
Abstract
The human body consists of hundreds of kinds of cells specified from a single genome overlaid with cell type-specific epigenetic information. Comprehensively profiling the body’s distinct epigenetic landscapes will allow researchers to verify cell types used in regenerative medicine and to determine the epigenetic effects of disease, environmental exposures and genetic variation. Key marks/factors that should be investigated include regions of nucleosome-free DNA accessible to regulatory factors, histone marks defining active enhancers and promoters, DNA methylation levels, regulatory RNAs, and factors controlling the three-dimensional conformation of the genome. Here we use the lung to illustrate the importance of investigating an organ’s purified cell epigenomes, and outline the challenges and promise of realizing a comprehensive catalog of primary cell epigenomes.
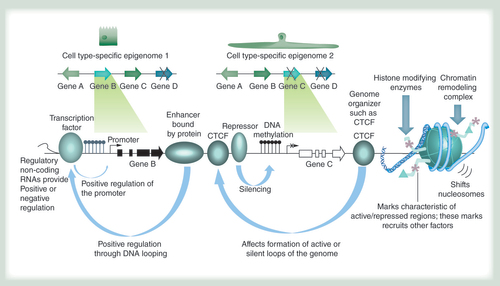
The epigenome allows cell types with identical genomes (schematically depicted at the top) to exhibit differential gene expression patterns through a variety of mechanisms. They include, from left to right: regulatory RNAs of different types such as long noncoding RNAs, miRNAs and others; recruitment of transcription factors that can activate gene expression; enhancer-binding proteins that can activate gene expression by looping to promoters in cis or in trans (when different chromosomes interact); proteins like CTCF that mediate looping interactions in concert with other proteins such as cohesin and thereby support the three-dimensional organization of the genome; the binding of repressor proteins that can silence genes; DNA methylation that is usually absent in active regulatory regions such as promoters and enhancers (hydroxymethylation, thought to occur during DNA methylation removal, and other modifications can also occur); a variety of histone modifying enzymes that lead to protein interactions associated with different chromatin states; nucleosome remodeling complexes that can lead to the removal or deposition of nucleosomes in active and repressed genomic regions, respectively.
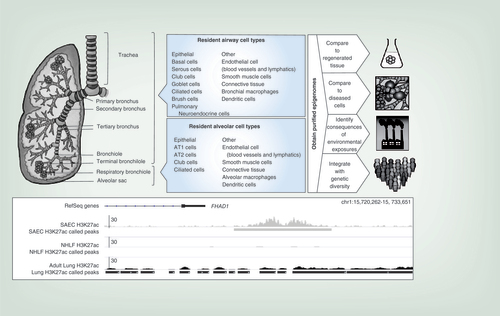
Only the right lung is shown. Airway and alveolar compartments and their cell types are indicated. The composition of human respiratory bronchioles has been poorly investigated but is thought to contain club cells and rare ciliated cells. At right, different applications for obtained epigenomes are indicated. At the bottom, an example is shown of epigenetic differences between lung cell types: histone 3 lysine 27 acetylation (H3K27Ac, a mark indicating an active enhancer) ChIP-seq data is compared between small airway epithelium, normal human lung fibroblasts and adult lung tissue. The presence of an enhancer is noted downstream of the FHAD gene in small airway epithelial cells but not in lung fibroblasts; the element is not clearly detectable in adult lung, which is a mix of many different cell types.
A single fertilized human egg, providing a single diploid genome, gives rise to a vast array of cells, recently estimated to be about 37 trillion [Citation1]. These cells are thought to represent over 210 histologically distinct cell types, most organized in complex higher order structures that form highly specialized organs [Citation2]. A key question in human biology is how these distinct cell types are derived and maintained during development and adulthood. Because the genomic information in all cells is identical (excepting specific genetic rearrangements such as immunoglobulin gene recombination in B cells), the distinct phenotypic properties of different cell types in an organ must be acquired by restricting accessibility to the genetic code in a cell type-specific and dynamic manner. This is accomplished by the epigenome, defined as the information overlaid on the genome that blocks access to certain regions and opens others to gene expression. Epigenomic information encompasses chemical modifications of DNA such as DNA methylation and hydroxymethylation, proteins bound to the DNA such as transcription factors that populate promoters and enhancers, modifications of the histones that make up the nucleosomes, nucleosome remodeling complexes, proteins that influence the three-dimensional organization of the genome such as lamins and CTCF, and numerous noncoding regulatory RNAs (). Together, these epigenetic factors constitute the epigenome and specify and maintain the different cell types in the body. The analysis of the epigenome can provide insight into disease risk and onset when affected cell types are compared with their healthy (reference) counterparts. While the cost of sequencing may necessarily limit which features can be profiled for many different cell types, the epigenetic marks in are widely accepted as defining regulatory regions of DNA. In the following sections we use the lung as an example to highlight the importance of understanding the epigenetic basis for cell phenotype specification, and describe specific instances in which cell type-specific epigenomic information might be leveraged: in tissue regeneration research, in determining disease states compared with healthy progenitor cells, in assessing the consequences of environmental exposures and in identifying functional noncoding variants associated with disease risk in genome-wide association studies (GWAS).
Examination of purified cell types is crucial for tissue regeneration research
The availability of human embryonic stem cells (ESCs) and induced pluripotent stem cells has opened avenues for in vitro differentiation of different tissues and cell types, so that repair and regeneration of all tissues is becoming a reality. However, derived cells and tissues must properly recapitulate the natural state and phenotype, or they may put patients at risk for tissue degeneration, malfunction, or possibly even cancer [Citation12]. Analysis of the epigenomes of numerous established human ESC lines has shown abundant epigenetic abnormalities, indicating that culture conditions might predispose cells to neoplastic transformation [Citation13]. Whether the proper phenotype of progenitors or differentiated cells derived from stem cells has been achieved can be assessed by examining whether the epigenome (and thereby the transcriptome) of the derived cells mirrors those of the desired target cell. However, this requires that the epigenomes of stem cells and target cells be well characterized. The Roadmap Epigenome project and the ENCODE project have made progress in this effort [Citation6,Citation13]. Among the 111 analyzed cell types in the Roadmap Epigenome project are induced pluripotent cells, human embryonic stem cells and several derived progenitor cells. However, the characterization of individual cell types that make up complex organs is in its very early stages. Due to the difficulties in purifying large numbers of individual cell types, initial efforts to elucidate organ-specific epigenomes have focused largely on the characterization of tissues samples consisting of mixed cell populations. While this provides a good starting point, the resulting data reflects an average epigenome that may be based on vastly differing epigenomes of distinct cell populations [Citation14–16]. A limited examination of purified cell populations has taken place, including numerous immortalized or cancerous human cell lines and several fully differentiated purified cell types, such as skin keratinocytes, skin fibroblasts and melanocytes [Citation17–19]. Although collectively these efforts have provided key insights into tissue-specific epigenomic differences, it remains largely unknown how the individual cell types within each organ contribute to the average epigenomes obtained for these tissues.
Identifying disease-associated epigenetic aberrations will require knowledge of progenitor cells & establishment of reference epigenomes
Numerous human diseases have been associated with epigenomic abnormalities, including diabetes [Citation20,Citation21], sepsis [Citation15], mental illnesses [Citation16,Citation22–25] and all types of cancer [Citation26]. While promising, challenges to future similar studies are at least threefold:
Difficulties related to obtaining pure populations of diseased cells from patient tissues;
Debate around true cells of origin for many diseases; and
The dearth of comprehensive reference or ‘baseline’ epigenomes even in scenarios in which the progenitor cells of disease are undisputed.
Studies of cancer illustrate all three examples. First, the simple objective of obtaining pure populations of ‘tumor’ and ‘normal’ cells is complicated by the fact that the tumor mass is comprised of a heterogeneous cell mixture comprising connective tissue, blood vessels and infiltrating lymphocytes. In addition, tumor samples are often compared with adjacent noncancerous tissue in the same patient, which can also be comprised of numerous cell types. Mathematical models for dealing with tissue heterogeneity using DNA methylation signatures have been developed and can address this problem to a certain extent [Citation14,Citation18,Citation19]. Heterogeneity can also be minimized by expert pathologists and accurate microdissection of clinical samples, however, this may come at the expense of sufficient starting material for downstream sequencing. Despite these efforts, it remains a challenge to obtain purified cell types, particularly where large epidemiological studies are concerned.
Secondly, the cell type of origin for many diseases may not be known. Moreover, even if the cell type of origin is known, such cells may be rare in samples of noncancerous tissue [Citation27,Citation28]. The latter would be true if the cell type of origin is a stem cell present in the tissue of interest, or if it represents a very small fraction of cells in normal tissue. A prime example is small-cell lung cancer (SCLC), a very aggressive cancer found predominantly in smokers. SCLC arises from rare pulmonary neuroendocrine cells which lie in small clusters along the airways, as discussed below [Citation29]. Even though epigenomic profiles of SCLC have been described [Citation30,Citation31], it is unclear which aspects are abnormal because the epigenomes of the healthy reference cell populations have not been obtained.
Measuring the effects of environmental exposures requires knowledge of the baseline epigenomes of unexposed cell types
Studies of mono- and dizygotic twins have been particularly informative in illustrating the profound effects of epigenomic variation on human health and the role of the environment in inducing this variation [Citation25,Citation32,Citation33]. In Alzheimer’s disease (AD), early life epigenetic changes may predispose to neurological disease at a later time, based on DNA methylation and oxidative status of certain gene promoters [Citation22,Citation23,Citation34]. Fetal alcohol exposure may also affect DNA methylation patterns and ensuing disease [Citation35]. Lung diseases are particularly associated with exposure to airborne environmental agents, including cigarette smoke and traffic-related air pollution such as diesel exhaust. These exposures are implicated in diseases like asthma, chronic obstructive pulmonary disease (COPD), emphysema and lung cancer. Exposures can occur in utero, in childhood or in adulthood, and the age at which exposures occur may affect the extent of epigenetic changes. The fact that some tissues, including lung and reproductive tissue, develop throughout adolescence and are affected by environmental influences, underscores the importance not only of determining the epigenomes of adult tissues, but also those of cells from embryos and children.
One very interesting example of environmental effects on the epigenome is the population-based analysis of DNA methylation patterns in smokers versus nonsmokers [Citation36,Citation37]. DNA methylation has been favored as the investigated epigenetic mark because it can be determined from purified DNA, which is often the only available biological material in epidemiological studies. However, instead of DNA obtained from lung cells, it is usually derived from blood cells that are frequently used as a surrogate; in many studies is too costly to obtain, store, and analyze multiple tissue types. Using white blood cell DNA, modest but reproducible DNA methylation changes have been observed in numerous studies [Citation36]. Some of these changes are rapidly reversible upon cessation of exposure, and others can persist for many years, as determined in studies from children exposed in utero and adults examined at different time points following smoking cessation [Citation38,Citation39]. Since the blood cells from which DNA was isolated were not usually sorted by cell type, an important question is whether the observed DNA methylation changes could largely be related to changes in the relative cell type composition of the white blood cells, as recently observed [Citation40]. Corrections for cell type composition are thus required [Citation18,Citation19]. Another key question is whether epigenetic changes seen in the blood reflect changes and disease risk in the lungs. We have recently investigated smoking-associated DNA methylation changes using samples of histologically normal lung from lung cancer patients (unpublished observations). Among others, we detected hypomethylation of cg05575921, which lies in an AHRR intron. This same hypomethylation event has also been observed in numerous studies of the white blood cell DNA of smokers [Citation36]. Because we were not able to analyze purified lung cell types we do not know which cells contributed to the observed DNA methylation changes. However, we were able to exclude the possibility that hypomethylation merely reflects changes in tissue composition (such as immune cell infiltration) rather than epigenetic alterations because we carried out functional studies. This illustrates the importance of measuring environmental responses in individual cell types and having clearly established baseline epigenomes in such cells.
Interpretations of the effects of genetic polymorphisms on disease risk requires knowledge of epigenetic marks in the risk-associated cell types
Genetic variants can affect the epigenome in many ways [Citation41–43]. For example, variants in the coding regions of genes involved in establishing or maintaining epigenetic features, such as DNA methyltransferases [Citation44–48] or histone-modifying enzymes [Citation49] have been reported to affect the epigenome on a broad scale, and are frequently detected as somatic mutations in tumors [Citation50–52]. Germline variants have been reported to affect the epigenome at discrete loci in more subtle ways [Citation43,Citation53–55].
Genome-wide association studies (GWAS) performed with microarrays containing millions of probes for SNPs have identified over 8000 variants associated with human illness [Citation56,Citation57]. Mechanistically connecting GWAS SNPs to disease processes is complicated by two main factors [Citation56,Citation58]:
Most are co-inherited with hundreds of variants in linkage disequilibrium (LD) that could be more causal than their representative or ‘index’ SNP on the array; and
Most GWAS and associated LD SNPs lie outside of coding regions of genes, making hypotheses about their function more elusive than for coding SNPs.
Several recent in-depth follow-up studies support the notion that functional noncoding SNPs reside in regulatory regions of the genome, such as gene enhancers, which orchestrate transcriptional programs underpinning health and disease. In all of these studies, the epigenomic features of disease-relevant cell types were profiled and integrated over the locations of GWAS and LD SNPs. This resulted in a shortlist of potential SNP candidates to prioritize in functional follow-up experiments that range from allele-specific ChIP and reporter gene assays [Citation53,Citation54] to genome-wide expression studies following CRISPR-driven deletion of enhancers overlapping risk SNPs [Citation55]. To date, noncoding functional SNPs have been reported to disrupt transcription factor binding sites and enhancer-promoter interactions [Citation53,Citation55], affect DNA methylation [Citation43,Citation59], modulate miRNA and lncRNAs expression and binding [Citation60,Citation61] and affect CTCF occupancy [Citation62].
The importance of evaluating GWAS SNPs in disease-relevant epigenomic contexts is evident in disorders of the respiratory tract. The lungs (as discussed in more detail in the section below) consist of many epithelial cell types that can be broadly categorized as ‘airway’ or ‘alveolar’. These broad categories harbor subgroups of highly specialized cell types with dramatically distinct phenotypes, epigenomes and disease predispositions [Citation1,Citation63–68]. For example, epithelial cell types lining the conducting airways and bronchioles are implicated in squamous cell lung cancer (SQLC) and disorders related to bronchitis [Citation27,Citation28,Citation69]. Conversely, AT2 cells of the alveolar compartment are thought to give rise both to lung adenocarcinoma (LUAD), the major subtype of lung cancer, and idiopathic pulmonary fibrosis (IPF), a deadly illness in which the alveoli are destroyed and replaced by scar tissue [Citation70,Citation71]. Interestingly, the C-allele of rs2736100, located in an intron of the telomerase-encoding TERT gene, is linked to increased risk of LUAD but not SQLC or other lung cancer subtypes in GWA studies [Citation72,Citation73]. In contrast, the rs2736100-A allele is the only risk SNP that replicates in GWA studies of IPF [Citation74]. Heightened telomerase activity is a well-documented phenomenon in tumor predisposition [Citation75], while decreased activity is linked to epithelial stem cell exhaustion and tissue remodeling seen in emphysema and IPF [Citation76,Citation77]. Not surprisingly, the rs2736100-C allele is associated with elevated TERT expression in lung tissue [Citation73], while the rs2736100-A allele is associated with shortened telomeres that are a hallmark of IPF [Citation78–80]. Epigenomic aberrations have been associated with LUAD and IPF at disparate loci in several reports [Citation81–83]. Animal models of both disorders exist [Citation84,Citation85], and epigenomically profiling labeled AT2 cells at different stages of disease progression might provide actionable insight into the mechanism whereby rs2736100 deregulates telomerase. Likewise, epigenomic profiles may elucidate the role of other causal SNPs elsewhere in the genome.
The complexities of determining comprehensive organ-specific epigenomes: the lung as an example
To address the challenges described in the above sections, the epigenomes of all cell types in the human body would need to be mapped. Ideally, this should be performed by experts in cell purification in an organ-by-organ fashion. In this section, we provide a brief overview of the types of cells found in the lung to illustrate the complexity of dissecting the epigenomes of cells comprising whole organs. In the lungs, there are over 40 different types of cells ranging from a variety of epithelial cells to smooth muscle cells, cells composing the vasculature and lymphatic system and immune cells defending the airways against foreign particulate matter and pathogens () [Citation1,Citation63–68]. As outlined in the sections of this review, the individual epigenomes of the different cell types could be used for numerous purposes, including comparison to regenerated tissues and diseased cells, to identify the consequences of environmental exposures and to gain insights into the effects of genetic diversity (, right side). An example illustrating the importance of characterizing cell type-specific epigenomes is given in the bottom panel of , which compares ChIP-seq data for histone 3 lysine 27 acetylation (H3K27Ac, a mark indicating an active enhancer) in small airway epithelium, normal human lung fibroblasts and unpurified adult lung tissue. It indicates the presence of an enhancer downstream of the FHAD gene in small airway epithelial cells but not in lung fibroblasts; the element is not clearly detectable in adult lung, which is a mix of many different cell types. This point is further underscored by studies of two epithelial populations in human peripheral lung, AT1 and AT2 cells; dramatic differences can be observed between their epigenomes even though they are derived from two cell types linked by a direct progenitor-progeny relationship and co-existing in close proximity in the same organ compartment [Citation86].
The airways begin with the trachea, which connects the throat to the primary bronchi. The latter start at the bifurcation to both lungs and branch out into the secondary bronchi connecting to the lung lobes (three on the right and two on the left in humans due to the presence of the heart). This is followed by the tertiary bronchi, which transport the air into lung segments. The bronchioles follow, transitioning into the terminal bronchioles, considered the smallest parts of the airways. The transition to the alveoli occurs through the respiratory bronchioles. Cell types found in the airways play numerous roles, and are often divided into categories based on their function [Citation63–68]. Basal cells lie on the basement membrane and are thought to be involved in airway regeneration upon injury. They are also implicated as progenitors of squamous cell carcinoma [Citation28]. A variety of secretory cells defend against pathogens and offer protection against contaminants. These include: serous cells, which produce lysozyme, albumin and antileukoprotease, and are implicated in lung repair upon injury; goblet cells, which secrete mucus and have numerous membrane-bound secretory granules; and club cells, which express detoxifying enzymes, the characteristic protein uteroglobin (CC10) and secrete surfactant. Other epithelial cell types found in the airways include ciliated cells, whose function is to move mucus up and out of the airways, thereby ridding the lungs of particulate matter and pathogens, and chemosensory cells, including pulmonary neuroendocrine cells (PNECs) and the pear-shaped brush cells. The latter two cell types are rare, each accounting for less than 1% of lung epithelial cells. PNECs are thought to function in oxygen sensing and as mentioned have been very strongly implicated as the cell of origin of SCLC [Citation29]. The function of brush cells remains enigmatic. It is interesting that they express proteins of the taste transduction pathways, and this may be related to the sensing of pathogens [Citation87]. In addition to the epithelial cells listed above, the airways are lined with smooth muscle, which allow the bronchioles in particular to expand and contract in response to exercise or inhaled particulates respectively. Connective tissue and the vascular and lymphatic systems surround the airways. Lastly, immune cells populate the airways, consisting of specialized macrophages and dendritic cells. The relative cell composition in the upper and lower airways differs, and it has been suggested that even single cell types like basal cells might differ phenotypically dependent on their proximal or distal location [Citation64].
The alveolar compartment has fewer epithelial cell types. The cellular composition of the human respiratory bronchiole has been poorly characterized but is thought to include club cells and rare ciliated cells. The large delicate AT1 cells constitute the respiratory surface (approximately 95% of the alveolar surface area) and are in intimate contact with the capillary network, while cuboidal AT2 cells produce surface tension-reducing surfactants that prevent the alveolar sacs from collapsing upon exhalation. In addition, AT2 cells are thought to play a role in peripheral lung regeneration upon injury and can transdifferentiate into AT1 cells [Citation88]. This process can be recapitulated in vitro over the course of 6–8 days and was used by us to obtain human AT1 cells for epigenomic profiling because the fragile primary human AT1 cells are very difficult to purify in large numbers [Citation86,Citation89]. AT2 cells are the presumed precursors of lung adenocarcinoma [Citation70]. As with the airways, numerous other cells are present: interstitial cells (fibroblasts), a large number of vascular and lymphatic cells, limited smooth muscle, and alveolar macrophages and dendritic cells. The latter are distinct from other macrophages in the body and should be independently studied [Citation90].
From these observations it is clear that samples of lung tissue will contain many different cell types. How much of each type is present in a piece of tissue depends on the sampled location and the natural abundance of each cell type. In an analysis of cell type numbers in an adult human lung, endothelial cells were most numerous (~140 billion), followed by AT2 cells (~70 billion), AT1 cells (~40 billion) and alveolar macrophages (~30 billion) [Citation1]. All airway cells are at least an order of magnitude less common, with the chemosensory cells in lowest abundance.
In order to determine the epigenomes for all the cell types in the lung, each must be separately purified and characterized, a process that can be challenging for delicate cells or cells of relatively low abundance. How this can be approached will be discussed in the next section. Given the technical difficulty and expense of such purifications, they do not lend themselves to be carried out for numerous subjects, for example, for epidemiological studies. Rather, the goal would be to use purified epigenomes from a limited number of individual lungs to identify regulatory elements in regions that may be implicated based on epidemiological studies.
New approaches to obtaining cell-type-specific epigenomes
When attempting to characterize purified human epigenomes, it is of course desirable to obtain primary cells, although for some fragile cell types such as AT1 cells, this can be challenging. In initial epigenomic studies of the peripheral lung, low-cell-number methods for epigenomic mapping were not yet available, requiring the purification tens of millions of cells to capture high quality data for each histone mark by chromatin immunoprecipitation-high throughput sequencing (ChIP-seq) [Citation86]. Such cell quantities can only be obtained from a deceased person. Because remnant transplant lung was used, the donors had often been on life support for several days. Despite the unnatural conditions, lungs that have been maintained in this way are successfully transplanted into patients, indicating that lung function is sufficiently maintained. For our experiments, remnant transplant lung that was disease-free was transported by air to the laboratory where it was lavaged to remove macrophages. These can be saved for independent profiling. The lung was then digested with elastase, using a procedure adapted from the Matthay laboratory [Citation86,Citation91]. The tissue was subsequently chopped, filtered and subjected to gradient centrifugation. Any remaining immune cells were then removed by panning on IgG-coated plates and AT2 cells were purified on epithelial cell adhesion molecule-carrying microbeads. This yielded very large numbers of AT2 cells that were approximately 90% pure. Because AT1 cells are too fragile to purify in large quantities, their in vitro equivalent was obtained by plating AT2 cells and allowing them to spontaneously differentiate [Citation86,Citation89]. Dramatic transcriptional changes occurred within two days of plating and by 6 days the cells had attained an AT1 cell-like phenotype and had formed tight junctions as measured by transepithelial resistance. Culturing cells to obtain derivative cell types introduces potential variables, but it may be the only way large numbers of some cells can be obtained. This type of approach has been successfully used to grow cells from organs such as the kidney, brain and colon with maintenance of the proper cell characteristics [Citation92–95]. In the case of peripheral lung, it allowed us to profile AT2 cells and AT1-like cells for DNA methylation, numerous histone marks and open chromatin, the latter by formaldehyde-assisted identification of regulatory elements (FAIRE)-seq, which allows one to use remnant material from the ChIP experiments. However, dealing with the large number of cells that was needed limited the ability to purify additional cell types simultaneously.
Fortunately, important progress has been made, not only in high throughput sequencing technologies but also in low-cell-number approaches. This means that FACS, which is too cumbersome and expensive to use for obtaining very large numbers of cells, is now a viable option for cell purification. Combined with the development of new low-cell-number methods for epigenomic profiling such as ATAC-seq to map accessible DNA (equivalent to DNase hypersensitive sites) [Citation4], and iChIP-seq [Citation7] or recovery via protection (RP)-ChIP-seq and favored amplification RP-ChIP-seq (FARP-ChIP-seq) [Citation8,Citation96], it will now be possible to carry out epigenomic profiling using limited numbers of many different kinds of cells. In fact, as technologies progress to single-cell approaches, cell numbers will be further reduced. A single-cell method for ATAC-seq has just been described [Citation96]. However, single cell methods always require a group of similar cells to obtain average signatures and this is expensive and computationally intense.
One important limitation of bead-based or FACS-based cell purification of distinct cell types is that antibodies to extracellular targets must be available to identify the desired cell types. Such antibodies are not in hand for many of the types of cells in the human body. This can be overcome by identifying cell-type specific markers for each cell type. To this end, single cell RNA-seq data for cell populations from all over the body would be extremely valuable [Citation3]. Principal component analysis of single cell populations will allow groups of cells with similar phenotypes to be identified. Such studies are being carried out for many tissues, including the lung and brain [Citation3,Citation97], and will help clarify whether similar cell types from different organs or from different regions of the same organ are distinct, such as epithelial cells found in the bronchi versus the terminal bronchioles, or airway versus alveolar macrophages [Citation98]. Single cell and low cell number techniques will also enable the epigenomic analysis of cells and tissues from living humans through biopsies or different types of noninvasive sampling. This will allow a closer approximation of the natural state of tissues.
What about model organisms? Would it not be much easier to use genetically engineered animals with cell type-specifically labeled nuclei to investigate the composite epigenomes of organs, and then to translate those findings to humans? Elegant procedures have been developed that allow nuclei to be labeled using animals engineered with cell type-specific promoters driving a nuclear envelope protein [Citation99,Citation100]. The tissues of interest can quickly be shredded to purify the labeled nuclei. Indeed, these model organism systems are very valuable, and can yield much information on the epigenetic encoding of tissues, even from organisms distantly related to humans like flies and worms [Citation17]. However, there are numerous differences even between human organs and their mammalian counterparts, and these differences will likely affect cellular epigenomes. In the lung, for example, the types of cells in different compartments can differ markedly between species [Citation63–68].
Future perspective
The epigenomic profiling of cell types within organs, and ultimately of all the cell types in the human body, will provide key information supporting biomedical investigations. In the short term, the focus should be placed on elucidating the collective epigenomes for a limited number of adult individuals of both genders. However, such studies would form just the very beginning of whole organism epigenomic analyses. We must build upon them by determining the comparative epigenomes from individuals of different ages, including embryos, infants, children and adolescents, different ethnicities and a wide range of environmental exposures. Only then will we truly begin to grasp the range of epigenetic variation within and between individuals. Cost and complexity would prevent the examination of the large numbers of individuals usually enrolled in epidemiological studies. Instead, a limited number of epigenomes would be used to provide a basis for the investigation of very specific genomic regions or epigenetic marks in a large number of individuals. We anticipate that single-cell techniques will further evolve to allow the analysis of all epigenetic marks of interest in single cells, and that sequencing approaches will improve to enable the determination of numerous epigenetic marks from each individual cell. This will, in turn, permit real-time sampling of dynamic epigenomes in patients to inform personalized medical strategies aimed at determination of disease risk, prevention and treatment.
Table 1. Recommended epigenetic factors to be examined.
Examination of purified cell types is crucial for tissue regeneration research
Epigenomic profiles are required to verify the integrity of stem cells and their derivatives used for cell-based therapies and tissue regeneration.
Identifying disease-associated epigenetic aberrations will require knowledge of progenitor cells & establishment of reference epigenomes
Within a tissue distinct cell types can be affected by a variety of diseases.
In order to establish how the epigenomes of diseased cells have been affected, the baseline epigenome of the cell of origin for each particular disease must be established.
Measuring the effects of environmental exposures requires knowledge of the baseline epigenomes of unexposed cell types
Environmental exposures can be measured in disease-prone and surrogate tissues.
Determination of epigenetic changes can inform prevention and therapeutic strategies.
Interpretations of the effects of genetic polymorphisms on disease risk requires knowledge of epigenetic marks in the risk-associated cell types
Most SNPs lie in introns or intergenic regions.
The biological function of SNPs, therefore, depends on whether they disrupt epigenetic regulatory elements in disease-associated cell types.
The complexities of determining comprehensive organ-specific epigenomes: the lung as an example
Each organ consists of a wide variety of different cell types with distinct functions and disease predispositions.
The epigenomes of closely related cells in the same organ compartment can differ dramatically.
Different cell types must be individually purified and epigenomically characterized.
New approaches to obtaining cell-type-specific epigenomes
FACS sorting and culture of purified cell types can provide cells for epigenomic analyses.
Low-cell-number and single cell techniques are lowering the numbers of cells needed for epigenomic profiling, facilitating analyses.
Future perspective
Efficient single-cell epigenome analysis will allow a personalized approach to assessment of genetic risk, environmental exposure and disease states.
Disclaimer
None of the funding agencies had any role in the content or timing of the publication of this manuscript; the content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies.
Acknowledgements
The authors acknowledge AN Laird and P Marconett for help with illustrations in .
Financial & competing interests disclosure
The authors were supported by the Hastings and Whittier Foundations, donations from C Pembroke and W Pembroke, J Glick, L Auerbach, NE Warner, HJ Norris, and the Canary and Thomas G. Labrecque Foundations to IA Laird-Offringa, Department of Defense Concept Award (W81XWH1410174) to IA Laird-Offringa, NIH grant (R01 HL114094) to IA Laird-Offringa and Z Borok, NIH grants (P30 H101258, R01HL112638 and 4 R37HL062569) to Z Borok and (1R01HL114959) to B Zhou, and the Norris Comprehensive Cancer Center core grant P30CA0189 from the National Cancer Institute. TR Stueve was supported by the National Institute of Environmental Health Sciences (NIEHS, NIH T32ES013678) and by the USC Provost’s Postdoctoral Scholar Research Grant. CN Marconett was supported by an ACS/Canary postdoctoral fellowship number PFTED-10-207-01-SIED, R01 HL114094 and the Department of Surgery. Z Borok is the Edgington Chair in Medicine. CN Marconett and IA Laird-Offringa are inventors listed on pending USC patent application 61/817,840, entitled “Accurate in Vitro Copying of DNA Methylation”. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Bianconi E , PiovesanA , FacchinFet al. An estimation of the number of cells in the human body . Ann. Hum. Biol.40 , 463 – 471 ( 2013 ).
- Alberts B , JohnsonA , LewisJet al. Molecular Biology of The Cell (6th Edition) . Garland Science , Hamden, CT, USA ( 2014 ).
- Wu AR , NeffNF , KaliskyTet al. Quantitative assessment of single-cell RNA-sequencing methods . Nat. Methods11 , 41 – 46 ( 2014 ).
- Buenrostro JD , WuB , ChangHY , GreenleafWJ . ATAC-seq: a method for assaying chromatin accessibility genome-wide . Curr. Protoc. Mol. Biol.109 , 21.29.1 – 9 ( 2015 ).
- Smallwood SA , LeeHJ , AngermuellerCet al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity . Nat. Methods11 , 817 – 820 ( 2014 ).
- Roadmap Epigenomics Consortium , KundajeA , MeulemanWet al.Integrative analysis of 111 reference human epigenomes . Nature518 , 317 – 330 ( 2015 ).
- Lara-Astiaso D , WeinerA , Lorenzo-VivasEet al. Immunogenetics. Chromatin state dynamics during blood formation . Science345 , 943 – 949 ( 2014 ).
- Zheng X , YueS , ChenH , WeberB , JiaJ , ZhengY . Low-cell-number epigenome profiling aids the study of lens aging and hematopoiesis . Cell Rep.13 , 1505 – 1518 ( 2015 ).
- Bonasio R , TuS , ReinbergD . Molecular signals of epigenetic states . Science330 , 612 – 616 ( 2010 ).
- Dowen JM , FanZP , HniszDet al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes . Cell159 , 374 – 387 ( 2014 ).
- Globisch D , MünzelM , MüllerMet al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates . PLoS ONE5 , e15367 ( 2010 ).
- Simonson OE , DomogatskayaA , VolchkovP , RodinS . The safety of human pluripotent stem cells in clinical treatment . Ann. Med.47 , 370 – 380 ( 2015 ).
- Werbowetski-Ogilvie TE , BosséM , StewartMet al. Characterization of human embryonic stem cells with features of neoplastic progression . Nat. Biotechnol.27 , 91 – 97 ( 2009 ).
- Jaffe AE , IrizarryRA . Accounting for cellular heterogeneity is critical in epigenome-wide association studies . Genome Biol.15 , R31 ( 2014 ).
- Bataille A , GalichonP , ZiliotisMJ , SadiaI , HertigA . Epigenetic changes during sepsis: on your marks . Crit. Care19 , 358 ( 2015 ).
- Maze I , ShenL , ZhangBet al. Analytical tools and current challenges in the modern era of neuroepigenomics . Nat. Neurosci.17 , 1476 – 1490 ( 2014 ).
- Brown JB , CelnikerSE . Lessons from modENCODE . Annu. Rev. Genomics. Hum. Genet.16 , 31 – 53 ( 2015 ).
- Houseman EA , AccomandoWP , KoestlerDCet al. DNA methylation arrays as surrogate measures of cell mixture distribution . BMC Bioinformatics13 , 86 ( 2012 ).
- Houseman EA , KimS , KelseyKT , WienckeJK . DNA methylation in whole blood: uses and challenges . Curr. Environ. Health. Rep.2 , 145 – 154 ( 2015 ).
- Jimenez-Chillaron JC , Ramon-KrauelM , RiboS , DiazR . Transgenerational epigenetic inheritance of diabetes risk as a consequence of early nutritional imbalances . Proc. Nutr. Soc.75 , 78 – 89 ( 2016 ).
- Jiménez-Chillarón JC , DíazR , MartínezDet al. The role of nutrition on epigenetic modifications and their implications on health . Biochimie94 , 2242 – 2263 ( 2012 ).
- Lahiri DK , MaloneyB , ZawiaNH . The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases . Mol. Psychiatry14 , 992 – 1003 ( 2009 ).
- Lahiri DK , MaloneyB . The “LEARn”(Latent Early-life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer’s disease, and proposes remedial steps . Exp. Gerontol.45 , 291 – 296 ( 2010 ).
- Maze I , WenderskiW , NohKMet al. Critical role of histone turnover in neuronal transcription and plasticity . Neuron87 , 77 – 94 ( 2015 ).
- Nestler EJ , PeñaCJ , KundakovicM , MitchellA , AkbarianS . Epigenetic basis of mental illness . Neuroscientist10.1177/1073858415608147 ( 2016 ) ( Epub ahead of print ).
- Baylin SB , JonesPA . A decade of exploring the cancer epigenome – biological and translational implications . Nat. Rev. Cancer11 , 726 – 734 ( 2011 ).
- Hanna JM , OnaitisMW . Cell of origin of lung cancer . J. Carcinog.12 , 6 ( 2013 ).
- Sutherland KD , BernsA . Cell of origin of lung cancer . Mol. Oncol.4 , 397 – 403 ( 2010 ).
- Sutherland KD , ProostN , BrounsI , AdriaensenD , SongJY , BernsA . Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung . Cancer Cell19 , 754 – 764 ( 2011 ).
- Kalari S , JungM , KernstineKH , TakahashiT , PfeiferGP . The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells . Oncogene32 , 3559 – 3568 ( 2013 ).
- Poirier JT , GardnerEE , ConnisNet al. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2 . Oncogene34 , 5869 – 5878 ( 2015 ).
- Fraga MF , BallestarE , PazMFet al. Epigenetic differences arise during the lifetime of monozygotic twins . Proc. Natl Acad. Sci. USA102 , 10604 – 10609 ( 2005 ).
- Busche S , ShaoX , CaronMet al. Population whole-genome bisulfite sequencing across two tissues highlights the environment as the principal source of human methylome variation . Genome Biol.16 , 290 ( 2015 ).
- Bennett DA , YuL , YangJ , SrivastavaGP , AubinC , De JagerPL . Epigenomics of Alzheimer’s disease . Transl. Res.165 , 200 – 220 ( 2015 ).
- Resendiz M , ChenY , OztürkNC , ZhouFC . Epigenetic medicine and fetal alcohol spectrum disorders . Epigenomics5 , 73 – 86 ( 2013 ).
- Gao X , JiaM , ZhangY , BreitlingLP , BrennerH . DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies . Clin. Epigenetics7 , 113 ( 2015 ).
- Shenker NS , PolidoroS , van VeldhovenKet al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking . Hum. Mol. Genet.22 , 843 – 851 ( 2013 ).
- Tsaprouni LG , YangTP , BellJet al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation . Epigenetics9 , 1382 – 1396 ( 2014 ).
- Novakovic B , RyanJ , PereiraN , BoughtonB , CraigJM , SafferyR . Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy . Epigenetics9 , 377 – 386 ( 2014 ).
- Bauer M , LinselG , FinkBet al. A varying T cell subtype explains apparent tobacco smoking induced single CpG hypomethylation in whole blood . Clin. Epigenetics7 , 81 ( 2015 ).
- Grundberg E , MeduriE , SandlingJKet al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements . Am. J. Hum. Genet.93 , 876 – 890 ( 2013 ).
- Hannon E , LunnonK , SchalkwykL , MillJ . Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes . Epigenetics10 , 1024 – 1032 ( 2015 ).
- Shi J , MarconettCN , DuanJet al. Characterizing the genetic basis of methylome diversity in histologically normal human lung tissue . Nat. Commun.5 , 3365 ( 2014 ).
- Arakawa Y , WatanabeM , InoueN , SarumaruM , HidakaY , IwataniY . Association of polymorphisms in DNMT1, DNMT3A, DNMT3B, MTHFR and MTRR genes with global DNA methylation levels and prognosis of autoimmune thyroid disease . Clin. Exp. Immunol.170 , 194 – 201 ( 2012 ).
- Kullmann K , DeryalM , OngMF , SchmidtW , MahlknechtU . DNMT1 genetic polymorphisms affect breast cancer risk in the central European Caucasian population . Clin. Epigenetics5 , 7 ( 2013 ).
- Saradalekshmi KR , NeethaNV , SathyanS , NairIV , NairCM , BanerjeeM . DNA methyl transferase (DNMT) gene polymorphisms could be a primary event in epigenetic susceptibility to schizophrenia . PLoS ONE9 , e98182 ( 2014 ).
- Wu H , ZhangK , GongPet al. A novel functional TagSNP Rs7560488 in the DNMT3A1 promoter is associated with susceptibility to gastric cancer by modulating promoter activity . PLoS ONE9 , e92911 ( 2014 ).
- Hamidi T , SinghAK , ChenT . Genetic alterations of DNA methylation machinery in human diseases . Epigenomics7 , 247 – 265 ( 2015 ).
- Cebrian A , PharoahPD , AhmedSet al. Genetic variants in epigenetic genes and breast cancer risk . Carcinogenesis27 , 1661 – 1669 ( 2006 ).
- Gonzalez-Perez A , Jene-SanzA , Lopez-BigasN . The mutational landscape of chromatin regulatory factors across 4,623 tumor samples . Genome Biol.14 , r106 ( 2013 ).
- Weirich S , KudithipudiS , KyciaI , JeltschA . Somatic cancer mutations in the MLL3-SET domain alter the catalytic properties of the enzyme . Clin. Epigenetics7 , 36 ( 2015 ).
- Scourzic L , MoulyE , BernardOA . TET proteins and the control of cytosine demethylation in cancer . Genome Med.7 , 9 ( 2015 ).
- Huang Q , WhitingtonT , GaoPet al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding . Nat. Genet.46 , 126 – 135 ( 2014 ).
- Rhie SK , CoetzeeSG , NoushmehrHet al. Comprehensive functional annotation of seventy-one breast cancer risk Loci . PLoS ONE8 , e63925 ( 2013 ).
- Yao L , TakYG , BermanBP , FarnhamPJ . Functional annotation of colon cancer risk SNPs . Nat. Commun.5 , 5114 ( 2014 ).
- Coetzee SG , RhieSK , BermanBP , CoetzeeGA , NoushmehrH . FunciSNP: an R/bioconductor tool integrating functional non-coding data sets with genetic association studies to identify candidate regulatory SNPs . Nucleic Acids Res.40 , e139 ( 2012 ).
- Welter D , MacArthurJ , MoralesJet al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations . Nucleic Acids Res.42 , D1001 – D1006 ( 2014 ).
- Tak YG , FarnhamPJ . Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome . Epigenetics Chromatin8 , 57 ( 2015 ).
- Liu Y , LiX , AryeeMJet al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease . Am. J. Hum. Genet.94 , 485 – 495 ( 2014 ).
- Jendrzejewski J , HeH , RadomskaHSet al. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type . Proc. Natl Acad. Sci. USA109 , 8646 – 8651 ( 2012 ).
- Hazelett DJ , RhieSK , GaddisMet al. Comprehensive functional annotation of 77 prostate cancer risk loci . PLoS Genet.10 , e1004102 ( 2014 ).
- Dai J , ZhuM , WangCet al. Systematical analyses of variants in CTCF-binding sites identified a novel lung cancer susceptibility locus among Chinese population . Sci. Rep.5 , 7833 ( 2015 ).
- Reynolds SD , PinkertonKE , MariassyAT . Epithelial cells of trachea and bronchi. In: Comparative Biology of the Normal Lung . ParentRA ( Ed ). Academic Press , San Diego, USA , 61 – 81 ( 2015 ).
- Rackley CR , StrippBR . Building and maintaining the epithelium of the lung . J. Clin. Invest.122 , 2724 – 2730 ( 2012 ).
- Mercer RR , CrapoJD . Architecture of the gas exchange region of the lungs . In : Comparative Biology of the Normal Lung (Second Edition) . ParentRA ( Ed. ). Academic Press , San Diego, USA , 93 – 104 ( 2015 ).
- Pinkerton KE , Van WinkleLS , PlopperCG , Smiley-JewellS , CovarrubiasEC , McBrideJT . Architecture of the Tracheobronchial Tree . In : Comparative Biology of the Normal Lung (Second Edition) . ParentRA ( Ed. ). Academic Press , San Diego, USA , 33 – 51 ( 2015 ).
- Pinkerton KE , Van WinkleLS , PlopperCG . Overview of diversity in the respiratory system of mammals . In : Comparative Biology of the Normal Lung (Second Edition) . ParentRA ( Ed. ). Academic Press , San Diego, USA , 3 – 5 ( 2015 ).
- Plopper CG , HydeDM . Epithelial Cells of the Bronchiole . In : Comparative Biology of the Normal Lung (Second Edition) . ParentRA ( Ed. ). Academic Press , San Diego, USA , 83 – 92 ( 2015 ).
- Thurlbeck WM . The pathology of small airways in chronic airflow limitation . Eur. J. Respir. Dis. Suppl.121 , 9 – 18 ( 1982 ).
- Desai TJ , BrownfieldDG , KrasnowMA . Alveolar progenitor and stem cells in lung development, renewal and cancer . Nature507 , 190 – 194 ( 2014 ).
- Willis BC , LieblerJM , Luby-PhelpsKet al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis . Am. J. Pathol.166 , 1321 – 1332 ( 2005 ).
- Landi MT , ChatterjeeN , YuKet al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma . Am. J. Hum. Genet.85 , 679 – 691 ( 2009 ).
- Wei R , CaoL , PuHet al. TERT polymorphism rs2736100-C is associated with EGFR mutation-positive non-small cell lung cancer . Clin. Cancer Res.21 , 5173 – 5180 ( 2015 ).
- Fingerlin TE , MurphyE , ZhangWet al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis . Nat. Genet.45 , 613 – 620 ( 2013 ).
- Kim NW , PiatyszekMA , ProwseKRet al. Specific association of human telomerase activity with immortal cells and cancer . Science266 , 2011 – 2015 ( 1994 ).
- Hao LY , ArmaniosM , StrongMAet al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity . Cell123 , 1121 – 1131 ( 2005 ).
- Armanios M , BlackburnEH . The telomere syndromes . Nat. Rev. Genet.13 , 693 – 704 ( 2012 ).
- Codd V , NelsonCP , AlbrechtEet al. Identification of seven loci affecting mean telomere length and their association with disease . Nat. Genet.45 , 422 – 427 , 427e1 ( 2013 ).
- Walsh KM , CoddV , SmirnovIVet al. Variants near TERT and TERC influencing telomere length are associated with high-grade glioma risk . Nat. Genet.46 , 731 – 735 ( 2014 ).
- Alder JK , ChenJJ , LancasterLet al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis . Proc. Natl Acad. Sci. USA105 , 13051 – 13056 ( 2008 ).
- Suzuki A , MakinoshimaH , WakaguriHet al. Aberrant transcriptional regulations in cancers: genome, transcriptome and epigenome analysis of lung adenocarcinoma cell lines . Nucleic Acids Res.42 , 13557 – 13572 ( 2014 ).
- Yang IV , PedersenBS , RabinovichEet al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis . Am. J. Respir. Crit. Care Med.190 , 1263 – 1272 ( 2014 ).
- Sanders YY , AmbalavananN , HalloranBet al. Altered DNA methylation profile in idiopathic pulmonary fibrosis . Am. J. Respir. Crit. Care Med.186 , 525 – 535 ( 2012 ).
- Kim CF , JacksonEL , KirschDGet al. Mouse models of human non-small-cell lung cancer: raising the bar . Cold Spring Harb. Symp. Quant. Biol.70 , 241 – 250 ( 2005 ).
- B Moore B , LawsonWE , OuryTD , SissonTH , RaghavendranK , HogaboamCM . Animal models of fibrotic lung disease . Am. J. Respir. Cell. Mol. Biol.49 , 167 – 179 ( 2013 ).
- Marconett CN , ZhouB , RiegerMEet al. Integrated transcriptomic and epigenomic analysis of primary human lung epithelial cell differentiation . PLoS Genet.9 , e1003513 ( 2013 ).
- Reid L , MeyrickB , AntonyVB , ChangLY , CrapoJD , ReynoldsHY . The mysterious pulmonary brush cell: a cell in search of a function . Am. J. Respir. Crit. Care Med.172 , 136 – 139 ( 2005 ).
- Adamson IY , BowdenDH . Derivation of type 1 epithelium from type 2 cells in the developing rat lung . Lab. Invest.32 , 736 – 745 ( 1975 ).
- Marconett CN , ZhouB , SiegmundKD , BorokZ , Laird-OffringaIA . Transcriptomic profiling of primary alveolar epithelial cell differentiation in human and rat . Genom. Data2 , 105 – 109 ( 2014 ).
- Hussell T , BellTJ . Alveolar macrophages: plasticity in a tissue-specific context . Nat. Rev. Immunol.14 , 81 – 93 ( 2014 ).
- Ballard PL , LeeJW , FangXet al. Regulated gene expression in cultured type II cells of adult human lung . Am. J. Physiol. Lung Cell. Mol. Physiol.299 , L36 – 50 ( 2010 ).
- Helbert MJ , DauweSE , De BroeME . Flow cytometric immunodissection of the human distal tubule and cortical collecting duct system . Kidney Int.59 , 554 – 564 ( 2001 ).
- Van der Hauwaert C , SavaryG , GnemmiVet al. Isolation and characterization of a primary proximal tubular epithelial cell model from human kidney by CD10/CD13 double labeling . PLoS ONE8 , e66750 ( 2013 ).
- Rozental R , GebhardD , PadinCet al. Purification of cell populations from human fetal brain using flow cytometric techniques . Brain Res. Dev. Brain Res.85 , 161 – 170 ( 1995 ).
- Jung P , SatoT , Merlos-SuárezAet al. Isolation and in vitro expansion of human colonic stem cells . Nat. Med.17 , 1225 – 1227 ( 2011 ).
- Buenrostro JD , WuB , LitzenburgerUMet al. Single-cell chromatin accessibility reveals principles of regulatory variation . Nature523 , 486 – 490 ( 2015 ).
- Darmanis S , SloanSA , ZhangYet al. A survey of human brain transcriptome diversity at the single cell level . Proc. Natl Acad. Sci. USA112 , 7285 – 7290 ( 2015 ).
- Van de Laar E , CliffordM , HasenoederSet al. Cell surface marker profiling of human tracheal basal cells reveals distinct subpopulations, identifies MST1/MSP as a mitogenic signal, and identifies new biomarkers for lung squamous cell carcinomas . Respir. Res.15 , 160 ( 2014 ).
- Deal RB , HenikoffS . The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana . Nat. Protoc.6 , 56 – 68 ( 2011 ).
- Steiner FA , TalbertPB , KasinathanS , DealRB , HenikoffS . Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling . Genome Res.22 , 766 – 777 ( 2012 ).