
Abstract
Recent advances in chromosome conformation capture technologies are improving the current appreciation of how 3D genome architecture affects its function in different cell types and disease. Long-range chromatin interactions are organized into topologically associated domains, which are known to play a role in constraining gene expression patterns. However, in cancer cells there are alterations in the 3D genome structure, which impacts on gene regulation. Disruption of topologically associated domains architecture can result in alterations in chromatin interactions that bring new regulatory elements and genes together, leading to altered expression of oncogenes and tumor suppressor genes. Here, we discuss the impact of genetic and epigenetic changes in cancer and how this affects the spatial organization of chromatin. Understanding how disruptions to the 3D architecture contribute to the cancer genome will provide novel insights into the principles of epigenetic gene regulation in cancer and mechanisms responsible for cancer associated mutations and rearrangements.
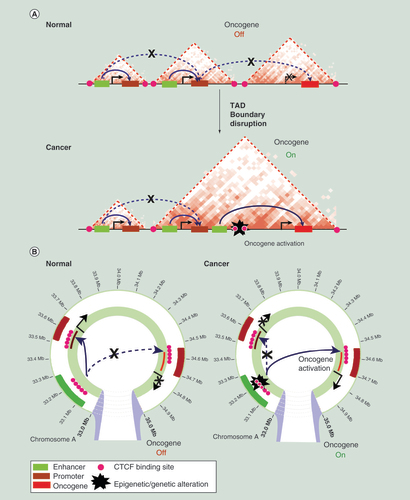
(A) A schematic representation of topologically associated domains (TADs) and enhancer–promoter interactions on a linear genome in normal and cancer cells is shown. In normal cells, enhancers regulate expression of genes only located within the same TAD. An example oncogene is shown that is not expressed due to the lack of enhancer interactions. The TAD boundary is disrupted in cancer cells, by genetic or epigenetic alterations at CTCF-binding sites, resulting in aberrant enhancer interactions within the new TAD and subsequent oncogene activation. (B) A schematic representation of enhancer–promoter interactions in normal and cancer cells. An example oncogene is shown that becomes activated in cancer cells due to disruption of enhancer–promoter interactions by genetic or epigenetic alterations at CTCF-binding sites.
Example of chromatin interactions visualized in the Rondo interactive analysis tool [Citation64]. Anchor points of differential interactions are visualized simultaneously with ChIP-seq (H3K27ac, H3K4me1, H3K4me3) and RefSeq genes inferring functionality (both active and repressive) of interactions. Hi-C data obtained from [Citation53] are presented at 100 kb resolution. Differential chromatin interactions in prostate cancer cells lymph node carcinoma of the prostate (LNCaP) and normal prostate cells primary prostate epithelial cells (PrEC) at an example region of long-range epigenetic activation and adjacent long-range epigenetic silencing are shown. Gray indicates shared interactions in normal and cancer cells. Blue indicates interactions that are lost in cancer. RNA-seq gene expression demonstrates that genes located at long-range epigenetic activation/long-range epigenetic silencing differential interactions have altered expression in cancer.
![Figure 2. Changes in long-range chromatin interactions in cancer results in aberrant long-range activation and repression of genes.Example of chromatin interactions visualized in the Rondo interactive analysis tool [Citation64]. Anchor points of differential interactions are visualized simultaneously with ChIP-seq (H3K27ac, H3K4me1, H3K4me3) and RefSeq genes inferring functionality (both active and repressive) of interactions. Hi-C data obtained from [Citation53] are presented at 100 kb resolution. Differential chromatin interactions in prostate cancer cells lymph node carcinoma of the prostate (LNCaP) and normal prostate cells primary prostate epithelial cells (PrEC) at an example region of long-range epigenetic activation and adjacent long-range epigenetic silencing are shown. Gray indicates shared interactions in normal and cancer cells. Blue indicates interactions that are lost in cancer. RNA-seq gene expression demonstrates that genes located at long-range epigenetic activation/long-range epigenetic silencing differential interactions have altered expression in cancer.](/cms/asset/52518754-09ee-4a8c-87b1-0180a67dff15/iepi_a_12325191_f0002.jpg)
First draft submitted: 1 September 2016; Accepted for publication: 24 October 2016; Published online: 12 December 2016
The 3D genome is a highly organized hierarchical structure
Cancer development and progression is accompanied by vast genetic and epigenetic alterations. The direct relationship between the cancer genome, epigenome and transcriptome however still remains to be defined. Architecture of the interphase genome is increasingly considered as a major player in the control of gene expression [Citation1]. Development of high-resolution chromatin conformation capture (3C) techniques has been instrumental in determining the principles of 3D nuclear organization. Studies using 3C (one loci to one loci) [Citation2], 4C-seq (one to all) [Citation3,Citation4] and 5C-seq (many to many) [Citation5] revealed that enhancers regulate their target genes by physically associating with their promoters in 3D via chromatin looping [Citation6,Citation7]. Subsequent genome-wide studies with Hi-C (high-throughput chromatin conformation capture sequencing; all to all) [Citation8] and ChIA-PET (chromatin interaction analysis by paired-end tag sequencing; all to all for candidate protein-bound DNA) [Citation9] showed that the eukaryotic genome is packaged into a highly organized hierarchical structure with three layers of organization. First, at the level of single genes, the genome is organized into local enhancer–promoter interactions. Second, the genome is organized into topologically associated domains (TADs) or contact domains that are hundreds of kilobases in size and encompass multiple genes and regulatory elements [Citation10,Citation11]. Finally at the higher level, intra- and inter-chromosomal interactions between TADs are organized into functionally distinct megabase compartments, which comprise large blocks of chromatin that are either A-type (open, gene rich) or B-type (closed, gene poor) [Citation8,Citation12]. TADs are characterized by preferential interactions within them and lack of interactions between different TADs, suggesting that TAD boundaries act as insulators by preventing communication between elements on either side of the boundary. TADs are conserved and reported to be largely invariant between different cell types [Citation10,Citation13], while the chromatin interactions or loops within TADs are more tissue specific [Citation10,Citation13–15]. Changes within TADs (sub-TADs) have also been reported to occur during cell differentiation [Citation13] and reprogramming [Citation16,Citation17] in embryonic stem cells. Together the data suggest that within the hierarchical genome structure there is a discreet level of 3D organization that displays more plasticity to give cell type specific expression.
Architectural protein CTCF is involved in 3D genome organization
Recent Hi-C studies revealed that architectural proteins play a critical role in determining the 3D organization of the genome. The insulator protein CCCTC-binding factor (CTCF) and SMC-family complex, cohesin, are the most well known architectural proteins that regulate genomic stability, expression and epigenetic homeostasis [Citation18]. CTCF is essential during development and has been long suggested to partition the genome into functional domains [Citation19]. CTCF binds frequently across the genome and a large proportion of the CTCF-binding sites are shared across tissues [Citation20]. Interestingly, CTCF-binding sites are commonly located at TAD boundaries and loop anchors [Citation10–11,Citation21].
Many studies have addressed the consequences of CTCF loss at these regions using knock-out or knock-down experiments in different cell models and provide evidence for the role of CTCF in organizing the genome into functional segments. A study of the mouse β-globin locus in erythroid cells showed that a targeted disruption of CTCF destabilizes long-range interactions around the locus and causes local loss of histone acetylation and gain of histone methylation, but no alterations were observed in expression of genes at the locus [Citation22]. The role of CTCF in domain boundary and loop formation was further confirmed by detailed genome editing studies using in vitro and in vivo models. CRISPR-mediated deletion of CTCF sites at domain boundary in embryonic stem cells altered the frequency of interactions and expression of genes within the domain [Citation23]. Similarly, it was shown that deletion of CTCF-binding sites within the HOX clusters results in the expansion of active chromatin into a repressive domain, supporting the role of CTCF in establishing and maintaining TAD boundaries [Citation24]. These studies serve to demonstrate the critical role of architectural proteins in partitioning the genome into functional domains and loop formation.
Alterations in CTCF-binding disrupt 3D genome organization of TADs
A recent high-resolution Hi-C study [Citation21] showed that CTCF sites at domain boundaries occur predominantly in a convergent orientation and that these sites frequently loop together to form domains. Further detailed functional analysis of the enhancer–promoter loop formation demonstrated that the orientation of CTCF sites restricts its choice of interacting partner and therefore predicts the 3D organization of the genome [Citation19,Citation25]. Using CRISPR-Cas9 genome editing Guo et al. inverted CTCF-binding sites at the topological domain boundary at the PCDH locus and showed that CTCF-binding direction is crucial for loop topology and gene expression [Citation25]. In another study, deletion of core CTCF-binding sites resulted in a loss of CTCF and cohesin recruitment to these sites and disruption of chromatin loops with distal, convergent CTCF sites [Citation26]. Finally, by manipulating 13 CTCF-binding sites at loop anchors, Sanborn et al. established that genome editing of CTCF motifs disrupts corresponding loops, consistent with the convergent rule [Citation27]. Together these important studies strongly suggest that genetic or epigenetic alteration of CTCF binding can lead to TAD disruption and therefore may result in aberrant cancer gene expression (A).
TAD organization plays an insulator role in gene regulation
Considering the importance of the topological organization of the genome, several studies have investigated the role of TADs in regulating gene expression by deleting, inverting or relocating TAD boundaries. Inversion around a TAD boundary between two domains, containing either the TFAP2C locus or BMP7 locus, resulted in relocation of the BMP7 enhancer into the TFAP2C domain and concomitant increase in TFAP2C expression and a decrease in BMP7 expression [Citation28]. In another study (Giorgio et al. [Citation29]), a 60 kb deletion encompassing a TAD boundary allowed for atypical interactions between lamin B1 gene promoter and enhancers that are normally located outside of lamin B1 domain. Using several hundred insertions of regulatory sensor in mouse embryos, Symmons et al. studied the distribution of gene regulatory activities along the mouse genome [Citation30]. The authors found that enhancers distribute their activities along broad genomic regions that correlate strongly with TADs, supporting the notion that TADs confine regulatory activities to specific regulatory domains, contributing to the establishment of specific gene expression profiles [Citation30].
Disease-associated structural variants located at TAD boundaries have also been shown to cause pathogenic rewiring of promoter–enhancer interactions leading to altered gene expression [Citation31]. Lupianez et al. showed that structural variants associated with limb malformation in humans alter the structure of WNT6/IHH/EPHA4/PAX3 locus located within a TAD [Citation31]. Genetic disruption of a CTCF-associated TAD boundary caused atypical interactions between promoters and enhancers resulting in aberrant expression of genes within that locus and different disease phenotypes. A recent study of genomic duplications showed that structural variants located at TAD boundaries can result in the formation of new chromatin domains (neo-TADs) and that this process determines their molecular pathology [Citation32]. Duplications within the mouse SOX9 TAD (intra-TAD) resulted in increased contact frequencies within that TAD, while inter-TAD duplications resulted in the formation of a novel TAD–neo-TAD. Incorporation of the adjacent KCNJ2 gene in the neo-TAD resulted in its consecutive misexpression via increased contacts with the duplicated part of the SOX9 regulatory region, leading to limb malformation phenotype [Citation32]. Together, these influential studies demonstrate the role of TADs and TAD boundaries in confining gene expression via restricting enhancer–promoter interactions and implicate a potential role that genetic disruption of TAD architecture may play in oncogene activation in tumorigenesis.
CTCF binding is altered in cancer
Both cohesin and CTCF-encoding genes are frequently mutated in cancer. Recent studies have shown that CTCF/cohesin-binding sites are often mutated in colorectal cancer [Citation33,Citation34] and accumulation of mutations was further confirmed in multiple different cancer subtypes [Citation34,Citation35]. CTCF-binding sites located at TAD boundaries are particularly enriched for somatic point mutations in many types of cancer [Citation35,Citation36], implicating that genetic alterations to a subset of CTCF-binding sites may affect aberrant cancer gene expression, and genomic instability. Additionally, CTCF binding can also be influenced by epigenetic modifications, including DNA methylation. CTCF occupancy is anticorrelated with DNA methylation of its binding sites in vivo [Citation37,Citation38] and its binding to DNA can be inhibited by increased methylation in vitro [Citation39]. A recent study by Maurano et al. showed that although DNA methylation is not a main regulator of CTCF binding genome-wide, a proportion of CTCF sites show methylation-dependent binding in vivo [Citation40]. Notably, a study of mutant gliomas demonstrated that treatment of cells with a demethylation agent reduces methylation at CTCF binding sites, allowing for CTCF binding and formation of a new TAD boundary, which in turn restores correct gene expression patterns in these cells [Citation41]. It is of interest to speculate that as DNA hypomethylation is a known hallmark of cancer, methylation-dependent changes to CTCF binding, may represent a novel mechanism of tumor development and progression through its genome-wide impact on 3D chromatin structure in many different types of cancer.
Alterations in chromatin interactions are present in cancer genomes
Alterations in local chromatin interactions have been a focus of many studies into long-range gene regulation and oncogene activation in cancer. Pomerantz et al. used 3C technology to evaluate the transcriptional landscape of 8q24 region, which harbors a cancer-associated risk variant, and demonstrated that the risk locus directly interacts with the MYC proto-oncogene in colon cancer [Citation42]. Subsequent study by Sotelo et al. showed that the 8q24 locus contains multiple enhancer elements that regulate MYC gene transcription via long-range interactions with the MYC promoter [Citation43]. This association was further confirmed in other types of cancer, including colorectal, prostate, breast and lung [Citation44,Citation45]. One of the more recent reports studying cancer-associated chromatin organization showed that overexpression of ERG, an oncogenic fusion protein, in normal prostate cells is associated with local changes in chromatin interactions [Citation46]. A study of the HOXA gene cluster in leukemia demonstrated that chromatin conformation data could be used to distinguish different subtypes of the disease. Rousseau et al. performed chromatin organization profiling of the HOXA cluster by 5C experiments in a panel of leukemia cell lines and used the data to develop a model that was able to accurately classify cell types based on the MLL-fusion status [Citation47].
By applying Capture-based Hi-C methods, multiple studies have shown examples of how local chromatin interactions can be disrupted by genetic variation in the cancer genome and result in aberrant gene expression. Studies in breast [Citation48], prostate [Citation49] and colorectal cancer [Citation50] have demonstrated that Capture Hi-C approach can be used to identify key long-range chromatin interactions associated with common GWAS cancer risk single nucleotide polymorphisms and to find their direct gene targets. Together, these studies suggest that genetic alterations, which are frequently observed in cancer cells, can cause a disruption of local chromatin interactions that drive altered expression of oncogenes and tumor suppressor genes (B). In the future Capture Hi-C could provide a valuable tool for the identification of novel drug targets and biomarkers.
TAD architecture can be disrupted in cancer
The extent of 3D chromosome alterations that occur in the cancer genome is still largely uncharted. This question is particularly pertinent as cancer development and progression is accompanied by vast genetic alterations, including copy number variation, mutations and translocations and epigenetic changes including differential DNA methylation and histone modifications. In a study of chromosomal rearrangements in AML cells, Groschel et al. [Citation51] have shown that an inversion on chromosome 3 disrupts two TADs and results in aberrant expression of genes (GATA2 and EVI1) located within these TADs, by repositioning the distal GATA2 enhancer to activate EVI1 and simultaneously silence the GATA2 gene [Citation51]. Similarly, a study of MYC translocations in myeloma demonstrated that TAD rearrangements might result in colocalization of oncogenes with super enhancers from developmentally important loci, resulting in increased expression of MYC and an aggressive disease phenotype [Citation52]. A recent comprehensive study in glioblastoma demonstrated that 3D changes in looping contribute to cancer progression by altering the local topology of enhancer–promoter interactions [Citation41]. Loss of CTCF-maintained insulation between TADs resulted in aberrant activation of genes, including the PDGFRA oncogene. Disruption of CTCF sites at the boundary, by genome editing, enabled atypical interactions between a constitutive enhancer and the PDGFRA oncogene leading to its upregulation. In a different study of chromatin organization in cancer, Hnisz et al. explored the role of CTCF/cohesin-mediated domains (insulated neighbourhoods) in regulating expression of oncogenes in T-cell acute lymphoblastic leukemia [Citation36]. The authors analyzed recurrent microdeletions frequently located at CTCF/cohesin-mediated TAD boundaries and found that perturbations of these boundaries resulted in activation of T-cell acute lymphoblastic leukemia proto-oncogenes in nonmalignant cells. Together these two studies of chromatin topology in cancer strongly suggest that disruption of TAD architecture may drive activation of oncogenes in cells (A).
Taberlay et al. was the first genome-wide study to show genetic and epigenetic disruption of TADs in cancer compared with normal cells, by directly comparing Hi-C data from normal prostate cells and two prostate cancer cell lines [Citation53]. In cancer cells, TADs were found to be smaller in size, more numerous and frequently located within the TAD-architecture of normal cells (sub-TADs). Notably, the formation of new TAD boundaries commonly occurred at regions of copy number variation, demonstrating that genomic alterations can impact on long-range chromatin interactions. Indeed, altered gene expression and long-range epigenetic reprogramming, known to occur in multiple cancers [Citation54–62], was associated with formation of new cancer-specific chromatin interactions. The ectopic interactions were frequently located at regulatory elements, especially enhancers and promoters (). Genome-wide analysis of chromatin interaction patterns in epithelial and breast cancer cells by Hi-C also show differences in overall interaction frequencies between two cell lines, especially in small, gene-rich chromosomes and in telomeric and subtelomeric regions of the genome [Citation63]. Barutcu et al. discovered that in cancer cells TADs were frequently ‘broken’ into multiple sub-TADs [Citation63]. Together, these studies provide detailed evidence of 3D chromatin organization disruption in cancer and provide a new insight into the functional basis of the cancer genome.
Future perspective
There is a growing interest into how the 3D architectural blueprint is altered in cancer. Importantly a key question is what is the role of genetic and epigenetic lesions in causing or being caused by a change in cancer 3D chromatin structure. Currently only a small number of studies have directly interrogated the role of chromatin interactions in cancer cells. In contrast many more studies have addressed the role of TAD architecture, architectural proteins or chromatin interactions in regulating gene expression. Recent work has shown that cancer cells maintain the overall structure of normal cells, namely the segmentation into TADs; however, discreet genetic changes in TAD boundaries and local interactions appear to be present. These studies provide compelling evidence that misregulation of the 3D genome architecture may indeed be a common hallmark for multiple types of cancer. Genome-wide 3D assays, however, are still technically challenging for most laboratories and are dependent on large numbers of homogenous populations of cells and great depth of sequencing coverage to gain the desired resolution of promoter/enhancer interactions. As tumors by nature are genetically and epigenetically heterogeneous, the ‘holy grail’ will be to eventually assess the 4D genome structure at the single cell level in clinical samples and follow how the 3D structure changes over time as the tumor progresses and gains more genetic and epigenetic alterations. In the mean time we predict that the development of novel chromatin conformation capture techniques for primary patient tumors will further clarify the role of localized differences in chromatin organization in tumor development. Future research into the 3D and 4D genome blueprint of different types of cancer cells will allow a novel spatial platform for the innovative development of cancer drug targets including epigenetic and gene therapies and genome editing based treatment strategies.
Recent developments in chromosome conformation capture techniques led to the identification of topologically associated domains (TADs) and long range chromatin interactions.
TADs play a critical role in gene regulation and disruption of TAD boundaries leads to altered gene expression.
Architectural protein CTCF is involved in formation of TADs and chromatin loops and can be disrupted by mutations or DNA methylation in cancer.
TAD organization in cancer cells is largely intact. Changes in TAD boundaries can however occur due to genetic variation in the cancer genome.
Alterations in long-range interactions in cancer lead to aberrant gene regulation.
Genome-editing technologies are used to study the pathogenicity of 3D chromatin organization disruption in cancer.
Future studies are required to further the current understanding of the role of 3D and 4D organization and the impact of the accumulation of genetic and epigenetic alterations in cancer.
Financial & competing interests disclosure
SJ Clark is supported by a National Health and Medical Research Council (NHMRC) Fellowship grant (#1088144). This work was partially supported by NHMRC Project Grants #1011447 and #1051757. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Dekker J , MirnyL . The 3D Genome as moderator of chromosomal communication . Cell164 ( 6 ), 1110 – 1121 ( 2016 ).
- Dekker J , RippeK , DekkerM , KlecknerN . Capturing chromosome conformation . Science295 ( 5558 ), 1306 – 1311 ( 2002 ).
- Zhao Z , TavoosidanaG , SjolinderMet al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions . Nat. Genet.38 ( 11 ), 1341 – 1347 ( 2006 ).
- Simonis M , KlousP , SplinterEet al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) . Nat. Genet.38 ( 11 ), 1348 – 1354 ( 2006 ).
- Dostie J , RichmondTA , ArnaoutRAet al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements . Genome Res.16 ( 10 ), 1299 – 1309 ( 2006 ).
- Gheldof N , SmithEM , TabuchiTMet al. Cell-type-specific long-range looping interactions identify distant regulatory elements of the CFTR gene . Nucleic Acids Res.38 ( 13 ), 4325 – 4336 ( 2010 ).
- Sanyal A , LajoieBR , JainG , DekkerJ . The long-range interaction landscape of gene promoters . Nature489 ( 7414 ), 109 – 113 ( 2012 ).
- Lieberman-Aiden E , Van BerkumNL , WilliamsLet al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome . Science326 ( 5950 ), 289 – 293 ( 2009 ).
- Fullwood MJ , LiuMH , PanYFet al. An oestrogen-receptor-alpha-bound human chromatin interactome . Nature462 ( 7269 ), 58 – 64 ( 2009 ).
- Dixon JR , SelvarajS , YueFet al. Topological domains in mammalian genomes identified by analysis of chromatin interactions . Nature485 ( 7398 ), 376 – 380 ( 2012 ).
- Nora EP , LajoieBR , SchulzEGet al. Spatial partitioning of the regulatory landscape of the X-inactivation centre . Nature485 ( 7398 ), 381 – 385 ( 2012 ).
- Zhang Y , MccordRP , HoYJet al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations . Cell148 ( 5 ), 908 – 921 ( 2012 ).
- Dixon JR , JungI , SelvarajSet al. Chromatin architecture reorganization during stem cell differentiation . Nature518 ( 7539 ), 331 – 336 ( 2015 ).
- Smith EM , LajoieBR , JainG , DekkerJ . Invariant TAD boundaries constrain cell-type-specific looping interactions between promoters and distal elements around the CFTR locus . Am. J. Hum. Genet.98 ( 1 ), 185 – 201 ( 2016 ).
- Phillips-Cremins JE , SauriaME , SanyalAet al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment . Cell153 ( 6 ), 1281 – 1295 ( 2013 ).
- Beagan JA , GilgenastTG , KimJet al. Local genome topology can exhibit an incompletely rewired 3D-folding state during somatic cell reprogramming . Cell Stem Cell18 ( 5 ), 611 – 624 ( 2016 ).
- Krijger PH , Di StefanoB , De WitEet al. Cell-of-origin-specific 3D genome structure acquired during somatic cell reprogramming . Cell Stem Cell18 ( 5 ), 597 – 610 ( 2016 ).
- Ong CT , CorcesVG . CTCF: an architectural protein bridging genome topology and function . Nat. Rev. Genet.15 ( 4 ), 234 – 246 ( 2014 ).
- Nichols MH , CorcesVG . A CTCF code for 3D genome architecture . Cell162 ( 4 ), 703 – 705 ( 2015 ).
- Li Y , HuangW , NiuL , UmbachDM , CovoS , LiL . Characterization of constitutive CTCF/cohesin loci: a possible role in establishing topological domains in mammalian genomes . BMC Genomics14 , 553 ( 2013 ).
- Rao SS , HuntleyMH , DurandNCet al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping . Cell159 ( 7 ), 1665 – 1680 ( 2014 ).
- Splinter E , HeathH , KoorenJet al. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus . Genes. Dev.20 ( 17 ), 2349 – 2354 ( 2006 ).
- Dowen JM , FanZP , HniszDet al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes . Cell159 ( 2 ), 374 – 387 ( 2014 ).
- Narendra V , RochaPP , AnDet al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation . Science347 ( 6225 ), 1017 – 1021 ( 2015 ).
- Guo Y , XuQ , CanzioDet al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function . Cell162 ( 4 ), 900 – 910 ( 2015 ).
- De Wit E , VosES , HolwerdaSJet al. CTCF binding polarity determines chromatin looping . Mol. Cell60 ( 4 ), 676 – 684 ( 2015 ).
- Sanborn AL , RaoSS , HuangSCet al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes . Proc. Natl Acad. Sci. USA112 ( 47 ), E6456 – 6465 ( 2015 ).
- Tsujimura T , KleinFA , LangenfeldK , GlaserJ , HuberW , SpitzF . A discrete transition zone organizes the topological and regulatory autonomy of the adjacent tfap2c and bmp7 genes . PLoS Genet.11 ( 1 ), e1004897 ( 2015 ).
- Giorgio E , RobyrD , SpielmannMet al. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD) . Hum. Mol. Gen.24 ( 11 ), 3143 – 3154 ( 2015 ).
- Symmons O , UsluVV , TsujimuraTet al. Functional and topological characteristics of mammalian regulatory domains . Genome Res.24 ( 3 ), 390 – 400 ( 2014 ).
- Lupianez DG , KraftK , HeinrichVet al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions . Cell161 ( 5 ), 1012 – 1025 ( 2015 ).
- Franke M , IbrahimDM , AndreyGet al. Formation of new chromatin domains determines pathogenicity of genomic duplications . Nature538 ( 7624 ), 265 – 269 ( 2016 ).
- Katainen R , DaveK , PitkanenEet al. CTCF/cohesin-binding sites are frequently mutated in cancer . Nat. Genet.47 ( 7 ), 818 – 821 ( 2015 ).
- Lawrence MS , StojanovP , MermelCHet al. Discovery and saturation analysis of cancer genes across 21 tumour types . Nature505 ( 7484 ), 495 – 501 ( 2014 ).
- Kaiser VB , TaylorMS , SempleCA . Mutational Biases Drive Elevated Rates of Substitution at Regulatory Sites across Cancer Types . PLoS Genet.12 ( 8 ), e1006207 ( 2016 ).
- Hnisz D , WeintraubAS , DayDSet al. Activation of proto-oncogenes by disruption of chromosome neighborhoods . Science351 ( 6280 ), 1454 – 1458 ( 2016 ).
- Phillips JE , CorcesVG . CTCF: master weaver of the genome . Cell137 ( 7 ), 1194 – 1211 ( 2009 ).
- Wang H , MauranoMT , QuHet al. Widespread plasticity in CTCF occupancy linked to DNA methylation . Genome Res.22 ( 9 ), 1680 – 1688 ( 2012 ).
- Filippova GN , FagerlieS , KlenovaEMet al. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes . Mol. Cell. Biol.16 ( 6 ), 2802 – 2813 ( 1996 ).
- Maurano MT , WangH , JohnSet al. Role of DNA methylation in modulating transcription factor occupancy . Cell Rep.12 ( 7 ), 1184 – 1195 ( 2015 ).
- Flavahan WA , DrierY , LiauBBet al. Insulator dysfunction and oncogene activation in IDH mutant gliomas . Nature529 ( 7584 ), 110 – 114 ( 2016 ).
- Pomerantz MM , AhmadiyehN , JiaLet al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer . Nat. Genet.41 ( 8 ), 882 – 884 ( 2009 ).
- Sotelo J , EspositoD , DuhagonMAet al. Long-range enhancers on 8q24 regulate c-Myc . Proc. Natl Acad. Sci. USA107 ( 7 ), 3001 – 3005 ( 2010 ).
- Wright JB , BrownSJ , ColeMD . Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells . Mol. Cell Biol.30 ( 6 ), 1411 – 1420 ( 2010 ).
- Ahmadiyeh N , PomerantzMM , GrisanzioCet al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC . Proc. Natl Acad. Sci. USA107 ( 21 ), 9742 – 9746 ( 2010 ).
- Rickman DS , SoongTD , MossBet al. Oncogene-mediated alterations in chromatin conformation . Proc. Natl Acad. Sci. USA109 ( 23 ), 9083 – 9088 ( 2012 ).
- Rousseau M , FerraiuoloMA , CrutchleyJLet al. Classifying leukemia types with chromatin conformation data . Genome Biol.15 ( 4 ), R60 ( 2014 ).
- Dryden NH , BroomeLR , DudbridgeFet al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C . Genome Res.24 ( 11 ), 1854 – 1868 ( 2014 ).
- Du M , TillmansL , GaoJet al. Chromatin interactions and candidate genes at ten prostate cancer risk loci . Sci. Rep.6 , 23202 ( 2016 ).
- Jager R , MiglioriniG , HenrionMet al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci . Nat. Commun.6 , 6178 ( 2015 ).
- Groschel S , SandersMA , HoogenboezemRet al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia . Cell157 ( 2 ), 369 – 381 ( 2014 ).
- Walker BA , WardellCP , BrioliAet al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients . Blood Cancer J.4 , e191 ( 2014 ).
- Taberlay PC , Achinger-KaweckaJ , LunATet al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations . Genome Res.26 ( 6 ), 719 – 731 ( 2016 ).
- Frigola J , SongJ , StirzakerC , HinshelwoodRA , PeinadoMA , ClarkSJ . Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band . Nat. Genet.38 ( 5 ), 540 – 549 ( 2006 ).
- Stransky N , VallotC , ReyalFet al. Regional copy number-independent deregulation of transcription in cancer . Nat. Genet.38 ( 12 ), 1386 – 1396 ( 2006 ).
- Hitchins MP , LinVA , BuckleAet al. Epigenetic inactivation of a cluster of genes flanking MLH1 in microsatellite-unstable colorectal cancer . Cancer Res.67 ( 19 ), 9107 – 9116 ( 2007 ).
- Novak P , JensenT , OshiroMM , WattsGS , KimCJ , FutscherBW . Agglomerative epigenetic aberrations are a common event in human breast cancer . Cancer Res.68 ( 20 ), 8616 – 8625 ( 2008 ).
- Seng TJ , CurreyN , CooperWAet al. DLEC1 and MLH1 promoter methylation are associated with poor prognosis in non-small cell lung carcinoma . Br. J. Cancer99 ( 2 ), 375 – 382 ( 2008 ).
- Dallosso AR , HancockAL , SzemesMet al. Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms’ tumor . PLoS Genet.5 ( 11 ), e1000745 ( 2009 ).
- Rafique S , ThomasJS , SproulD , BickmoreWA . Estrogen-induced chromatin decondensation and nuclear re-organization linked to regional epigenetic regulation in breast cancer . Genome Biol.16 , 145 ( 2015 ).
- Bert SA , RobinsonMD , StrbenacDet al. Regional activation of the cancer genome by long-range epigenetic remodeling . Cancer Cell23 ( 1 ), 9 – 22 ( 2013 ).
- Coolen MW , StirzakerC , SongJZet al. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity . Nat. Cell Biol.12 ( 3 ), 235 – 246 ( 2010 ).
- Barutcu AR , LajoieBR , MccordRPet al. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells . Genome Biol.16 , 214 ( 2015 ).
- Rondo: Visual Analytics of Chromosome Structure . www.rondo.ws .