
Abstract
LSD1 (KDM1A; BHC110; AOF2) was the first protein reported to exhibit histone demethylase activity and has since been shown to have multiple essential roles in mammalian biology. Given its enzymatic activity and its high-level expression in many human malignancies, a significant recent focus has been the development of pharmacologic inhibitors. Here we summarize structural and biochemical knowledge of this important epigenetic regulator, with a particular emphasis on the functional and preclinical studies in oncology that have provided justification for the evaluation of tranylcypromine derivative LSD1 inhibitors in early phase clinical trials.
(A) Schematic illustrates the core components of the CoREST transcription repressor complex recruited to chromatin in association with TF. (B) Biochemical steps in the FAD-dependent demethylation of monomethyl lysine by LSD1 (C) x-ray structure representation of LSD1:RCOR1 bound to a peptide substrate. Domains are highlighted in colors. (D) Details of the catalytic site from the LSD1:CoREST structure highlighting the flavin moiety of FAD (gray carbon atoms), the amine oxidase domain (cyan) and the histone H3 peptide (pink). The position of LSD1 lysine 661 is indicated, as is that of lysine 4 of the histone peptide, here shown as MET-4. For purposes of the crystallization, lysine was replaced by methionine in view of the higher binding affinity of the pLys4Met peptide. Images were generated using PyMOL (Molecular Graphics System, Schrödinger, LLC) and a published structure [Citation12].
FAD: Flavin-adenine dinucleotide; TF: Transcription factor.
![Figure 1. LSD1 protein complex, structure and enzymatic activity. (A) Schematic illustrates the core components of the CoREST transcription repressor complex recruited to chromatin in association with TF. (B) Biochemical steps in the FAD-dependent demethylation of monomethyl lysine by LSD1 (C) x-ray structure representation of LSD1:RCOR1 bound to a peptide substrate. Domains are highlighted in colors. (D) Details of the catalytic site from the LSD1:CoREST structure highlighting the flavin moiety of FAD (gray carbon atoms), the amine oxidase domain (cyan) and the histone H3 peptide (pink). The position of LSD1 lysine 661 is indicated, as is that of lysine 4 of the histone peptide, here shown as MET-4. For purposes of the crystallization, lysine was replaced by methionine in view of the higher binding affinity of the pLys4Met peptide. Images were generated using PyMOL (Molecular Graphics System, Schrödinger, LLC) and a published structure [Citation12].FAD: Flavin-adenine dinucleotide; TF: Transcription factor.](/cms/asset/efa3d946-6506-4b2f-a1a8-008f452719e9/iepi_a_12325173_f0001.jpg)
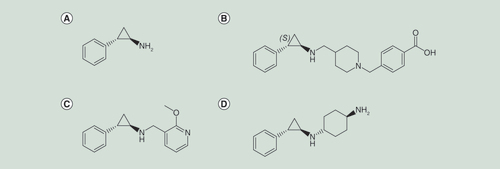
Chemical structures of (A) tranylcypromine; (B) GSK2879552; (C) OG86 (also known as Compound B) and (D) ORY1001. Images were generated using ChemDraw (PerkinElmer Inc).
First draft submitted: 25 January 2016; Accepted for publication: 27 May 2016; Published online: 1 August 2016
Gene expression & chromatin modifications
Transcription factors and chromatin modifiers are important in the programming and reprogramming of cellular states during development as well as for maintaining the correct cellular transcriptional profile. After binding to DNA, transcription factors recruit co-activator or co-repressor complexes, comprised in part by chromatin-modifying enzymes that may facilitate or impair transcription through covalent modification of histone tails. Methylation is one such modification which may occur on both arginine and lysine residues. Unlike acetylation it does not change the net charge of chromatin but instead creates new docking sites for recognition and binding by so-called reader proteins [Citation1]. Both promoter and enhancer regions are subject to lysine methylation which is often positively associated with active transcription. Typically H3K4 trimethylation is found at actively transcribed promoters, H3K4 dimethylation at both active promoters and enhancers and H3K4 monomethylation in particular at primed enhancer elements [Citation2]. Histone tail methylation is a dynamic process with the prevailing methylation state reflecting the sum of histone methyltransferase and histone demethylase activities. There are two families of histone demethylases: the larger Jumonji domain family and the smaller lysine-specific demethylase (LSD) family. LSD1 (also known as KDM1A, BHC110 or AOF2), the subject of this review, was the first histone demethylase to be identified [Citation3] and belongs to the flavin-adenine dinucleotide (FAD)-dependent family of amine oxidases.
LSD1 & its protein complexes
LSD1 was first isolated as an interaction partner of the histone deacetylase HDAC2 in HeLa cells and noted to have sequence homology with polyamine oxidase [Citation4]. Subsequent independent analyses (using HDAC1, HDAC2, RCOR1 or HMG20B immunoaffinity purification strategies) identified a protein complex consisting of LSD1, RCOR1 (also known as CoREST), HDAC1, HDAC2, ZNF217, PHF21A and HMG20B, often termed the CoREST transcription repressor complex (A) [Citation5–7]. LSD1 has also been identified in complex with ZMYM2, ZMYM3, GSE1 and GTF2I [Citation7], CTBP1, HMG20A, HSPA1A, PHF21B, RCOR3 and RREB1 [Citation8,Citation9], members of the Mi-2/nucleosome remodeling and deacetylase (NuRD) complex [Citation10] and the lysine methyltransferase mixed lineage leukemia (MLL) coactivator complex [Citation11].
The functional role of some of the interactions of LSD1 has been uncovered. For example, the incorporation of LSD1 into protein complexes such as CoREST or NuRD is essential for the ability of LSD1 to demethylate nucleosomes: it cannot demethylate nucleosomes alone and requires association with RCOR1 or MTA2 in each respective complex [Citation9–10,Citation13]. The LSD1:RCOR1 interaction also protects LSD1 from proteasomal degradation. The association of HDAC1 or HDAC2 in complex with LSD1 may remove local histone acetylation marks which inhibit the demethylase activity of LSD1 [Citation14]. PHF21A, another member of the complex, binds unmethylated H3K4 and stabilizes the association of LSD1 with chromatin, consequently promoting the activity of LSD1 as a transcription repressor [Citation15]. Nevertheless, the function of many of the different LSD1 complex members remains to be fully elucidated. For instance, HMG20A and HMG20B have been proposed to be alternative mutually exclusive subunits of the CoREST complex during neuronal differentiation [Citation16], as are RCOR1 homologues RCOR2 and RCOR3. These alternate subunits appear to confer different capacities to promote transcription repression [Citation17,Citation18]. Post-translational modification also seems important. For example, RCOR1 acts as a bridging factor between LSD1 and the ubiquitin-related modifier protein SUMO2 [Citation19]. Post-translational modification by SUMO regulates diverse cellular processes: in most cases SUMO conjugation alters the localization and/or activity of the substrate by providing a new protein–protein interaction interface [Citation20]. The depletion of SUMO-2/3 CoREST complex conjugates led to derepression and loss of promoter occupancy by RCOR1 and LSD1 at some but not all neuronal target genes [Citation19]. Bringing even more complexity, the CoREST complex is linked to Polycomb Repressive Complex 2 by the long noncoding RNA HOTAIR to assist in their recruitment to the HOXD locus [Citation21].
LSD1 is highly expressed in many cancer types including breast, prostate, esophageal, bladder and lung cancer, neuroblastoma and acute myeloid leukemia (AML) [Citation22]. It is likely that different mechanisms control LSD1 levels in each cell type. For example, LSD1 transcripts are negatively regulated by mir137, a brain-specific miRNA, to regulate neural stem cell proliferation and differentiation [Citation23]. LSD1 protein is ubiquitinated by JADE2, a novel E3-ubiquitin ligase, to facilitate its proteasome degradation which triggers embryonic stem cell differentiation toward the neural lineage [Citation24]. This process is antagonized by the deubiquitinase USP28 which stabilizes LSD1 protein levels, and whose expression is correlated with the overabundant levels of LSD1 in multiple cancer cell lines and breast tumor samples [Citation25].
Structure & enzymatic activity of LSD1 & its homolog LSD2
LSD1 is not the only member of the LSD/KDM1 demethylase protein family: it has a homolog called LSD2 (KDM1B; AOF1). Both enzymes are characterized by the presence of an amine oxidase (AO)-like domain (shared with several metabolic enzymes [Citation26]) and a Swi3p, Rsc8p and Moira (SWIRM) domain, which is unique to chromatin-associated proteins [Citation27]. Other than these two domains, LSD1 and LSD2 exhibit different structural architectures facilitating association with different protein complexes and different genomic loci. LSD1 contains a coiled-coil Tower domain protruding from the AO domain which is not found in any other monoamine oxidase [Citation28], while LSD2 contains an aminoterminal zinc finger domain of unknown function [Citation29].
The enzymatic activity of LSD1 was first demonstrated in a seminal study from the Shi laboratory in 2004 where it was found to demethylate mono- or di-methyl-lysine 4 of histone H3 (H3K4m1/me2), but not trimethyl-H3K4 or methyl-H3K9 [Citation3]. The catalytic activity of LSD1 (and LSD2) resides in the AO domain and is dependent on its cofactor flavin-adenine dinucleotide (FAD). The chemical reaction involves the stepwise conversion of methylated lysine to an iminium cation via abstraction of a hydride anion by the oxidized FAD prosthetic group. The cation is then hydrolyzed to give a carbinol amine which then decomposes to formaldehyde and the demethylated residue. The reduced FAD produced in the initial two electron reaction step is rapidly reoxidized by molecular oxygen to give a molecule of hydrogen peroxide and regenerated oxidized FAD. The demethylation mechanism relies on a lone electron pair present on the lysine ε-nitrogen atom, which is the reason why LSD enzymes can only demethylate mono- and di-methyl lysine but not trimethylated H3K4 (B) [Citation3,Citation26,Citation28,Citation30].
The AO domain has two lobes: one forms a noncovalent FAD-binding site and the other a substrate binding and recognition site. FAD sits in the deepest part of the pocket and is orientated in the correct plane through its interaction with lysine 661 [Citation31]. The FAD-binding subdomain shows considerable similarity to that of other amine oxidases, but the substrate-binding one is larger than that of other members and is able to accommodate not just the demethylation target but also its surrounding residues. This large pocket allows the recognition of several residues near the target lysine. Indeed histone H3 tail peptides greater than 16 amino acid length are necessary to achieve high demethylase efficiency [Citation30]. The AO rim is lined with negatively charged residues which likely facilitate electrostatic LSD1:substrate interactions (e.g., with positively charged histone tails). Furthermore, between the SWIRM and AO domains there is a large surface cleft which may provide additional interactions with substrates.
The differentiating structural domain of LSD1 (e.g., vs LSD2), the Tower domain hairpin, originates from the catalytic site raising a possibility that partner protein binding provides allosteric regulation of catalysis or substrate recognition. Indeed the RCOR1:LSD1 interaction occurs through the inter-SANT linker sequence and SANT2 domain of RCOR1, and the Tower domain and AO-substrate-binding lobe of LSD1 (C). The SANT2 interaction with the Tower domain is required for the demethylase activity of LSD1, likely through the former’s interaction with nucleosomal DNA [Citation32]. Based on molecular dynamics studies, LSD1/CoREST has been hypothesized to function as a flexible binding clamp, with the distance between its SANT domains being highly variable and its binding pocket having a capacity to change its volume by more than twofold. Substrate binding is predicted to occur through an induced fit mechanism which leads to allosteric regulation of the inter-SANT distance and nucleosome binding [Citation33].
The LSD1-SWIRM domain lacks the DNA-binding properties found in other SWIRM domain proteins and instead participates in protein–protein interactions [Citation34], maintaining protein structural integrity by the apposition of the SWIRM domain to the AO domain across a large hydrophobic interface. It is responsible for the association of LSD1 with androgen receptor, an interaction which has been argued to switch the lysine target specificity of LSD1 from H3K4 to H3K9 [Citation35]. The SWIRM domain of LSD2, which is slightly different to that of LSD1, is implicated in the interaction with glyoxylate reductase homolog 1GLYR1 (or NPAC) and positively stimulates its demethylase activity [Citation29]. Mutational analyses have demonstrated that the ability of LSD1 to demethylate histone tails is dependent upon multiple residues within the active site and SWIRM/AO interface. Some of these affect the catalysis, some the protein structure and some the substrate interaction. Mutations in the substrate (e.g., R2 of histone H3) also abolish the ability of LSD1 to physically interact with and demethylate its target [Citation28,Citation32,Citation36]. Therefore, the ability of LSD1 to demethylate H3K4 is influenced by the presence of nearby post-translational modifications. For example, as previously discussed, hyperacetylated nucleosomes are less amenable to demethylation by LSD1 than hypoacetylated ones [Citation9], and K9 acetylation reduces and S10 phosphorylation abolishes K4 demethylation [Citation30]. Interestingly, binding of the histone tail peptide is regulated separately from the demethylation reaction. Following substrate demethylation, substrate release is not required [Citation37] and both unmodified and K4 trimethylated histone tail peptides, and unmodified total histone H3, can bind LSD1 and function as competitive inhibitors [Citation28,Citation30,Citation38].
In addition to histone H3, LSD1 has also been reported to demethylate TP53, E2F1 and DNMT1 [Citation39–41], regulating their protein interactions, activity or stability. Surprisingly there are no sequence similarities between nonhistone and histone substrates, perhaps supporting the idea of the role of interaction partners on the demethylation process.
Biologic roles of LSD1
LSD1 is an essential gene in mammalian biology and many different specific roles have been reported. Germline murine knockouts exhibit embryonic lethality before E7.5: the egg cylinder fails to elongate and gastrulate resulting in development arrest [Citation41,Citation42]. This phenotype is due, at least in part, to failure of suppression of endogenous retrotransposable elements [Citation43]. Interestingly LSD1 mRNA and protein is highly expressed in undifferentiated human embryonic stem cells and is progressively downregulated during differentiation, suggesting a significant role in development. Indeed, in embryonic stem cell cultures it has been reported to be a key player in the maintenance of pluripotency through the control of H3K4 methylation levels at genes exhibiting bivalent chromatin domains (i.e., marked by both H3K4me2/3 and H3K27me3), such as FOXA2, EOMES and BMP2 [Citation44] thus contributing to repression of lineage-specific developmental programs [Citation44,Citation45]. LSD1 is also involved at the first emergence of hematopoiesis in vertebrates, where it controls hemangioblast formation (the progenitor cell capable of giving rise to both hematopoietic and endothelial progenitors) and generation of hematopoietic stem cells [Citation46,Citation47].
LSD1 is an important regulator of neural stem cell proliferation [Citation48] and neuronal development. The recruitment of LSD1 (and the full CoREST complex) to RE1 sites by the REST/NRSF transcription factor regulates transition from pluripotent to neural stem cells and from progenitor to mature neurons. LSD1/RCOR1 (as part of a complex identified as BRAF–histone deacetylase complex) mediates the repression of neuronal-specific genes [Citation49] and its disassembly is needed for differentiation into mature neurons [Citation50]. Interestingly, during embryonic development (E14.5–E17.5) RCOR1 is required for the development of dendrites and the migration of pyramidal neurons, but this migration is impaired following LSD1 knockdown independently of the interaction with the transcription factor REST [Citation51]. Further, three de novo variants of LSD1 have been identified in children with similar phenotypes including intellectual disabilities. The three mutations are located in the AO domain, although in different sites and have as yet unknown functions [Citation52]. Further information about the association of LSD1 with neurologic disease has recently been reviewed by Maes et al. [Citation53].
Tissue-specific knockouts have shown that LSD1 is required for proper differentiation of endocrine cells of the anterior pituitary [Citation42], for proper function of oocytes [Citation54] and for normal bone marrow function [Citation55]. Conditional deletion of LSD1 in adult bone marrow leads to rapid failure of multilineage hematopoiesis, although a functionally impaired immunophenotypic long-term hematopoietic stem cell compartment is retained to an extent, with cells entering the cell cycle [Citation55]. In vitro and in vivo knockdown approaches confirm that normal granulopoiesis, erythropoiesis and thrombopoiesis are all dependent upon LSD1 [Citation56,Citation57]. The C. elegans homolog spr-5 regulates Notch signaling [Citation58,Citation59], and maintains transgenerational epigenetic memory and fertility [Citation60]; and the yeast spLsd1/2 and Drosophila Su(var)3-3 homologs regulate heterochromatic gene silencing [Citation61,Citation62] and the size of the germline stem cell niche in Drosophila ovary [Citation63]. LSD1 has also been reported to have a role in the DNA damage response [Citation64], the circadian cycle [Citation65] and in repression of a mitochondrial metabolism and lipid oxidation energy expenditure program in adipocytes or liver cancer cells [Citation66,Citation67].
LSD1 as a transcription repressor
Multiple studies have demonstrated physical interaction of LSD1/CoREST with transcription regulators to promote repression. Examples include the binding of LSD1 to a proline-rich domain of the transcription factor PRDM1 (also known as BLIMP1) which contributes to suppression of the mature B-cell gene expression program (including regulation of CIITA, PAX5 and SPIB) during plasma cell differentiation [Citation68]; and the binding of LSD1 to a 40 amino acid sequence in TAL1 (also known as SCL) to effect repression of TAL1 targets during erythroid differentiation [Citation69]. The nuclear receptor TLX also recruits the LSD1-corepressor complex to promote neural stem cell proliferation [Citation48]. The capacity of LSD1 to act as a transcription repressor is illustrated both by GAL4-LSD1 fusions experiments [Citation3] and by approaches where LSD1 is fused to transcription activator-like effector repeat domains, or CRISPR-associated protein 9, which can then be selectively targeted to particular genomic sequences. These latter approaches lead to reduction of dimethyl-H3K4 and acetyl-H3K27 at the targeted enhancers with concomitant reduction of transcription of nearby genes, further supporting the role of LSD1 in controlling a repressive chromatin state on enhancers [Citation70,Citation71].
An essential additional example of LSD1’s transcription repressor activity is its physical interaction with members of the SNAG domain transcription factor family (e.g., SNAI1, SNAI2, GFI1, GFI1B, INSM1). These transcription factors feature at their N-terminal the SNAG (for SNAIL/GFI) domain sequence which mimics that of the histone H3 tail [Citation72]. LSD1 interacts with the SNAG domain sequence through its binding cleft analogous to its interaction with histone H3. The formation of a ternary complex between SNAI1/LSD1/RCOR1 is required for the stabilization of the three proteins and the repression of CDH1 (also known as E-cadherin), which impairs an epithelial-to-mesenchymal transition in several cancer types [Citation72,Citation73]. Endogenous LSD1/RCOR1/HDAC2 coimmunoprecipitates with GFI1 and GFI1B, and over 80% of GFI1B ChIP-on-chip targets in murine erythroid cells were co-occupied by LSD1 [Citation56]. The specific nature of these interactions is exemplified by several studies demonstrating that LSD1 is the effector of their repressive activity. When the interaction of GFI1 with LSD1 is completely abolished by a P2A amino acid substitution in GFI1, it renders the transcription factor inactive as a repressor [Citation56]. Similar findings were shown for SNAI1: here mutations of P2, R3, S4, F5, R8 and K9 in SNAI1 led to loss of the interaction with LSD1 which also results in loss of its ability to inhibit CDH1 promoter activity in luciferase assays [Citation72]. Moreover, deletion of the INSM1 SNAG domain and the INSM1 null mutation have indistinguishable effects on the development of endocrine cells of the pituitary gland [Citation74]. Interestingly, in competitive assays, addition of a SNAG peptide was much more effective in displacing the LSD1:SNAI1 interaction than a histone H3 peptide, suggesting a much higher affinity of LSD1 for the SNAG domain than the histone H3 tail [Citation75]. This intriguing finding for the mode of action of SNAG-transcription factors and their high affinity for LSD1 makes them inherently able to inhibit LSD1, because SNAG-residues must be released from the LSD1 cavity for LSD1 to be catalytically active against histone H3.
Despite its well-supported activity as a transcription repressor, LSD1 binds promiscuously across the genome at actively transcribed promoters and enhancers [Citation76,Citation77] suggesting that either it is inactive at most of its genomic-binding sites or that its activity is dynamically counterbalanced by co-located transcription activators and chromatin marks. Indeed, it has been proposed that distinct methyltransferases and demethylases (including LSD1) are utilized in a promoter-specific fashion, regulated by activation programs such as, for example, estrogen receptor signaling in MCF7 breast cancer cells [Citation76]. During pituitary organogenesis LSD1 is recruited to the Gh promoter as a component of the activator MLL1 complex, while the later expression of the zinc-finger repressor ZEB1 switches LSD1 to mediate repression as part of the LSD1-CoREST/CtBP complex [Citation42]. Another study analyzing embryonic stem cells found LSD1 associated with the NuRD complex occupying OCT4-regulated active enhancers, but without substantial demethylation activity on histone H3K4. Following the initiation of embryonic stem cell differentiation, OCT4 and EP300 levels decrease leading to reduced histone tail acetylation and consequent release from inhibition of LSD1 demethylase activity [Citation14,Citation30] which in turn permits the demethylation of H3K4 [Citation77]. These studies exemplify how cell context may control chromatin modifying enzymes, including LSD1 and its associated cofactors.
The fact that LSD1 targets H3K4me1, a key epigenetic mark of enhancers is itself interesting. Enhancers are dynamically utilized regions of the genome, and are identified as regions of 200–500 bp in length that show H3K4me1 and H3K27ac histone marks when active, and with clustered recognition sites for multiple transcription factors. De novo H3K4me1 establishment in hematopoiesis occurs in parallel with an increase in chromatin accessibility and correlates with chromatin reorganization during hematopoietic cell differentiation [Citation78]. LSD1 activity could therefore potentially block formation of new enhancers, which might, for example, contribute to a differentiation block in the setting of leukemia.
It is not completely clear whether H3K4me1 is associated with active or only primed enhancers, as both situations have been described. As concluded by Calo and Wysocka [Citation79] H3K4me1 perhaps broadly defines or correlates with a ‘window of opportunity’ within which enhancer activation can occur, for example through facilitating nucleosomal mobility or binding of pioneer transcription factors [Citation80]. In that scenario, the demethylation activity of LSD1 will lead to ‘silent enhancers’. One such example is FOXA1, a pioneer transcription factor for androgen receptor-regulated enhancers. In the LNCaP prostate cancer cell line FOXA1 knockdown markedly decreased basal LSD1 and RCOR1 binding to a series of androgen receptor/LSD1 regulated enhancers. Indeed LSD1 siRNA also decreases FOXA1 binding to these enhancers [Citation81] suggesting that FOXA1 may initially recruit the LSD1/CoREST complex to androgen-regulated enhancers, where FOXA1-LSD1 interaction may then stabilize the binding of both proteins to chromatin. This interesting finding suggests LSD1 may have additional roles at enhancers other than targeting H3K4me1: the stabilization of pioneer transcription factor binding to chromatin.
LSD1 as a transcription activator
LSD1 has also been suggested to be a coactivator in androgen receptor-dependent transcription through demethylation of the repression-associated trimethyl-H3K9 mark in prostate cancer cells [Citation35]. However, the mechanistic basis for the demethylation of H3K9 has been unclear: while the crystal structures of LSD1 indicate a mechanism for H3K4 demethylation [Citation12,Citation28,Citation32,Citation36], they do not for H3K9 demethylation. That said, recently a splice variant of LSD1 has been identified that arises from the retention of exon 8a, coding for an additional four amino acids positioned internal to the AO domain. This is one of four full-length isoforms of LSD1 arising from combinatorial retention of exons 2a and 8a. LSD1-8a is dynamically expressed during mammalian brain development and regulates neurite morphogenesis [Citation82]. The inclusion of exon 8a has been reported to create a docking site that facilitates an interaction between supervillin and LSD1 to convert LSD1 into an H3K9 demethylase during neuronal differentiation, where LSD1-E8a is enriched at promoters [Citation83]. A separate study reported LSD1-E8a to function as an H4K20 demethylase (and not as an H3K9 demethylase) in cortical neurons (where supervillin is expressed at very low level). In this context LSD1-8a promotes transcriptional initiation and elongation of neuronal activity-regulated genes including NPAS4, ARC and EGR1 by removing H4K20 methylation in transcribed coding regions. This function of LSD1-8a is required for spatial learning and memory [Citation84]. Moreover, another study reported that phosphorylation of the T369 residue in LSD1-8a generates a conformational change that provokes the detachment of the RCOR1 and HDAC1/2 corepressors from LSD1, which also impairs its repressive activity in neurons, and consequent function as a dominant negative regulator [Citation85]. While the inclusion of exon 2a can occur in all tissues, expression of LSD1-8a seems to be essentially restricted to the nervous system. It will be interesting to determine which LSD1 isoforms operate in each cancer type, specifically in prostate and androgen-receptor-mediated tumors where LSD1 is reported to have H3K9 demethylation capacities [Citation81] or in testis where LSD1-8a and LSD1-2a are both expressed [Citation82].
Pharmacologic inhibitors of LSD1
Given its enzymatic activity and its importance in cancer and neurologic diseases, there has been much interest in developing potent and specific pharmacologic inhibitors of LSD1. The nonselective monoamine oxidase inhibitor tranylcypromine (trans-2-phenylcyclopropylamine) (A) was the first drug reported to inhibit LSD1 [Citation86], functioning through a suicide-inactivation mechanism involving covalent modification of the essential cofactor FAD [Citation87]. Given the relative lack of potency and specificity of tranylcypromine versus LSD1, derivatives of tranylcypromine were subsequently developed which also function as suicide inactivators [Citation88]. Enhanced specificity to LSD1 versus monoamine oxidases A and B (MAOA/B) is achieved by addition of side groups to the phenyl ring or N-alkylation, and this is effective because the catalytic site of LSD1 is substantially larger than that of MAOA/B. These modifications also enhance affinity of the drug to LSD1 [Citation88]. Although achieving potent reversible inhibition of LSD1 has proved more challenging, several reversible LSD1 inhibitors have been reported to selectively compete with LSD1 substrate. Most of these remain at an early phase of development [Citation88,Citation89] but nevertheless have the potential to establish a novel type of LSD1 inhibition with alleviation of some of the potential side effects that tranylcypromine derivatives exert on erythropoiesis [Citation90]. Multiple compounds with irreversible or reversible LSD1 inhibitory activity have been reported: these include derivatives of the monoamine oxidase inhibitor phenelzine, peptide inhibitors based on the structure of the histone H3 tail or the SNAG domain, polyamine-based molecules, nonpeptide chemical histone tail mimics, small molecule amidoximes, phenyl oxazole derivatives, nonpeptidic propargylamines, aminothiazoles, thiazole sulfonamides, triazole dithiocarbamate hybrids, namoline, geranylgeranoic acid, benzohydrazides and pyrimidine thiourea hybrids [Citation91–101]. A summary of the most advanced LSD1 inhibitors is provided in , with additional details in recent reviews by Mould et al. and Zhang et al. [Citation88,Citation89].
Potential clinical use of LSD1 inhibitors
The observation of high-level LSD1 expression in malignancies such as those of prostate, breast and neuroblastoma, and its prediction for inferior prognosis, has led to a particular focus on cancer as a potential indication for LSD1 inhibition [Citation104–106]. Screening of a panel of 165 cell lines with the tranylcypromine derivative inhibitor GSK2879552 (B) revealed that AML and small-cell lung cancer (SCLC) lines are particularly sensitive [Citation91], although with typically cytostatic rather than cytocidal responses. In vivo, GSK2879552 effectively prevents growth of xenoengrafted NCI-H1417 SCLC cells without inducing hematologic toxicities, such as thrombocytopenia [Citation91].
In keeping with these observations, the tranylcypromine derivative LSD1 inhibitor OG86 (also known as Compound B) (C) promoted differentiation of murine and human primary AML cells and cell lines, with those exhibiting translocations targeting MLL being particularly sensitive. In vivo, drug treatment blocked accumulation of blast cells in the blood and selectively impaired the proliferative potential of AML cells but not normal hematopoietic stem and progenitor cells, which retained their ability to initiate secondary grafts [Citation90]. RN-1, another tranylcypromine derivative inhibitor, has also been shown to be effective across a large panel of AML and acute lymphoblastic leukemia cell lines. AML cells harboring the t(8;21) and MLL-rearrangements were more sensitive, and RN-1 completely abolished Kasumi-1 xenograft growth at well-tolerated doses [Citation102]. SP2509, a reversible inhibitor, has single agent efficacy in a xenograft model of Ewing sarcoma [Citation93] and SOX2-expressing squamous cell lung carcinoma cells show sensitivity to CBB1007/CBB1003 nonpeptide LSD1 substrate mimic inhibitors [Citation96]. LSD1 inhibitors may also have possible roles in the management of hemoglobinopathies such as sickle cell disease, because LSD1 inhibition enhances fetal hemoglobin production [Citation107], and in the prevention of reactivation from latency of Herpes virus infections [Citation108].
Also in AML, tranylcypromine or tranylcypromine derivatives have at least additive effects with all-trans retinoic acid to potentiate differentiation and loss of clonogenic potential [Citation109,Citation110], and the reversible LSD1 inhibitor SP2509 and the HDAC-inhibitor panobinostat together potentiate cell death in AML cell lines and xenograft mouse models [Citation103]. HDAC and LSD1 inhibition (using vorinostat and tranylcypromine) have also shown response in glioblastoma cell lines and patient-derived xenografts [Citation111], and synergy between RN-1 and cytarabine, as well as RN-1 and EZH2 inhibitors, has been shown in vitro using AML cell lines [Citation112].
Two tranylcypromine derivatives are currently undergoing evaluation in early phase clinical trials and results are awaited. Oryzon Genomics (partnered with Roche) is investigating ORY1001 (D) in the setting of acute leukemia [Citation53] and Glaxo Smith Kline is investigating GSK2879552 in the settings of AML and SCLC [Citation91]. Patents are in place for other LSD1 inhibitors from Takeda [Citation113], Imago Biosciences [Citation114], Incyte Corporation [Citation115], The University of Nevada [Citation116], Epitherapeutics [Citation117] and Quanticel Pharmaceuticals [Citation118]. Oryzon Genomics also has a dual LSD1 MAOB inhibitor (ORY2001) which is intended for evaluation in neurodegenerative disease [Citation53].
While the phenotypic consequences of these inhibitors in preclinical studies are presumed related to inhibition of the demethylase activity of LSD1, it is important to emphasize that much of the data are correlative. For example, it cannot immediately be concluded that an accumulation of dimethyl-H3K4 marks at a genomic LSD1-binding site following treatment of cells with an LSD1 inhibitor is directly due to pharmacologic LSD1 inhibition. It is known that certain LSD1 inhibitors displace the enzyme from chromatin [Citation112] and also that other enzymes demethylate dimethyl-H3K4 [Citation1]. As already mentioned, a SNAG peptide is more effective in displacing the interaction of LSD1 with SNAI1 than a histone H3 peptide suggesting that the LSD1:SNAG interaction may be of higher affinity than the LSD1:histone H3 interaction [Citation72]. Coupled with the observation that tranylcypromine abrogates the LSD1:SNAI2 interaction [Citation73], the possibility arises that the consequence of pharmacologic inhibition of LSD1 may at least in part be the inactivation of SNAG domain transcription factors, with genome-wide changes in histone methylation being downstream consequences of transcription factor inactivation. More generally, the biologic importance of the enzymatic activity of LSD1 versus its interactions with SNAG domain transcription factors remains unclear. It is also very interesting to note that, in the setting of AML cell lines, both reversible and irreversible LSD1 inhibitors (i.e., RN-1 and GSK690) led to a marked loss of LSD1 occupancy at a number of genomic locations (e.g., LY96, ID3 and RGBM) with concomitant increase in dimethyl-H3K4 levels. The eviction of LSD1, together with its cofactors RCOR1 and HMG20B, was only observed using potent inhibitors and not the weaker GSK690 compound, suggesting a connection to enzyme inhibition [Citation112]. However, these changes were not observed in SCLC models upon LSD1 inhibition [Citation91], potentially indicating that cell-specific contexts are important. In summary, in terms of its therapeutic potential as a drug target and its biologic roles and activities, there remains much more to be learned about LSD1.
Future perspective
From its initial discovery as a component of a protein complex with transcription repressing activity, and the subsequent discovery of its demethylase activity, a key question has been: can the biologic activities of LSD1 be targeted by small molecules for therapeutic benefit? With the transition of tranylcypromine derivative inhibitors into early phase trials in AML and SCLC, the answer to this question will soon become apparent. The insights arising from exposure of humans to LSD1 inhibition in the setting of clinical trials are, moreover, anticipated to feedback into further understanding of both the mechanism of action of these inhibitors and the role of LSD1 in mammalian biology. Clarity is required as to the various roles of LSD1 in cell-context-specific settings, including in which tissues its structural activities might be more important than its enzymatic activities. How the pharmacologic inhibition of LSD1 alters its interactions with cofactors, and its association with local histone modifications and chromatin structure, will be highly interesting questions to address in the near future.
Table 1. Pharmacologic inhibitors of lysine-specific demethylase 1 with proven cellular or functional effects.
LSD1 protein complexes, structure & enzymatic activity
LSD1 is an essential component of the repressive CoREST complex.
LSD1 is an FAD-dependent amine oxidase able to demethylate mono- or dimethyl-H3K4.
Interactions with protein partners stabilize LSD1 and facilitate its enzymatic activities.
Interactions of LSD1 with transcription regulators
LSD1/CoREST interacts with transcription factors to promote transcription repression.
SNAG domain transcription factors interact through their N-terminal sequences which mimic the structure of the histone H3 N-terminal tail.
The neuron-specific LSD1 splice variant (LSD1-8a) is an H3K9 and/or H4K20 demethylase functioning as a transcription activator.
Biologic role of LSD1
LSD1 is an essential gene in mammalian biology.
LSD1 is essential for proper differentiation of mature blood cells.
LSD1 is highly expressed in multiple malignancies.
LSD1 is a candidate therapeutic target in acute myeloid leukemia and small-cell lung cancer.
Pharmacologic inhibitors of LSD1 & potential clinical use
Tranylcypromine is a suicide inactivator inhibitor of LSD1, the first to be reported.
Tranylcypromine derivatives ORY1001 and GSK2879552 are now in early phase clinical trials.
Acknowledgements
The authors thank B Waszkowycz and D Mould for their assistance with figures; and G Spencer and H Rienhoff for a critical reading of the manuscript.
Financial & competing interests disclosure
A Maiques-Diaz and T Somervaille are supported by Cancer Research UK Grant Number C5759/A12328. T Somervaille has ongoing research collaborations with Oryzon Genomics and consults for Imago Biosciences. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Kouzarides T . Chromatin modifications and their function . Cell128 ( 4 ), 693 – 705 ( 2007 ).
- H⊘jfeldt JW , AggerK , HelinK . Histone lysine demethylases as targets for anticancer therapy . Nat. Rev. Drug Discov.12 ( 12 ), 917 – 930 ( 2013 ).
- Shi YY , LanF , MatsonCet al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1 . Cell119 ( 7 ), 941 – 953 ( 2004 ).
- Tong JK , HassigCA , SchnitzlerGR , KingstonRE , SchreiberSL . Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex . Nature395 ( 6705 ), 917 – 921 ( 1998 ).
- Humphrey GW , WangY , RussanovaVRet al. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1 . J. Biol. Chem.276 ( 9 ), 6817 – 6824 ( 2001 ).
- You A , TongJK , GrozingerCM , SchreiberSL . CoREST is an integral component of the CoREST–human histone deacetylase complex . Proc. Natl Acad. Sci. USA98 ( 4 ), 1454 – 8 ( 2001 ).
- Hakimi MA , DongY , LaneWS , SpeicherDW , ShiekhattarR . A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes . J. Biol. Chem.278 ( 9 ), 7234 – 7239 ( 2003 ).
- Shi Y , SawadaJ , SuiGet al. Coordinated histone modifications mediated by a CtBP co-repressor complex . Nature422 ( 6933 ), 735 – 738 ( 2003 ).
- Shi YJY , MatsonC , LanF , IwaseS , BabaT , ShiYJY . Regulation of LSD1 histone demethylase activity by its associated factors . Mol. Cell19 ( 6 ), 857 – 864 ( 2005 ).
- Wang Y , ZhangH , ChenYet al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer . Cell138 ( 4 ), 660 – 672 ( 2009 ).
- Nakamura T , MoriT , TadaSet al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation . Mol. Cell10 ( 5 ), 1119 – 1128 ( 2002 ).
- Forneris F , BindaC , AdamoA , BattaglioliE , MatteviA . Structural basis of LSD1-CoREST selectivity in histone H3 recognition . J. Biol. Chem.282 ( 28 ), 20070 – 20074 ( 2007 ).
- Lee MG , WynderC , CoochN , ShiekhattarR . An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation . Nature437 ( 7057 ), 432 – 435 ( 2005 ).
- Lee MG , WynderC , BocharDA , HakimiMA , CoochN , ShiekhattarR . Functional interplay between histone demethylase and deacetylase enzymes . Mol. Cell. Biol.26 ( 17 ), 6395 – 6402 ( 2006 ).
- Lan F , CollinsRE , De CegliRet al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression . Nature448 ( 7154 ), 718 – 722 ( 2007 ).
- Rivero S , Ceballos-ChávezM , BhattacharyaSS , ReyesJC . HMG20A is required for SNAI1-mediated epithelial to mesenchymal transition . Oncogene34 ( 41 ), 5264 – 5276 ( 2015 ).
- Upadhyay G , ChowdhuryAH , VaidyanathanB , KimD , SalequeS . Antagonistic actions of Rcor proteins regulate LSD1 activity and cellular differentiation . Proc. Natl Acad. Sci. USA111 ( 22 ), 8071 – 8076 ( 2014 ).
- Barrios ÁP , GómezAV , SáezJEet al. Differential properties of transcriptional complexes formed by the CoREST family . Mol. Cell. Biol.34 ( 14 ), 2760 – 2770 ( 2014 ).
- Ouyang J , ShiY , ValinA , XuanY , GillG . Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex . Mol. Cell34 ( 2 ), 145 – 154 ( 2009 ).
- Geiss-Friedlander R , MelchiorF . Concepts in sumoylation: a decade on . Nat. Rev. Mol. Cell Biol.8 ( 12 ), 947 – 956 ( 2007 ).
- Tsai M-C , ManorO , WanYet al. Long noncoding RNA as modular scaffold of histone modification complexes . Science329 ( 5992 ), 689 – 693 ( 2010 ).
- Lynch JT , HarrisWJ , SomervailleTC . LSD1 inhibition: a therapeutic strategy in cancer?Expert Opin. Ther. Targets16 ( 12 ), 1239 – 1249 ( 2012 ).
- Sun G , YeP , MuraiKet al. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells . Nat. Commun.2 , 529 ( 2011 ).
- Han X , GuiB , XiongCet al. Destabilizing LSD1 by Jade-2 promotes neurogenesis: an antibraking system in neural development . Mol. Cell55 ( 3 ), 482 – 494 ( 2014 ).
- Wu Y , WangY , YangXHet al. The deubiquitinase USP28 stabilizes LSD1 and confers stem-cell-like traits to breast cancer cells . Cell Rep.5 ( 1 ), 224 – 236 ( 2013 ).
- Culhane JC , ColePA . LSD1 and the chemistry of histone demethylation . Curr. Opin. Chem. Biol11 ( 5 ), 561 – 568 ( 2007 ).
- Aravind L , IyerLM . The SWIRM domain: a conserved module found in chromosomal proteins points to novel chromatin-modifying activities . Genome Biol.3 ( 8 ), research0039.1–research0039.7 ( 2002 ).
- Stavropoulos P , BlobelG , HoelzA . Crystal structure and mechanism of human lysine-specific demethylase-1 . Nat. Struct. Mol. Biol.13 ( 7 ), 626 – 632 ( 2006 ).
- Fang R , ChenF , DongZet al. LSD2/KDM1B and its cofactor NPAC/GLYR1 endow a structural and molecular model for regulation of H3K4 demethylation . Mol. Cell49 ( 3 ), 558 – 570 ( 2013 ).
- Forneris F , BindaC , VanoniMA , BattaglioliE , MatteviA . Human histone demethylase LSD1 reads the histone code . J. Biol. Chem.280 ( 50 ), 41360 – 41365 ( 2005 ).
- Polticelli F , BasranJ , FasoCet al. Lys300 plays a major role in the catalytic mechanism of maize polyamine oxidase . Biochemistry44 ( 49 ), 16108 – 16120 ( 2005 ).
- Yang M , GockeCB , LuoXet al. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase . Mol. Cell23 ( 3 ), 377 – 387 ( 2006 ).
- Baron R , VelloreN . LSD1/CoREST is an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition . Proc. Natl Acad. Sci. USA109 ( 31 ), 12509 – 12514 ( 2012 ).
- Yoneyama M , TochioN , UmeharaTet al. Structural and functional differences of SWIRM domain subtypes . J. Mol. Biol.369 ( 1 ), 222 – 238 ( 2007 ).
- Metzger E , WissmannM , YinNet al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription . Nature437 ( 7057 ), 436 – 439 ( 2005 ).
- Yang M , CulhaneJC , SzewczukLMet al. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation . Nat. Struct. Mol. Biol.14 ( 6 ), 535 – 539 ( 2007 ).
- Forneris F , BindaC , Dall’AglioA , FraaijeMW , BattaglioliE , MatteviA . A highly specific mechanism of histone H3-K4 recognition by histone demethylase LSD1 . J. Biol. Chem.281 ( 46 ), 35289 – 35295 ( 2006 ).
- Burg JM , GonzalezJJ , MaksimchukKR , McCaffertyDG . Lysine-specific demethylase 1A (KDM1A/LSD1): product recognition and kinetic analysis of full-length histones . Biochemistry55 ( 11 ), 1652 – 1662 ( 2015 ).
- Huang J , SenguptaR , EspejoABet al. p53 is regulated by the lysine demethylase LSD1 . Nature449 ( 7158 ), 105 – 108 ( 2007 ).
- Kontaki H , TalianidisI . Lysine methylation regulates E2F1-induced cell death . Mol. Cell39 ( 1 ), 152 – 160 ( 2010 ).
- Wang J , HeviS , KurashJKet al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation . Nat. Genet.41 ( 1 ), 125 – 129 ( 2009 ).
- Wang J , ScullyK , ZhuXet al. Opposing LSD1 complexes function in developmental gene activation and repression programmes . Nature446 ( 7138 ), 882 – 887 ( 2007 ).
- Macfarlan TS , GiffordWD , AgarwalSet al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A . Genes Dev.25 ( 6 ), 594 – 607 ( 2011 ).
- Adamo A , SeséB , BoueSet al. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells . Nat. Cell Biol.13 ( 6 ), 652 – 659 ( 2011 ).
- Foster CT , DoveyOM , LezinaLet al. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability . Mol. Cell. Biol.30 ( 20 ), 4851 – 4863 ( 2010 ).
- Takeuchi MM , FuseY , WatanabeMet al. LSD1/KDM1A promotes hematopoietic commitment of hemangioblasts through downregulation of Etv2 . Proc. Natl Acad. Sci. USA112 ( 45 ), 13922 – 13927 ( 2015 ).
- Thambyrajah R , MazanM , PatelRet al. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1 . Nat. Cell Biol.18 ( 1 ), 21 – 32 ( 2015 ).
- Sun G , AlzayadyK , StewartRet al. Histone demethylase LSD1 regulates neural stem cell proliferation . Mol. Cell. Biol.30 ( 8 ), 1997 – 2005 ( 2010 ).
- Hakimi MA , BocharDA , ChenowethJ , LaneWS , MandelG , ShiekhattarR . A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes . Proc. Natl Acad. Sci. USA99 ( 11 ), 7420 – 7425 ( 2002 ).
- Ballas N , GrunseichC , LuDD , SpehJC , MandelG . REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis . Cell121 ( 4 ), 645 – 657 ( 2005 ).
- Fuentes P , CánovasJ , BerndtFA , NoctorSC , KukuljanM . CoREST/LSD1 control the development of pyramidal cortical neurons . Cereb. Cortex22 ( 6 ), 1431 – 1441 ( 2012 ).
- Chong JX , YuJ-H , LorentzenPet al. Gene discovery for Mendelian conditions via social networking: de novo variants in KDM1A cause developmental delay and distinctive facial features . Genet. Med. doi:10.1038/gim.2015.161 ( 2015 ) ( Epub ahead of print ).
- Maes T , MascaróC , OrtegaAet al. KDM1 histone lysine demethylases as targets for treatments of oncological and neurodegenerative disease . Epigenomics7 ( 4 ), 609 – 626 ( 2015 ).
- Kim J , SinghAK , TakataYet al. LSD1 is essential for oocyte meiotic progression by regulating CDC25B expression in mice . Nat. Commun.6 , 10116 ( 2015 ).
- Kerenyi MA , ShaoZ , HsuY-JJet al. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation . Elife2 , e00633 ( 2013 ).
- Saleque S , KimJ , RookeHM , OrkinSH . Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1 . Mol. Cell27 ( 4 ), 562 – 572 ( 2007 ).
- Sprussel A , SchulteJH , WeberSet al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation . Leukemia26 ( 9 ), 2039 – 2051 ( 2012 ).
- Jarriault S , GreenwaldI . Suppressors of the egg-laying defective phenotype of sel-12 presenilin mutants implicate the CoREST corepressor complex in LIN-12/Notch signaling in C. elegans . Genes Dev.16 ( 20 ), 2713 – 2728 ( 2002 ).
- Eimer S , LakowskiB , DonhauserR , BaumeisterR . Loss of spr-5 bypasses the requirement for the C. elegans presenilin sel-12 by derepressing hop-1 . EMBO J.21 ( 21 ), 5787 – 5796 ( 2002 ).
- Katz DJ , EdwardsTM , ReinkeV , KellyWG . A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory . Cell137 ( 2 ), 308 – 320 ( 2009 ).
- Lan F , ZaratieguiM , VillénJet al. S. pombe LSD1 homologs regulate heterochromatin propagation and euchromatic gene transcription . Mol. Cell26 ( 1 ), 89 – 101 ( 2007 ).
- Rudolph T , YonezawaM , LeinSet al. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3-3 . Mol. Cell26 ( 1 ), 103 – 115 ( 2007 ).
- Eliazer S , PalaciosV , WangZ , KolliparaRK , KittlerR , BuszczakM . Lsd1 restricts the number of germline stem cells by regulating multiple targets in escort cells . PLoS Genet.10 ( 3 ), e1004200 ( 2014 ).
- Mosammaparast N , KimH , LaurentBet al. The histone demethylase LSD1/KDM1A promotes the DNA damage response . J. Cell Biol.203 ( 3 ), 457 – 470 ( 2013 ).
- Nam HJ , BooK , KimDet al. Phosphorylation of LSD1 by PKCα is crucial for circadian rhythmicity and phase resetting . Mol. Cell53 ( 5 ), 791 – 805 ( 2014 ).
- Hino S , SakamotoA , NagaokaKet al. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure . Nat. Commun.3 , 758 ( 2012 ).
- Sakamoto A , HinoS , NagaokaKet al. Lysine demethylase LSD1 coordinates glycolytic and mitochondrial metabolism in hepatocellular carcinoma cells . Cancer Res.75 ( 7 ), 1445 – 1456 ( 2015 ).
- Su S-T , YingH-Y , ChiuY-K , LinF-R , ChenM-Y , LinK-I . Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation . Mol. Cell. Biol.29 ( 6 ), 1421 – 1431 ( 2009 ).
- Hu X , LiX , ValverdeKet al. LSD1-mediated epigenetic modification is required for TAL1 function and hematopoiesis . Proc. Natl Acad. Sci. USA106 ( 25 ), 10141 – 10146 ( 2009 ).
- Mendenhall EM , WilliamsonKE , ReyonDet al. Locus-specific editing of histone modifications at endogenous enhancers . Nat. Biotechnol.31 ( 12 ), 1133 – 1136 ( 2013 ).
- Kearns NA , PhamH , TabakBet al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion . Nat. Methods12 ( 5 ), 401 – 403 ( 2015 ).
- Lin Y , WuY , LiJet al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1 . EMBO J.29 ( 11 ), 1803 – 1816 ( 2010 ).
- Ferrari-Amorotti G , FragliassoV , EstekiRet al. Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion . Cancer Res.73 ( 1 ), 235 – 245 ( 2013 ).
- Welcker JE , Hernandez-MirandaLR , PaulFEet al. Insm1 controls development of pituitary endocrine cells and requires a SNAG domain for function and for recruitment of histone-modifying factors . Development140 ( 24 ), 4947 – 4958 ( 2013 ).
- Lin T , PonnA , HuX , LawBK , LuJ . Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial–mesenchymal transition . Oncogene29 ( 35 ), 4896 – 4904 ( 2010 ).
- Garcia-Bassets I , KwonY-S , TeleseFet al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors . Cell128 ( 3 ), 505 – 518 ( 2007 ).
- Whyte WA , BilodeauS , OrlandoDAet al. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation . Nature482 ( 7384 ), 221 – 225 ( 2012 ).
- Lara-Astiaso D , WeinerA , Lorenzo-VivasEet al. Chromatin state dynamics during blood formation . Science345 ( 6199 ), 943 – 949 ( 2014 ).
- Calo E , WysockaJ . Modification of enhancer chromatin: what, how, and why?Mol. Cell49 ( 5 ), 825 – 837 ( 2013 ).
- Zaret KS , CarrollJS . Pioneer transcription factors: establishing competence for gene expression . Genes Dev.25 ( 21 ), 2227 – 2241 ( 2011 ).
- Cai C , HeHH , GaoSet al. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity . Cell Rep.9 ( 5 ), 1618 – 1627 ( 2014 ).
- Zibetti C , AdamoA , BindaCet al. Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system . J. Neurosci.30 ( 7 ), 2521 – 2532 ( 2010 ).
- Laurent B , RuituL , MurnJet al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation . Mol. Cell57 ( 6 ), 957 – 970 ( 2015 ).
- Wang J , TeleseF , TanYet al. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control . Nat. Neurosci.18 ( 9 ), 1256 – 1264 ( 2015 ).
- Toffolo E , RusconiF , PaganiniLet al. Phosphorylation of neuronal lysine-specific demethylase 1LSD1/KDM1A impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2 . J. Neurochem.128 ( 5 ), 603 – 616 ( 2014 ).
- Lee MG , WynderC , SchmidtDM , McCaffertyDG , ShiekhattarR . Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications . Chem. Biol.13 ( 6 ), 563 – 567 ( 2006 ).
- Schmidt DMZ , McCaffertyDG . Trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1 . Biochemistry46 ( 14 ), 4408 – 4416 ( 2007 ).
- Zheng Y-C , MaJ , WangZet al. A systematic review of histone lysine-specific demethylase 1 and its inhibitors . Med. Res. Rev.35 ( 5 ), 1032 – 1071 ( 2015 ).
- Mould DP , McGonagleAE , WisemanDH , WilliamsEL , JordanAM . Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date . Med. Res. Rev.35 ( 3 ), 586 – 618 ( 2015 ).
- Harris WJ , HuangX , LynchJTet al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells . Cancer Cell21 ( 4 ), 473 – 487 ( 2012 ).
- Mohammad HP , SmithemanKN , KamatCDet al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC . Cancer Cell28 ( 1 ), 57 – 69 ( 2015 ).
- Cui S , LimK-C , ShiLet al. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice . Blood126 ( 3 ), 386 – 396 ( 2015 ).
- Sankar S , TheisenER , BearssJet al. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth . Clin. Cancer Res.20 ( 17 ), 4584 – 4597 ( 2014 ).
- Schmitt ML , HauserA-T , CarlinoLet al. Nonpeptidic propargylamines as inhibitors of lysine specific demethylase 1 (LSD1) with cellular activity . J. Med. Chem.56 ( 18 ), 7334 – 7342 ( 2013 ).
- Wang J , LuF , RenQet al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties . Cancer Res.71 ( 23 ), 7238 – 7249 ( 2011 ).
- Zhang X , LuF , WangJet al. Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells . Cell Rep.5 ( 2 ), 445 – 457 ( 2013 ).
- Hitchin JR , BlaggJ , BurkeRet al. Development and evaluation of selective, reversible LSD1 inhibitors derived from fragments . MedChemComm4 ( 11 ), 1513 ( 2013 ).
- Culhane JC , SzewczukLM , LiuX , DaG , MarmorsteinR , ColePA . A mechanism-based inactivator for histone demethylase LSD1 . J. Am. Chem. Soc.128 ( 14 ), 4536 – 4537 ( 2006 ).
- Kumarasinghe IR , WosterPM . Synthesis and evaluation of novel cyclic peptide inhibitors of lysine-specific demethylase 1 . ACS Med. Chem. Lett.5 ( 1 ), 29 – 33 ( 2014 ).
- Murray-Stewart T , WosterPM , CaseroRA . The re-expression of the epigenetically silenced e-cadherin gene by a polyamine analogue lysine-specific demethylase-1 (LSD1) inhibitor in human acute myeloid leukemia cell lines . Amino Acids46 ( 3 ), 585 – 594 ( 2014 ).
- Hazeldine S , PachaiyappanB , SteinbergsNet al. Low molecular weight amidoximes that act as potent inhibitors of lysine-specific demethylase 1 . J. Med. Chem.55 ( 17 ), 7378 – 7391 ( 2012 ).
- McGrath JP , WilliamsonKE , BalasubramanianSet al. Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes . Cancer Res.76 ( 7 ), 1975 – 1988 ( 2016 ).
- Fiskus W , SharmaS , ShahBet al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells . Leukemia28 ( 11 ), 2155 – 2164 ( 2014 ).
- Kahl P , GullottiL , HeukampLCet al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence . Cancer Res.66 ( 23 ), 11341 – 11347 ( 2006 ).
- Schulte JH , LimS , SchrammAet al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy . Cancer Res.69 ( 5 ), 2065 – 2071 ( 2009 ).
- Lim S , JanzerA , BeckerAet al. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology . Carcinogenesis31 ( 3 ), 512 – 520 ( 2010 ).
- Shi L , CuiS , EngelJD , TanabeO . Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction . Nat. Med.19 ( 3 ), 291 – 294 ( 2013 ).
- Liang Y , VogelJL , NarayananA , PengH , KristieTM . Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency . Nat. Med.15 ( 11 ), 1312 – 1317 ( 2009 ).
- Binda C , ValenteS , RomanenghiMet al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2 . J. Am. Chem. Soc.132 ( 19 ), 6827 – 6833 ( 2010 ).
- Schenk T , ChenWC , GollnerSet al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia . Nat. Med.18 ( 4 ), 605 – 611 ( 2012 ).
- Singh MM , JohnsonB , VenkatarayanAet al. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma . Neuro. Oncol.17 ( 11 ), 1463 – 1473 ( 2015 ).
- McGrath JP , WilliamsonKE , BalasubramanianSet al. Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes . Cancer Res.76 ( 7 ), 1975 – 1982 ( 2016 ).
- Tomita N , KajiiS , CaryDRet al. WO 2013/022047 AI ( 2013 ).
- McCall JM , ReinhoffHYJr , ClareM . Patent WO 2015021128 A1 ( 2015 ).
- Incyte Corporation , WO2015123437 ( 2015 ).
- The Board of Regents of the Nevada System of Higher Education . WO2014194280 ( 2014 ).
- Epitherapeutics Aps . US9221801 ( 2015 ).
- Quanticel Pharmaceuticals . WO2015168466 A1 ( 2015 ).