
Abstract
Aim: Epigenetic comparisons within monozygotic twin pairs have enhanced our understanding of nongenetic mechanisms underlying disease etiology. We present epigenetic findings for a unique case of doubly exchanged Colombian male monozygotic twins raised in extremely different environments. Results: Using genome-wide DNA methylation data from cheek swabs from which blood-specific differentially methylated probes had been removed, the individuals grouped by shared genetics rather than shared environment, except for one twin who presented as an outlier. Closer inspection of DNA methylation differences within both reared-apart twin pairs revealed several genes and genetic pathways likely to be influenced by the rearing environment. Conclusion: Together with our previous findings, we suggest that genetics, pre- and postnatal environments contribute to the epigenetic profile, although additional studies are needed to quantify these effects.
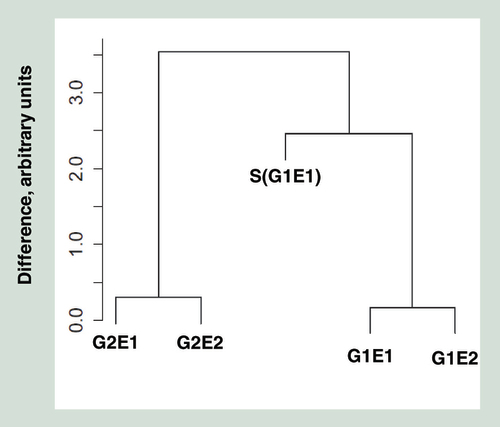
These probes are highly polymorphic SNPs, intended for analyzing relatedness between individuals. The y-axis represents arbitrary units of differences between individuals. Monozygotic pairs are coded as in the text.
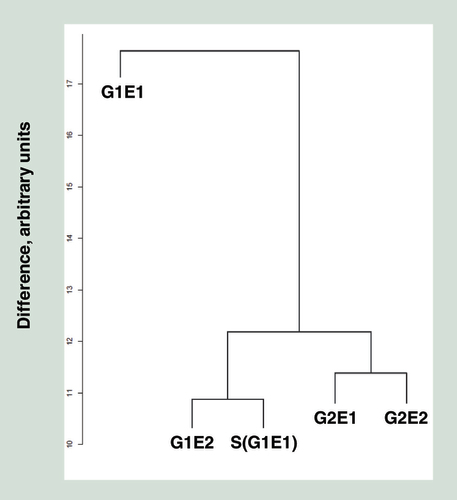
Individuals are labeled the same way as .
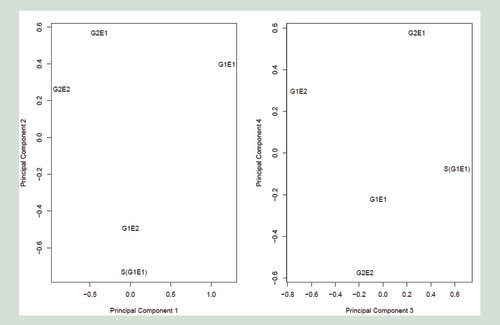
These are based on M values of 150,678 probes. Although we have a low sample size, genetics appears to be best represented by PC1 and rearing environment by PC3. Individuals are labeled the same say as .
Background
The classic monozygotic (MZ)-dizygotic (DZ) twin comparison is an informative tool for investigating genetic and environmental influences on human health and behavior. Greater resemblance within MZ compared with DZ twin pairs is consistent with genetic components to the condition under study [Citation1]. Studying MZ twins reared apart from birth (MZA) is an especially powerful variant of the classic twin design. Similarities within MZA twin pairs yield direct estimates of heritability, given the absence of a shared rearing environment [Citation2]. MZ twins can be epigenetically different at birth [Citation3,Citation4], most likely because they can experience different prenatal environments.
Epigenetics describes a number of biological processes whereby gene expression is regulated and maintained through cell division and includes the addition of small molecules such as the methyl group (CH3) to DNA and its packaging proteins. Recently, epigenetic analyses of disease-discordant MZ twins have drawn attention to mechanisms relevant to the differential expression of relevant genes [Citation5,Citation6], barring the presence of rare postzygotic mutation [Citation7]. DNA methylation at the dinucleotide CpG is an epigenetic mechanism which is often associated with repression of gene expression.
To date, no epigenetic studies of MZA twins have been conducted. Such studies, especially those using co-twins raised in dramatically different environments, can potentially identify key postnatal environmental factors associated with within-pair differences in epigenetic profiles. Furthermore, some studies, but not all, have concluded that MZ reared-together (MZT) twins may ‘drift apart’ or diverge in their epigenetic profiles over time [Citation3,Citation8–11]. It has been suggested that epigenetic drift may be driven by discordance in the rearing environments of MZT pairs [Citation12]. However, MZT and MZA pairs show comparable phenotypic similarity across a range of measures, including EEG, height, heart rate, blood pressure, electrodermal response, personality and well-being [Citation13]. Reconciling these sets of findings remains an important goal for future implications of environmental and genetic influences on development.
Here we present epigenetic findings, specifically DNA methylation, from the only known case of doubly exchanged adult MZ male twins, from Colombia, South America, raised in extremely different environments. The twins discovered at 25 years of age that one member from each of the two pairs had been inadvertently exchanged at birth with a member from the other set, generating two same-age unrelated pairs, known as virtual twins [Citation14]. The study also includes a biological sister who is four years older than a twin in one pair with whom she was raised. This situation is of great interest because she shares both genes and the postnatal environment with one biological brother, only the postnatal environment with her nonbiological brother and only genes with her reared-apart biological brother.
The inadvertent exchange of two MZ male twin pairs came to the attention of the second author in October 2014. Following a series of contacts from the second author, the twins agreed to participate in a psychological and life history study, scheduled for March–April 2015. IRB approval was granted from California State University, Fullerton where the first author directs the Twin Studies Center. In this manuscript we have coded twin names based on shared genetics (twin pairs G1 and G2) and shared rearing environments (E1 and E2).
MZ twins G1E1 and G1E2 were born in a major city, and MZ twins, G2E1 and G2E2, were approximately 150 miles away, in a rural village. Newborn health issues necessitated G2E1’s immediate transport to a hospital in the major city, the same one at which G1E1 and G1E2 were being cared for in the premature nursery. It is here that G2E1 and G1E2 were unintentionally exchanged, although the precise events surrounding this error are unknown. As a result, G1E1 and G2E1 grew up together for 25 years, believing that they were nonidentical or DZ twins. G1E1 and G2E1 had access to rich educational and cultural resources, and both twins were completing graduate degrees at the time of the study. G1E2 and G2E2 grew up together in the countryside, also believing that they were DZ twins. G1E2 and G2E2 were raised in a remote farm home without modern conveniences, and like other children in that area, neither twin had advanced beyond the fifth grade (although G1E2 completed a high school equivalency course in 2010).
In 2014, the four young men were living in different areas of the same city when G1E1 and G1E2 learned of each other’s existence, due to mistaken identity on the part of a friend. This chance interaction led to their inspection of pictures displaying G1E2 and G2E2 together, with the realization that G2E1 and G2E2 could also be MZ twin brothers because G2E2 looked exactly like G2E1. Thus, the brothers began to suspect that an exchange had occurred at the hospital.
Both sets of twins met for the first time in 2014 at the age of 25. The physical similarities between the members of each MZA set were striking. DNA testing was then arranged to establish the zygosity of the two separated pairs.
In October 2014, the monozygosity of the two twin pairs (G1E1-G1E2 and G2E1-G2E2) was confirmed (see ‘Methods’ section for more details). Dermatoglyphic analyses later completed at the University of Minnesota were consistent with these findings (data not shown).
The first and second authors visited the two MZA twin pairs and sister [S(G1E1)] in March and April 2014. Following informed consent, the four brothers completed a comprehensive life history interview, general intelligence tests (one prior to the research visit) and a battery of personality and interest questionnaires.
Here we present findings from the methylation analysis.
Methods
Buccal swab processing
Buccal samples were obtained in Colombia using ORAcollect•DNA swab kits containing preservative (ORC-100, DNA Genotek, Ottawa, Canada) and swabs were immediately mailed to Australia. Genomic DNA (gDNA) were extracted using Macherey-Nagel NucleoBond CB20 kits (Scientifix Pty. Ltd., Cheltenham, Australia), and sent to the Department of Pathology, University of Melbourne for bisulfite conversion and microarray analysis.
Genome-wide methylation data processing
Genome-wide methylation data were obtained from Infinium Human Methylation450 (HM450) BeadChip arrays according to manufacturer’s instructions [Citation15]. Raw intensity data (IDAT) files were uploaded to R studio and processed using the minfi package [Citation16] from Bioconductor as previously described [Citation17]. Data from all five samples had average detection p < 0.001. Between- and within-array normalization was performed using subset quantile normalization [Citation18] in the minfi package. Probes with detection p-value greater than 0.01, which are known to hybridize to multiple genomic locations [Citation19], were removed, along with those on the X and Y chromosomes (to allow for comparison of males and females). This resulted in 429,594 analyzable probes remaining. Because samples collected using ORAcollect•DNA swabs contain a mixture of blood and buccal cells [unpublished data], we used published datasets [Citation20] to remove all probes with >1% difference in DNA methylation between buccal epithelial and blood cells (tissue-specific differentially methylated probes [tDMPs]). This stringent cut-off, which left us with data from 150,678 Infinium probes, effectively removed all probes for which buccal/blood cellular heterogeneity could confound our results. Due to our very small sample size, we were not able to adjust our data computationally for cellular heterogeneity. Beta values (β; intensity of the methylated allele/[intensity of methylated allele + intensity of unmethylated allele]) [Citation15] were calculated for each remaining probe.
Zygosity confirmation
The twins’ zygosity was determined by 21 short tandem repeat markers at the Servicios Médicos Yunis Turbay and Associates, Instituto de Genética (Medical Services of Yunis Turbay and Associates, Genetics Institute), in Bogotá and the beta values of 65 SNP probes from Infinium HumanMethylation450 BeadChip (HM450) arrays (Illumina; ) [Citation21].
Statistical analysis
Statistical analysis was performed using R Studio software and Microsoft Excel. To investigate the correlation of methylation data within twin pairs, Pearson correlation plots and intraclass correlation coefficients (ICC) were generated using the logit transformed beta values (M values) of the 150,678 probes. ICC is the proportion of between-pair methylation variability to the total variability [Citation22], where a value close to +1 indicates high between-pair epigenetic variability (low within-pair variability) relative to total pair methylation variation [Citation23]. To identify the extent of within-pair DNA methylation discordance, unsupervised clustering and multidimensional scaling (MDS) plots were generated. Unsupervised clustering was performed based on the default ‘complete linkage clustering’ using hclust command in R, which calculates the maximum possible distance (square root of sum of squares of methylation differences) between individuals. MDS and principal component plots were constructed based on the 1000 probes with largest standard deviations between samples using the plotMDS command in the limma package. To identify probes that were most differentially methylated within both MZA pairs, absolute methylation differences within MZA pairs were calculated and ranked from largest to smallest within each set of twins, and ranks were added up to give the ‘sum of ranks’ for both MZA pairs. Enrichment analysis of genes annotated within the 1000 top-ranked probes were conducted using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) [Citation24,Citation25] and Reduce and Visualize Gene Ontology (REVIGO) [Citation26] bioinformatics tools. To compare ontologies and pathways with the genes associated with top DMPs, we focused only on genes associated with two or more top DMPs clustered within 1kb genomic regions to minimize type 1 errors. Enrichment analysis of functional genomic regions associated with the top 1000 DMPs were calculated based on observed/expected frequency to identify any specific functional regions where the top DMPs are more likely to be annotated. The Genomic Regions Enrichment of Annotations Tool (GREAT) [Citation27] was used for enrichment analysis specific for regions in relation to distance to translational start site.
Results
Low detection p-values for each sample (Supplementary Figure 1A) indicated that our methylation data were reliable, as signals from probes were significantly different from background. The methylation data quality of HM450 probes was then assessed by the data preprocessing method previously described [Citation11], resulting in the removal of 45,503 probes. Removal of tDMPs resulted in 150,678 probes remaining, and the density plot of beta values from these remaining probes showed uniformity (Supplementary Figure 1C), minimizing the potential of technical variation affecting the data analysis. All subsequent analyses were based on these probes.
Using logit transformed beta values (M values) of 150,678 probes obtained, Pearson correlation plots of the twins (Supplementary Figure 2) and ICC () showed that the correlation was the same for the MZA and for the unrelated pairs reared in the same family environment. Both Pearson and ICCs were on average 0.990, with ICCs ranging from 0.987 to 0.992. We also found no evidence for a difference in average (global, across all probes) DNA methylation between the twins raised in different environments (Supplementary Table 1). All individuals had an average global methylation value of 0.33.
Results from unsupervised cluster analysis of the five HM450 datasets are shown in . Similar to the clustering by genetics (), MZ twins G2E1 and G2E2 clustered together, as did G1E2 with S(G1E1), his reared-apart sister S(G1E1). However, G1E1 clustered well away from his twin G1E2 and biological sister S(G1E1). MDS plots allow the observation of the largest source of variation in methylation data. is a compilation of two such MDS plots, where different principal components (PC1 to PC4 ranked by magnitude, Supplementary Figure) are plotted against each other. PC1 appeared to separate individuals by their shared genetics (A), whereas PC3 appeared to separate individuals by their environment (B). This suggests that both genetic and rearing environment have a strong influence on genome-wide DNA methylation, with the former having a larger magnitude
To investigate the specific genomic locations at which shared rearing environment had the largest effect on DNA methylation in the MZA twins, we ranked probe data by the sum of the absolute methylation difference within the two MZA pairs. We then looked at the top 1000 DMPs (Supplementary Table 3) with respect to rearing environment. The average absolute methylation differences were higher between G1E1 and G1E2 (25.6%) than between G2E1 and G2E2 (12.1%), with a positive correlation in the within-pair methylation values of both pairs (Supplementary Figure 4). The top 1000 rearing environment-specific DMPs were enriched in regions outside regulatory regions, for example, CpG Island ‘shelves’ and ‘open seas’ and in enhancers, which are regulatory regions often at a distance from gene promoters (Supplementary Figure 5A). DMPs were underrepresented in CpG islands and promoters (Supplementary Figure 5A) and close to transcriptional start sites (Supplementary Figure 5B), all of which play a role in gene regulation [Citation17].
Genes associated with rearing environment-specific DMPs were enriched for biological pathways related to ‘immune response’, ‘protein complex assembly’, ‘cell death’, ‘death’ and ‘negative regulation of macromolecule metabolism’ (). To minimize type 1 errors in such a small sample size, we focused further on only those DMPs that clustered within 1 kb of genomic space. We found 32 such gene-associated clusters, of which, six (GABRA5, CD300LB, PROKR1, CXCR2, HLA-L, TNFRSF10C, Supplementary Table 2) were associated with gene ontology terms ‘cell death’, ‘cell surface receptor linked signal transduction’, ‘defense’, ‘immune response’ and ‘transcription’.
Discussion
This is the first study to analyze the genome-wide methylation profiles of MZA twins. From the correlation plots and ICC values, methylation data for both twin pairs seemed to correlate almost perfectly, in agreement with a previous study of monozygotic twins reared together [Citation28,Citation29]. This is not surprising as a large number of probes do largely increase the magnitude and stability of the correlation [Citation30]. From the MDS plots, genetic factors appear to be a greater influence on DNA methylation than the rearing environment. This is also not surprising as previous findings reported that region-specific methylation profiles of MZ twins are more similar compared with the methylation profiles of DZ twins (reviewed by Bell and Spector [Citation31]). An analysis of regions variably methylated in unrelated individuals estimated a 25% contribution of genotype, no contributions from environment only and a 75% contribution from interactions between genetic and environment factors [Citation32]. Coupled with a recent report from Gaunt et al. [Citation33] that showed a strong genetic influence on DNA methylation across the lifespan, these findings highlight the importance of genetic components on DNA methylation [Citation34,Citation35].
In our analyses, G1E1 was epigenetically different from all other individuals including his MZA twin. One possible reason for this is that the intrauterine environment differed between G1E1 and G1E2, although we have no way of testing this. However, we know from our previous studies that nonshared (twin-specific) intrauterine environmental events have a greater effect on DNA methylation in multiple tissues at birth than genetics and shared intrauterine environment [Citation3,Citation36,Citation37].
The evidence we generated for the effect of shared rearing environment on DNA methylation came from PC3, which divided individuals according to the postnatal environment. Furthermore, we showed that the genes that were most highly differentially methylated within both pairs reared in different environments were enriched within ontologies related to apoptosis and immune function, which have previously been linked to socioeconomic status [Citation38,Citation39]. Differential expression of these gene ontologies has also been linked to exposure to UV radiation [Citation40] and pesticides [Citation41], which will have differed greatly between the two environments in which the twins were raised (city – G1E1 and G2E1 vs country – G2E2 and G1E2). Pesticide exposure has also been found be linked with neurodegenerative disorders (reviewed in [Citation42]). Furthermore, epigenetic changes in genes linked to inflammation have been linked with later life chronic conditions such as cardiovascular [Citation43] and neurobiological diseases [Citation44], warranting a further investigation into these links in longitudinal studies.
MZA twins are an ideal model for investigating the influence of the rearing environment on epigenetics because they remove genetics as a confounding factor. The large differences in the rearing environment (city vs country) for twins in this report have facilitated our analysis. The major limitation of this study is the small sample size (2 MZA twin pairs), which made adjustment of cell heterogeneity and natural variability unfeasible. Cellular heterogeneity confounds many epigenetic studies, but in this study we stringently controlled for this by removing regions of the genome that were in any way associated with differences in DNA methylation between buccal and potentially contaminating blood cells. However, the consequence of this stringency is the low number of remaining probes, which is a limitation in comparison to other genome-wide methylation studies. To avoid reporting findings that were due to natural variability and not environment, we did not narrow our findings to just one or a few DMPs, but we looked at ontologies that were associated with our top 1000 DMPs. We also compared our data with a study of adolescent MZ twin pairs [Citation28], which reported that probes with the highest within-pair differences in DNA methylation were enriched in gene ontologies related to development and cell growth. The lack of overlap between our two studies, in gene ontologies associated with within-pair differences in DNA methylation, reduces the likelihood that our findings were due to general within-pair variability and supports the idea that we are looking at true effects of rearing environment. Another limitation of our study is the lack of buccal samples taken soon after birth, which would test our hypothesis about the effects of the intrauterine environment in both pairs. This emphasizes the importance of collecting birth samples for epigenetic studies.
Conclusion
Both genetics and intrauterine environment appeared to have more of an influence than rearing environment on DNA methylation in early adulthood, as assessed in two pairs of MZA twins reared in extremely different environments. Nevertheless, our findings, albeit from a case study, suggest that prenatal and postnatal environments contribute to the epigenetic profile in different ways. However, we are currently unable to conclude whether these results extend to other MZA twin pairs and to singletons.
Table 1. Between individual intraclass correlation coefficients analysis of twins across 150,678 probes.
Table 2. REVIGO terms with log10p value less than -1.3 and associated GO terms.
Monozygotic twins reared apart (MZA) are good models for studying the influence of pre- and postnatal environments on epigenetics, while controlling for shared genetics.
Genetic and intrauterine environmental factors appeared to have a stronger influence on DNA methylation than rearing environment, with the latter being very different for the two twin pairs.
The largest effects of the rearing environment on DNA methylation within our two MZA pairs involve genes relevant to immune response and cell death.
Author contributions
N Segal had the idea for the study, gathered the saliva samples from the five participants and forwarded them to the lab of JM Craig and YJ Loke. Y Montoya assisted in the collection of the saliva samples. YJ Loke and JM Craig performed the analyses and data interpretations. All authors contributed written sections of the manuscript based on their areas of expertise. All authors read and approved the final manuscript.
Ethics approval and participant consent
This project was approved by the IRB of California State University, Fullerton, where the first author is a faculty member. The five human subjects all signed consent forms prior to this research, as required by the university. As indicated above, the five participants in this case study signed consent forms agreeing to the research. The consent form was approved in advance by the IRB at CSU Fullerton.
Supplemental Figure
Download MS Power Point (518.7 KB)Supplemental tables
Download MS Word (96 KB)Supplemental information
Download MS Excel (841 KB)Acknowledgements
The authors wish to thank the five individuals in this case report for their contributions and JH Eric Joo from the Department of Pathology, University of Melbourne, for generating Infinium array data.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: http://www.tandfonline.com/doi/full/10.2217/nnm-2016-0104
Financial & competing interests disclosure
This study was supported by an intramural research grant and an Open Access Publication Fund Grant to the first author (CSU Fullerton), a book advance to the first two authors (St. Martin’s Press, NY, USA) and by the Victorian State Government’s Operational Infrastructure Support Program (YJ Loke and JM Craig). The funding bodies had no role in the study design, analysis, data interpretation or writing of this manuscript. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Schulz AC , StressigR , RitgenJet al. A classic twin study of isolated gastroschisis . Fetal Pediatr. Pathol.31 ( 5 ), 324 – 330 ( 2012 ).
- Segal NL . Born Together-Reared Apart: The Landmark Minnesota Twin Study . Harvard University Press , MA, USA ( 2012 ).
- Gordon L , JooJE , PowellJEet al. Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence . Genome Res.22 ( 8 ), 1395 – 1406 ( 2012 ).
- Ollikainen M , SmithKR , JooEJet al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome . Hum. Mol. Genet.19 ( 21 ), 4176 – 4188 ( 2010 ).
- Liu F , SunQ , WangL , NieS , LiJ . Bioinformatics analysis of abnormal DNA methylation in muscle samples from monozygotic twins discordant for type 2 diabetes . Mol. Med. Rep.12 ( 1 ), 351 – 356 ( 2015 ).
- Craig JM . Epigenetics in twin studies . Medical Epigenetics1 ( 1 ), 78 – 87 ( 2013 ).
- Ballestar E . Epigenetics lessons from twins: prospects for autoimmune disease . Clin. Rev. Allergy Immunol.39 ( 1 ), 30 – 41 ( 2010 ).
- Oh G , EbrahimiS , WangSCet al. Epigenetic assimilation in the aging human brain . Genome Biol.17 ( 1 ), 76 ( 2016 ).
- Teschendorff AE , WestJ , BeckS . Age-associated epigenetic drift: implications, and a case of epigenetic thrift?Hum. Mol. Genet.22 ( R1 ), R7 – R15 ( 2013 ).
- Issa JP . Aging and epigenetic drift: a vicious cycle . J. Clin. Invest.124 ( 1 ), 24 – 29 ( 2014 ).
- Martino D , LokeYJ , GordonLet al. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance . Genome Biol.14 ( 5 ), R42 ( 2013 ).
- Fraga MF , BallestarE , PazMFet al. Epigenetic differences arise during the lifetime of monozygotic twins . Proc. Natl Acad. Sci. USA102 ( 30 ), 10604 – 10609 ( 2005 ).
- Bouchard TJ Jr , LykkenDT , McgueM , SegalNL , TellegenA . Sources of human psychological differences: the Minnesota Study of Twins Reared Apart . Science250 ( 4978 ), 223 – 228 ( 1990 ).
- Segal NL , McGuireSA , StohsJH . What virtual twins reveal about general intelligence and other behaviors . Pers. Individ. Dif.53 ( 4 ), 405 – 410 ( 2012 ).
- Bibikova M , BarnesB , TsanCet al. High density DNA methylation array with single CpG site resolution . Genomics98 ( 4 ), 288 – 295 ( 2011 ).
- Aryee MJ , JaffeAE , Corrada-BravoHet al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays . Bioinformatics30 ( 10 ), 1363 – 1369 ( 2014 ).
- Martino D , SafferyR . Characteristics of DNA methylation and gene expression in regulatory features on the Infinium 450k Beadchip . bioRxiv doi:http://dx.doi.org/10.1101/032862 ( 2015 ) ( Epub ahead of print ).
- Touleimat N , TostJ . Complete pipeline for Infinium((R)) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation . Epigenomics4 ( 3 ), 325 – 341 ( 2012 ).
- Chen YA , LemireM , ChoufaniSet al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray . Epigenetics8 ( 2 ), 203 – 209 ( 2013 ).
- Lowe R , GemmaC , BeyanHet al. Buccals are likely to be a more informative surrogate tissue than blood for epigenome-wide association studies . Epigenetics8 ( 4 ), 445 – 454 ( 2013 ).
- Pidsley R , ChloeCYW , VoltaM , LunnonK , MillJ , SchalkwykLC . A data-driven approach to preprocessing Illumina 450K methylation array data . BMC Genomics14 , 293 ( 2013 ).
- Zidan M , ThomasRL , SlovisTL . What you need to know about statistics, part II: reliability of diagnostic and screening tests . Pediatr. Radiol.45 ( 3 ), 317 – 328 ( 2015 ).
- Kaminsky ZA , TangT , WangSCet al. DNA methylation profiles in monozygotic and dizygotic twins . Nat. Genet.41 ( 2 ), 240 – 245 ( 2009 ).
- Huang Da W , ShermanBT , LempickiRA . Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists . Nucleic Acids Res.37 ( 1 ), 1 – 13 ( 2009 ).
- Huang DW , ShermanBT , TanQet al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists . Nucleic Acids Res.35 ( Suppl. 2 ), W169 – W175 ( 2007 ).
- Supek F , BosnjakM , SkuncaN , SmucT . REVIGO summarizes and visualizes long lists of gene ontology terms . PLoS ONE6 ( 7 ), e21800 ( 2011 ).
- Mclean CY , BristorD , HillerMet al. GREAT improves functional interpretation of cis-regulatory regions . Nat. Biotechnol.28 ( 5 ), 495 – 501 ( 2010 ).
- Levesque ML , CaseyKF , SzyfMet al. Genome-wide DNA methylation variability in adolescent monozygotic twins followed since birth . Epigenetics9 ( 10 ), 1410 – 1421 ( 2014 ).
- Van Dongen J , EhliEA , SliekerRCet al. Epigenetic variation in monozygotic twins: a genome-wide analysis of DNA methylation in buccal cells . Genes (Basel)5 ( 2 ), 347 – 365 ( 2014 ).
- Fan J , HanF , LiuH . Challenges of big data analysis . Natl Sci. Rev.1 ( 2 ), 293 – 314 ( 2014 ).
- Bell JT , SpectorTD . DNA methylation studies using twins: what are they telling us?Genome Biol.13 ( 10 ), 172 ( 2012 ).
- Teh AL , PanH , ChenLet al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes . Genome Res.24 ( 7 ), 1064 – 1074 ( 2014 ).
- Gaunt TR , ShihabHA , HemaniGet al. Systematic identification of genetic influences on methylation across the human life course . Genome Biol.17 ( 1 ), 61 ( 2016 ).
- Mcrae AF , PowellJE , HendersAKet al. Contribution of genetic variation to transgenerational inheritance of DNA methylation . Genome Biol.15 ( 5 ), R73 ( 2014 ).
- Busche S , ShaoX , CaronMet al. Population whole-genome bisulfite sequencing across two tissues highlights the environment as the principal source of human methylome variation . Genome Biol.16 ( 1 ), 290 ( 2015 ).
- Loke YJ , GalatiJC , MorleyRet al. Association of maternal and nutrient supply line factors with DNA methylation at the imprinted IGF2/H19 locus in multiple tissues of newborn twins . Epigenetics8 ( 10 ), 1069 – 1079 ( 2013 ).
- Loke YJ , GalatiJC , SafferyR , CraigJM . Association of in vitro fertilization with global and IGF2/H19 methylation variation in newborn twins . J. Dev. Orig. Health Dis.6 ( 2 ), 115 – 124 ( 2015 ).
- Stringhini S , PolidoroS , SacerdoteCet al. Life-course socioeconomic status and DNA methylation of genes regulating inflammation . Int. J. Epidemiol.44 ( 4 ), 1320 – 1330 ( 2015 ).
- Needham BL , SmithJA , ZhaoWet al. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: the multi-ethnic study of atherosclerosis . Epigenetics10 ( 10 ), 958 – 969 ( 2015 ).
- Misovic M , MilenkovicD , MartinovicT , CiricD , BumbasirevicV , Kravic-StevovicT . Short-term exposure to UV-A, UV-B, and UV-C irradiation induces alteration in cytoskeleton and autophagy in human keratinocytes . Ultrastruct. Pathol.37 ( 4 ), 241 – 248 ( 2013 ).
- Benedetti D , NunesE , SarmentoMet al. Genetic damage in soybean workers exposed to pesticides: evaluation with the comet and buccal micronucleus cytome assays . Mutat. Res.752 ( 1–2 ), 28 – 33 ( 2013 ).
- Maloney B , LahiriDK . Epigenetics of dementia: understanding the disease as a transformation rather than a state . Lancet Neurol.15 ( 7 ), 760 – 774 ( 2016 ).
- Pasquier J , Hoarau-VechotJ , FakhroK , RafiiA , Abi KhalilC . Epigenetics and cardiovascular disease in diabetes . Curr. Diab. Rep.15 ( 12 ), 108 ( 2015 ).
- Lahiri DK , MaloneyB , ZawiaNH . The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases . Mol. Psychiatry14 ( 11 ), 992 – 1003 ( 2009 ).