbidgeCM, BeauchaineTP. Neural responses to monetary incentives among self-injuring adolescent girls. Dev. Psychopathol.28, 277–291 (2016).
- Luking KR , PagliaccioD, LubyJL, BarchDM. Reward processing and risk for depression across development. Trends Cogn. Sci.20, 456–468 (2016).
- Gotlib IH , HamiltonJP, CooneyRE, SinghMK, HenryML, JoormannJ. Neural processing of reward and loss in girls at risk for major depression. Arch. Gen. Psychiatry67, 380–387 (2010).
- Skuse DH . Endophenotypes and child psychiatry. Brit. J. Psychiatry178, 395–396 (2001).
- Biederman J , BallSW, MonuteauxMCet al. New insights into the comorbidity between ADHD and major depression in adolescent and young adult females. J. Am. Acad. Child Adolesc. Psychiatry47, 426–434 (2008).
- Pearson-Fuhrhop KM , DunnEC, MorteroSet al. Dopamine genetic risk score predicts depressive symptoms in healthy adults and adults with depression. PLoS ONE9, e93772 (2014).
- Nobrega JN , HedayatmofidiPS, LoboDS. Strong interactions between learned helplessness and risky decision-making in a rat gambling model. Sci. Rep.6, e37304 (2016).
- Vidal-Ribas P , BrotmanMA, ValdiviesoI, LeibenluftE, StringarisA. The status of irritability in psychiatry: a conceptual and quantitative review. J. Am. Acad. Child Adolesc. Psychiatry55, 556–570 (2016).
- Gatt JM , BurtonKLO, WilliamsLM, SchofieldPR. Specific and common genes implicated across major mental disorders: a review of meta-analysis studies. J. Psychiatry Res.60, 1e13 (2015).
- Cross-Disorder Group of the Psychiatric Genomics Consortium . Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet381, 1371–1379 (2013).
- Forbes EE , BrownSM, KimakM, FerrellRE, ManuckSB, HaririAR. Genetic variation in components of dopamine neurotransmission impacts ventral striatal reactivity associated with impulsivity. Mol. Psychiatr.14, 60–70 (2009).
- ADHDgene Database . http://adhd.psych.ac.cn/advancedCNVSearch.do.
- Degenhardt F , PriebeL, HermsSet al. Association between copy number variants in 16p11.2 and major depressive disorder in a German case–control sample. Am. J. Med. Genet. B159B, 263–273 (2012).
- Elia J , GaiX, XieHMet al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatr.15, 637–646 (2015).
- Gatzke-Kopp LM . The canary in the coalmine. Sensitivity of mesolimbic dopamine to environmental adversity during development. Neurosci. Biobehav. Rev.35, 794–803 (2011).
- Hermans EJ , BosPA, OssewaardeL, RamseyNF, FernándezG, van HonkJ. Effects of exogenous testosterone on the ventral striatal BOLD response during reward anticipation in healthy women. NeuroImage52, 277–283 (2010).
- Peters AT , Van MeterA, PruittPJet al. Acute cortisol reactivity attenuates engagement of fronto-parietal and striatal regions during emotion processing in negative mood disorders. Psychoneuroendocrinology73, 67–78 (2016).
- Beauchaine TP , ZisnerAR. Motivation, emotion regulation, and the latent structure of psychopathology. An integrative and convergent historical perspective. Int. J. Psychophysiol.199, 108–118 (2017).
- Sacher J , NeumannJ, FünfstückT, SolimanA, VillringerA, SchroeterML. Mapping the depressed brain: a meta-analysis of structural and functional alterations in major depressive disorder. J. Affect. Disorders140, 142–148 (2012).
- Schardt DM , ErkS, NüsserCet al. Volition diminishes genetically mediated amygdala hyperreactivity. NeuroImage53, 943–951 (2010).
- Jones AP , LaurensKR, HerbaCM, BarkerGJ, VidingE. Amygdala hypoactivity to fearful faces in boys with conduct problems and callous-unemotional traits. Am. J. Psychiatry166, 95–102 (2009).
- Cho YT , ErnstM, FudgeJL. Cortico–amygdala–striatal circuits are organized as hierarchical subsystems through the primate amygdala. J. Neurosci.33, 14017–14030 (2013).
- Corr PJ , McNaughtonN. Neural mechanisms of low trait anxiety and risk for externalizing behavior. In : The Oxford Handbook of Externalizing Spectrum Disorders. BeauchaineTP, HinshawSP( Eds). Oxford University Press, NY, USA, 220–238 (2016).
- Cohen BM . Embracing complexity in psychiatric diagnosis, treatment, and research. JAMA Psychiatry73, 1211–1212 (2016).
- Girirajan S , RosenfeldJA, CoeBPet al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med.367, 1321–1331 (2012).
- Beauchaine TP , NeuhausE, ZalewskiM, CrowellSE, PotapovaN. Effects of allostatic load on neural systems subserving motivation, mood regulation, and social affiliation. Dev. Psychopathol.23, 975–999 (2011).
- Neuhaus E , BeauchaineTP. Impulsivity and vulnerability to psychopathology. In : Child and Adolescent Psychopathology. BeauchaineTP, HinshawSP( Eds). Wiley, NJ, USA, 178–212 (2017).
- Caspi A , HoutsRM, BelskyDWet al. The p factor. One general psychopathology factor in the structure of psychiatric disorders? Clin. Psychol. Sci. 2, 119–137 (2015).
- Heatherton TF . Neuroscience of self and self-regulation. Annu. Rev. Psychol.62, 363–390 (2011).
- Tone EB , GarnCL, PineDS. Anxiety regulation: a developmental psychopathology perspective. In : Developmental Psychopathology. Volume 2: Developmental Neuroscience. CicchettiD( Ed.). Wiley, NJ, USA, 523–556 (2016).
- Cubillo A , HalariR, EckerC, GiampietroV, TaylorE, RubiaK. Reduced activation and inter-regional functional connectivity of fronto-striatal networks in adults with childhood ADHD and persisting symptoms during tasks of motor inhibition and cognitive switching. J. Psychiatr. Res.44, 629–639 (2010).
- Gold AL , ShechnerT, FarberMJet al. Amygdala–cortical connectivity: associations with anxiety, development, and threat. Depress. Anxiety33, 917–926 (2016).
- Hilt LM , HansonJL, PollakSD. Emotion dysregulation. In : Encyclopedia of Adolescence Volume 3. BrownBB, PrinsteinMJ( Eds). Elsevier, NY, USA, 160–169 (2011).
- Beauchaine TP . Future directions in emotion dysregulation and youth psychopathology. J. Clin. Child Adolesc.44, 875–896 (2015).
- Constantino JN , CharmanT. Diagnosis of autism spectrum disorder: reconciling the syndrome, its diverse origins, and variation in expression. Lancet Neurol.15, 279–291 (2016).
- Constantino JN , FrazierTW. The observed association between autistic severity measured by the Social Responsiveness Scale (SRS) and general psychopathology – a response to Hus et al. (2013). J. Child Psychol. Psychiatry54, 695–697 (2013).
- Constantino JN . The quantitative nature of autistic social impairment. Pediatr. Res.69, R55–R62 (2011).
- Constantino JN , LajonchereC, LutzMet al. Autistic social impairment in the siblings of children with pervasive developmental disorders. Am. J. Psychiatry163, 294–296 (2006).
- Robinson EB , KoenenKC, McCormickMCet al. Evidence that autistic traits show the same etiology in the general population and at the quantitative extremes (5%, 2.5%, and 1%). Arch. Gen. Psychiatry68, 1113–1121 (2011).
- Constantino JN . How continua converge in nature. Cognition, social competence, and autistic syndromes. J. Am. Acad. Child Adolesc. Psychiatry48, 97–98 (2009).