
Abstract
An important line of postgenomic research seeks to understand how genetic factors can influence epigenetic patterning. Here we review epigenetic effects of chromosomal aneuploidies, focusing on findings in Down syndrome (DS, trisomy 21). Recent work in human DS and mouse models has shown that the extra chromosome 21 acts in trans to produce epigenetic changes, including differential CpG methylation (DS-DM), in specific sets of downstream target genes, mostly on other chromosomes. Mechanistic hypotheses emerging from these data include roles of chromosome 21-linked methylation pathway genes (DNMT3L and others) and transcription factor genes (RUNX1, OLIG2, GABPA, ERG and ETS2) in shaping the patterns of DS-DM. The findings may have broader implications for trans-acting epigenetic effects of chromosomal and subchromosomal aneuploidies in other human developmental and neuropsychiatric disorders, and in cancers.
Diagram of chromosome 21 (Hsa21), which is trisomic in Down syndrome. Effector genes on the triplicated Hsa21 act on downstream target genes, mostly on other chromosomes, both by acute transcriptional effects and via epigenetic effects, including alterations in DNA methylation that can propagate to daughter cells in growing and self-renewing tissues, to produce biological phenotypes.
DS-DM: Differential CpG methylation.
(A) Heatmap showing reproducibility of DS-DM loci in whole blood from the study by Bacalini et al. [Citation57] and T cells from our study [Citation59]. Raw data from each dataset were processed separately using the same pipeline: after normalization by β-mixture quantile normalization using the ChAMP Bioconductor package, probes with more than 20% of missing values or detection p-value > 0.05, probes mapping to the XY chromosomes and probes querying CpGs that overlap with common single-nucleotide polymorphisms (dbSNP138, minor allele frequency >1%) were filtered out. Raw data were downloaded from NCBI-GEO (GSE74486 for T cells and GSE52588 for whole-blood data). Differentially methylated CpGs were defined for all datasets as nominal p-value <0.005, absolute methylation difference >0.15. In our meta-analysis, we only considered strong DS-DM CpGs, with at least two DS-DM CpGs in 500 bp windows. We identified 1083 DS-DM CpGs in PBL and 1300 in T cells, with 21% overlap. Although all enrichment analyses were done using these entire DS-DM CpG sets, for clarity, the heatmap only shows the top ranked DS-DM CpGs (based on p-values), all with an absolute methylation difference >0.2. There are more hypermethylated loci (red color in DS) than hypomethylated loci (blue color in DS) in the whole blood cells and T cells, although a greater bias toward hypermethylation is found in brain tissues [Citation59]. (B) Scatter plots showing strong DS-DM in RUNX1, SH3BP2, TMEM131 and ZDHHC14 in both whole blood and T cells. For each locus, fractional methylation values for the strongest DS-DM CpG are shown. The horizontal lines represent the average fractional methylation. T-test p-values for DM in DS versus controls are indicated. (C) Map and Bis-Seq validations of DS-DM in ZDHHC14. The bis-seq amplicons cover the DS-DM CpGs identified by Illumina 450K BeadChip array and flanking regions. The data reveal a discrete DS-DM region (amplicons 2 & 3) spanning 600 bp and overlapping an enhancer region marked by H3K4me1, immediately adjacent to a CTCF-bound insulator element.
C: Control; DS: Down syndrome; DS-DM: Differential CpG methylation; PBL: Peripheral blood leukocyte.
![Figure 2. Altered DNA methylation in whole blood cells and T lymphocytes with Ts21: consistency in independent studies and examples of biologically relevant genes with differential CpG methylation. (A) Heatmap showing reproducibility of DS-DM loci in whole blood from the study by Bacalini et al. [Citation57] and T cells from our study [Citation59]. Raw data from each dataset were processed separately using the same pipeline: after normalization by β-mixture quantile normalization using the ChAMP Bioconductor package, probes with more than 20% of missing values or detection p-value > 0.05, probes mapping to the XY chromosomes and probes querying CpGs that overlap with common single-nucleotide polymorphisms (dbSNP138, minor allele frequency >1%) were filtered out. Raw data were downloaded from NCBI-GEO (GSE74486 for T cells and GSE52588 for whole-blood data). Differentially methylated CpGs were defined for all datasets as nominal p-value <0.005, absolute methylation difference >0.15. In our meta-analysis, we only considered strong DS-DM CpGs, with at least two DS-DM CpGs in 500 bp windows. We identified 1083 DS-DM CpGs in PBL and 1300 in T cells, with 21% overlap. Although all enrichment analyses were done using these entire DS-DM CpG sets, for clarity, the heatmap only shows the top ranked DS-DM CpGs (based on p-values), all with an absolute methylation difference >0.2. There are more hypermethylated loci (red color in DS) than hypomethylated loci (blue color in DS) in the whole blood cells and T cells, although a greater bias toward hypermethylation is found in brain tissues [Citation59]. (B) Scatter plots showing strong DS-DM in RUNX1, SH3BP2, TMEM131 and ZDHHC14 in both whole blood and T cells. For each locus, fractional methylation values for the strongest DS-DM CpG are shown. The horizontal lines represent the average fractional methylation. T-test p-values for DM in DS versus controls are indicated. (C) Map and Bis-Seq validations of DS-DM in ZDHHC14. The bis-seq amplicons cover the DS-DM CpGs identified by Illumina 450K BeadChip array and flanking regions. The data reveal a discrete DS-DM region (amplicons 2 & 3) spanning 600 bp and overlapping an enhancer region marked by H3K4me1, immediately adjacent to a CTCF-bound insulator element.C: Control; DS: Down syndrome; DS-DM: Differential CpG methylation; PBL: Peripheral blood leukocyte.](/cms/asset/25ba520f-0144-4ebe-83be-42b24ff8decf/iepi_a_12325220_f0002.jpg)
(A) Bar graph showing enrichment in TF motifs among the whole blood hypomethylated DS-DM CpGs. Coordinates of occurrences of 90 motifs were downloaded from ENCODE to identify TF motifs within 100 bp of each CpG queried by the 450K BeadChips. Unlike in where only the top-ranked DM-CpGs are shown, we assessed enrichment using logistic regressions on all DS-DM CpGs in clusters of at least two DS-DM CpGs within 500 bp passing nominal p-value < 0.005 and fractional methylation differences <0.15. Only significant enrichment (nominal p-value < 0.05) of TF motifs with more than five occurrences in the DS-DM regions are shown. (B) Bar graph showing enrichment in TF motifs among the T-cell hypomethylated DS-DM CpGs. Methods as in (A). (C) List of pan and multitissue DS-DM genes. Raw data from five public datasets were processed separately using the same pipeline and criteria as described in . Raw data were downloaded from NCBI-GEO (GSE74486, GSE63347, GSE73747, GSE52588 and GSE66210). For each study, only tissues with at least three control and three DS samples were assessed. DM CpGs were defined for all datasets as CpGs in clusters of at least two DS-DM CpGs within 500 bp passing nominal p-value < 0.005 and methylation difference < 0.15. We merged the DM sets into five tissue sets: the brain datasets include samples from adult frontal cortex (FC) and cerebellum; fetal brain datasets samples from fetal cerebrum, FC and temporal cortex (TC); epithelial dataset samples from buccal epithelial cells; and blood dataset samples from T cells and PBL. Hyper and hypomethylated CpG sets from each tissue were overlapped using 1 kb windows, since differential methylation at adjacent CpGs often reflects methylation changes in the same regulatory element. Multitissue DS-DM CpGs were defined as CpGs with DS-DM within 500 bp present in at least three different tissues. Overall, we found 157 multitissue DS-DM CpGs, located in two genes (8 CpGs) with pan-tissue hypermethylation, 22 genes (141 CpGs) with multitissue hypermethylation and one gene (8 CpGs) with multitissue hypomethylation. (D) Bar graph showing enrichment in TF motifs among the pan or multitissues hypermethylated DS-DM CpGs. Methods were as in (A).
DS-DM: Differential CpG methylation; PBL: Peripheral blood leukocyte; TF: Transcription factor.

(A) The Hsa21 syntenic regions triplicated in major mouse models of DS are shown, which can serve as models for dissecting the mechanisms and biological consequences of DS-DM. The indicated regions of Hsa21 are syntenically conserved with three genomic regions separately located on mouse chromosome 10 (Mmu10), Mmu16 and Mmu17. Dp(16)1Yey and Ts65Dn mice carry more triplicated Hsa21 gene orthologs than any other triplication mouse models of DS. The genomic locations of methylation pathway genes and transcription factor genes, and their presence or absence in each of these models, are indicated. (B) Enrichment of Hsa21-orthologous TF gene consensus-binding motifs among Ts65Dn DS-DM CpGs. Enrichment analysis was performed using HOMER software applied to whole-genome bisulfite sequencing (WGBS) data from cerebral gray matter of Ts65Dn versus wt littermate mice [do c et al. 2016, manuscript in preparation]. DS-DM CpGs were defined as in [Citation59]. As discussed in the main text, the WGBS data from these mouse brains showed a significant excess of hypermethylated DM regions, but revealed a somewhat larger percentage of hypomethylated loci than were seen in the 450K Beadchip data from human brains. Strong enrichment is observed in sites recognized by TFs encoded by genes in the Mmu16 duplicated regions (bold font). Enrichment among hypomethylated DS-DM is suggestive of passive demethylation due to the TF occupancy, while enrichment among hypermethylated DS-DM is suggestive of active methylation by recruitment of DNMTs, as described in other settings [Citation74]. Significant, though weaker, enrichment in the E26 transformation-specific family of transcription factor (ETS) motif was also observed in our prior WGBS data [Citation59] from whole cerebrums of Dp(16) (p-value = 10-03) but not of Dp(10). Enrichment in transcription factor-binding sites (TFBSs) was defined as FDR <0.05 and fold-enrichment >1.2. The 10 top-ranked enriched TFBS sites are shown for hypermethylated and hypomethylated DS-DM CpGs. The finding of enrichment of three of these TFBS classes (all ETS family) among the hypermethylated DM loci and one (not ETS family) among the hypomethylated DM loci further supports a nonrandom mechanistic connection.
DS-DM: Differential CpG methylation; TF: Transcription factor.
![Figure 4. Mouse models for dissecting the mechanisms and biologicalconsequences of differential CpG methylation. (A) The Hsa21 syntenic regions triplicated in major mouse models of DS are shown, which can serve as models for dissecting the mechanisms and biological consequences of DS-DM. The indicated regions of Hsa21 are syntenically conserved with three genomic regions separately located on mouse chromosome 10 (Mmu10), Mmu16 and Mmu17. Dp(16)1Yey and Ts65Dn mice carry more triplicated Hsa21 gene orthologs than any other triplication mouse models of DS. The genomic locations of methylation pathway genes and transcription factor genes, and their presence or absence in each of these models, are indicated. (B) Enrichment of Hsa21-orthologous TF gene consensus-binding motifs among Ts65Dn DS-DM CpGs. Enrichment analysis was performed using HOMER software applied to whole-genome bisulfite sequencing (WGBS) data from cerebral gray matter of Ts65Dn versus wt littermate mice [do c et al. 2016, manuscript in preparation]. DS-DM CpGs were defined as in [Citation59]. As discussed in the main text, the WGBS data from these mouse brains showed a significant excess of hypermethylated DM regions, but revealed a somewhat larger percentage of hypomethylated loci than were seen in the 450K Beadchip data from human brains. Strong enrichment is observed in sites recognized by TFs encoded by genes in the Mmu16 duplicated regions (bold font). Enrichment among hypomethylated DS-DM is suggestive of passive demethylation due to the TF occupancy, while enrichment among hypermethylated DS-DM is suggestive of active methylation by recruitment of DNMTs, as described in other settings [Citation74]. Significant, though weaker, enrichment in the E26 transformation-specific family of transcription factor (ETS) motif was also observed in our prior WGBS data [Citation59] from whole cerebrums of Dp(16) (p-value = 10-03) but not of Dp(10). Enrichment in transcription factor-binding sites (TFBSs) was defined as FDR <0.05 and fold-enrichment >1.2. The 10 top-ranked enriched TFBS sites are shown for hypermethylated and hypomethylated DS-DM CpGs. The finding of enrichment of three of these TFBS classes (all ETS family) among the hypermethylated DM loci and one (not ETS family) among the hypomethylated DM loci further supports a nonrandom mechanistic connection.DS-DM: Differential CpG methylation; TF: Transcription factor.](/cms/asset/c45d1094-7b15-4fac-b7db-ac230f6545e5/iepi_a_12325220_f0004.jpg)
Map of the PCDHGA cluster. Differential CpG methylation in human DS (DS-DM) CpGs in the human 450K data and DM CpGs in the whole-genome bisulfite sequencing (WGBS) data from the mouse models were defined as in [Citation59]. WGBS for Dp(10)1Yey and Dp(16)1Yey was performed on whole cerebrums, while WGBS for Ts65Dn was performed on macrodissected cerebral gray matter. The Y-axes show fractional differences in CpG methylation. WGBS median read depth = 30 in all experiments; a total of 1074 CpGs were covered at >20 read depth in this chromosomal segment; only the CpGs passing our published criteria for significant differential methylation [Citation59] are indicated by the bars. DS-DM regions coincide with CTCF ChIP-seq peaks – consistent with the enrichment in CTCF motif sites that we and others have found among DS-DM loci. CTCF ChIP-seq data from human astrocytes and mouse whole brain were downloaded from ENCODE.
C: Control; DS: Down syndrome; WGBS: Whole-genome bisulfite sequencing; wt: Wild-type.
![Figure 5. Similarities between differential CpG methylation in human Down syndrome versus control brains and differential methylation in brains from the chromosomally engineered mouse models of Down syndrome illustrated by results in the PCDHGA2/Pcdhga2 gene clusters.Map of the PCDHGA cluster. Differential CpG methylation in human DS (DS-DM) CpGs in the human 450K data and DM CpGs in the whole-genome bisulfite sequencing (WGBS) data from the mouse models were defined as in [Citation59]. WGBS for Dp(10)1Yey and Dp(16)1Yey was performed on whole cerebrums, while WGBS for Ts65Dn was performed on macrodissected cerebral gray matter. The Y-axes show fractional differences in CpG methylation. WGBS median read depth = 30 in all experiments; a total of 1074 CpGs were covered at >20 read depth in this chromosomal segment; only the CpGs passing our published criteria for significant differential methylation [Citation59] are indicated by the bars. DS-DM regions coincide with CTCF ChIP-seq peaks – consistent with the enrichment in CTCF motif sites that we and others have found among DS-DM loci. CTCF ChIP-seq data from human astrocytes and mouse whole brain were downloaded from ENCODE.C: Control; DS: Down syndrome; WGBS: Whole-genome bisulfite sequencing; wt: Wild-type.](/cms/asset/05d0c1fa-950f-4db2-a226-6e8e779efe6f/iepi_a_12325220_f0005.jpg)
First draft submitted: 7 October 2016; Accepted for publication: 14 November 2016; Published online: 2 December 2016
Background
The topic of genetic–epigenetic interactions is attracting increasing research attention in our postgenomic era. Broadly speaking, effects of genetics on epigenetic patterning can be grouped into those that occur in cis, such as short-range effects of SNPs and haplotypes, reviewed elsewhere [Citation1,Citation2], and those that occur in trans, including the effects of chromosomal aneuploidies on CpG methylation and chromatin states, which we review here. Another important class of trans-acting genetic effects are due to point mutations in epigenetic ‘reader’ and ‘writer’ enzymes, which have been previously reviewed [Citation3,Citation4] and which we do not discuss here.
Research on chromosomal aneuploidies in a range of model organisms has a long and interesting history, which has been comprehensively reviewed by Amon and colleagues [Citation5]. As early as 1902, Theodor Boveri manipulated sea urchin embryos and found that only those that received the species-typical chromosomal complement developed into normal larva. He concluded that chromosome gain or loss leads to abnormal development and embryonic lethality. In experimental yeast models aneuploidies interfere with cell proliferation, and in mice all whole-chromosome aneuploidies are embryonic lethal at various stages of development, with cardiovascular, neurological and craniofacial defects as well as growth retardation commonly occurring as phenotypes. In humans, the best studied chromosomal aneuploidy is trisomy 21 (Ts21). This numerical chromosomal abnormality is compatible with viability in only a fraction of conceptuses, with a 30–40% rate of spontaneous fetal demise occurring in studies of pregnancy outcomes after diagnostic chorionic villus sampling at 9–14 weeks [Citation6,Citation7], and it leads to the phenotypic constellation of Down syndrome (DS) in those that survive to term. Since work on trans-acting genetic–epigenetic interactions in our laboratories has focused mostly on genetic and epigenetic effects of Ts21 in humans and mouse models, and since several other research groups working in parallel have contributed significantly to this area, some general principles are starting to emerge. We, therefore, devote most of our discussion to this topic. However, the findings have obvious potential relevance beyond Ts21, so we conclude our review by considering possible trans effects on epigenetic patterning in other human developmental and psychiatric disorders due to chromosomal or subchromosomal aneuploidies, and in cancers.
DS: phenotypic consequences of a simple chromosomal aneuploidy
It has been known for five decades that the etiology of DS is Ts21, but the pathogenesis of the syndrome, which includes intellectual disability, cardiac defects and a range of blood cell related phenotypes including increased autoimmunity and recurrent infections as well as predisposition to childhood acute leukemias, is still not fully understood [Citation8–10]. DS also confers resistance to the development of solid tumors such as neuroblastoma and breast cancer, which is being actively studied in humans and model systems but is still not fully explained at the genetic level [Citation11–13]. Later in life, adults with DS often develop an early onset form of Alzheimer’s disease (AD), with the increased susceptibility largely attributable to the extra copy of the amyloid precursor protein (APP) gene on human chromosome 21 (hereafter abbreviated as Hsa21), but with amyloid β-peptide levels and age-at-onset of AD influenced by modifier genes on various chromosomes [Citation14–17]. Similarities and differences in clinical and pathological features of AD in DS compared with this disease in the general population have recently been reviewed [Citation18]. Given these medically important components of the syndrome, a better understanding of DS pathogenesis will have multiple benefits: it can potentially guide therapies to benefit people with this disorder and their families; as an ‘experiment of nature’ DS can give insights into clinically significant phenotypes, including those that we have listed above, which are also prevalent in the general (non-DS) population; and research on DS can reveal basic principles of how cells respond to chromosomal aneuploidies – principles that may turn out to apply to other genetic disorders and to cancers.
Mouse models for dissecting the contributions of single genes & groups of genes in DS
As reviewed previously [Citation8,Citation19–21], much research effort has gone into attempts to identify genomic regions on Hsa21 that contain the critical genes associated with the above-mentioned DS phenotypes. Human genetic approaches have been used to analyze the phenotypes of rare individuals with segmental trisomies, and to complement such efforts, the mouse has been utilized as a model based on the evolutionary conservation between the regions on Hsa21 and three regions in the mouse genome, located on chromosome 10 (Mmu10), Mmu16 and Mmu17. Mouse models for identifying genes contributing to DS phenotypes have included YAC and BAC-transgenic (Tg) lines carrying single or multiple genes orthologous to those on Hsa21 ([Citation22,Citation23]; references therein), and several types of mice that carry large chromosomal duplications. The Ts65Dn mouse line, which was generated more than 20 years ago and is currently the most widely used model for DS, arose in progeny from a radiation mutagenesis experiment that caused unbalanced chromosomal translocations [Citation24]. The unbalanced derivative chromosome in Ts65Dn consists of a genomic fragment of approximately 13 Mb, extending from Mrpl39 to the telomere of Mmu16. Thus, approximately 49% of the syntenic regions and approximately 55% of the Hsa21 gene orthologs are triplicated in this model, but the Ts65Dn mice lack triplication of the Mmu10 and Mmu17 regions that contain the remainder of the Hsa21 orthologous genes. In addition, useful ‘transchromosomal’ mouse lines have been made that carry parts of human Chr21 [Citation25,Citation26]. Last, Cre-loxP-mediated chromosomal engineering has been utilized by the Yu lab to create mouse lines with precise duplications of each of the three regions of conserved synteny [Citation20,Citation27–29].
For evaluating the roles of single genes in DS phenotypes, the most definitive approach is to test for genetic rescue, in which a knockout allele is introgressed into these lines to normalize the dosage of a single gene and the animals are assessed for normalization of the phenotype of interest [Citation30,Citation31]. Based on experiments to date, none of the major DS phenotypes have been assigned to a single gene acting alone, but the combination of positive and negative findings from mouse models have been informative regarding the chromosomal regions and genes that do or do not contribute to specific DS phenotypes including cardiac defects [Citation27,Citation30,Citation32,33], hematological abnormalities [Citation34–36] and brain-related phenotypes including deficits in learning and memory ([Citation22,Citation29,Citation37]; references therein).
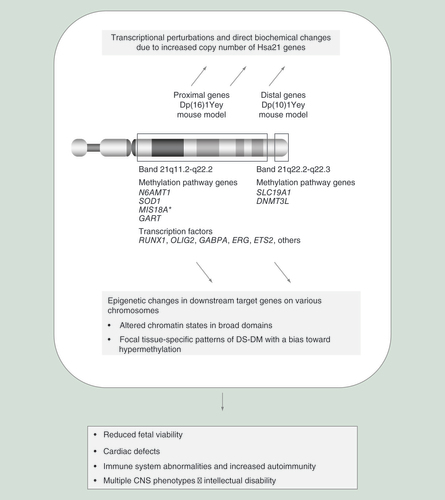
This important line of research comprises a genetic dissection of the roles of the effector genes on Hsa21 that cause DS when they are present in three copies. As diagrammed in , the products of these Hsa21-linked effector genes can exert their activities either directly via nongenomic effects or indirectly by controlling the expression of downstream target genes. Examples of direct effects are APP gene overexpression leading to toxic A-β peptide production in the brain [Citation38,Citation39] and overexpression of the gene encoding the DYRK1A kinase leading to abnormal protein phosphorylation events [Citation40]. Examples of indirect effects are overexpression of the ETS2 transcription factor gene leading to altered expression of ETS2-target genes [Citation41], and overexpression of the DNMT3L gene, coding for a methyltransferase component, potentially leading to DNA methylation events that can, in turn, affect gene expression (see next section). Nongenomic events such as altered signaling pathways can also feed back into the genome, for example, overexpression of the RCAN1 gene leads to altered Ca++/calcineurin signaling that in turn affects NFAT-mediated transcriptional programs in lymphocytes and calcium-dependent long-term potentiation in neurons [Citation22,Citation42].
Chromosomal aneuploidies & gene expression: linear & nonlinear effects
The most obvious hypothesis for DS pathogenesis is that it is accounted for by gene dosage effects leading to perturbations of transcriptional networks. Consistent with this idea, evidence from model organisms and humans has revealed a definite correlation between chromosomal copy numbers and gene expression. Yeast strains containing an extra chromosome show a corresponding increase in transcript levels, without evidence of dosage compensation, while diploid yeast strains lacking a chromosome show a matching decrease [Citation43,Citation44]. In microarray-based and RNA-Seq studies we and others have found that transcript levels of many of the genes encoded on chromosome 21 roughly parallel the increase in gene copy number in patients with DS, with the same dosage effect seen in mice with partial trisomies [Citation45–52]. However, an early study of gene expression in Ts21 versus control fibroblasts and fetal hearts found evidence for strong (i.e., nonlinear) overexpression of a small number of genes on Hsa21, while conversely some other Hsa21 genes lacked detectable increases in expression compared with the euploid control samples [Citation50]. Similar findings of a few genes that have >1.5× increased expression have been made in the other gene expression profiling studies of Ts21 referenced above. Nonlinear overexpression can in some cases be explained by gene–gene interactions when more than one gene in the same signaling or transcriptional pathway are both present on Hsa21. Examples are multiple chromosome 21-linked genes in the interferon pathway, which can explain the nonlinear overexpression of the interferon target gene MX1 [Citation50], and an interaction between the chromosome 21-linked ETS2 transcription factor and the APP gene promoter, leading to nonlinear overexpression of APP mRNA [Citation41]. Conversely, while not always featured in paper titles and abstracts, examination of the primary data typically shows that some genes on the trisomic chromosome are not differentially expressed, with the identities of these genes depending on the cell type. Thus, whether some genes on Hsa21 might be dosage compensated at the transcriptional level in specific cellular contexts remains an open question. Last but not least, all of these studies have shown that cells with Ts21 have many quantitative changes in the expression of genes on the other (nontrisomic) chromosomes, consistent with the chromosome 21 aneuploidy leading to perturbations of downstream transcriptional networks.
Chromosomal aneuploidies & epigenetic patterning: effects of Ts21 on CpG methylation & histone modifications
Going beyond simple transcriptional effects, an additional and more recent hypothesis for DS pathogenesis invokes epigenetics: the presence of the extra Hsa21 could act in trans to produce changes in DNA methylation and chromatin states, which can propagate to daughter cells and ‘lock in’ certain changes in gene expression in developing tissues (). Our initial work on this topic was motivated by the hypothesis that epigenetic changes might act on the extra copy of Hsa21 to produce a partial dosage compensation, analogous to the role of DNA methylation in X chromosome inactivation. Microarray-based DNA methylation profiling (Illumina 27K) in peripheral blood leukocytes (PBL) from individuals with DS and age-matched controls revealed gains and losses of DNA methylation, strongly affecting approximately 100 genes, as a consistent epigenetic response to Ts21 in these cells [Citation53]. However, the set of loci with differential methylation in DS versus controls (DS-DM) turned out not to be enriched for Hsa21 genes; rather, genes with DS-DM were found to be roughly evenly distributed on all of the chromosomes.
Since epigenetic patterning is often cell type-specific, methylation profiles of blood cells will not necessarily generalize to other organs. In fact, several labs took up this topic and examined other tissue types: genes with DS-DM in Ts21 placentas and fibroblasts overlap partially, but not extensively, with the set of DS-DM genes initially identified in blood cells, and when multiple labs analyzed blood cells they found DS-DM gene lists with substantial overlap [Citation54–62]. As shown in , the similarities between independent studies of DS versus control blood cells, from series of individuals seen in two different countries (USA and Italy), are quite striking and indicate that, in a given tissue, DS-DM is a highly recurrent and consistent response to the chromosomal trisomy.
Because moderate-to-severe intellectual disability is a highly penetrant phenotype in DS, attention quickly turned to profiling epigenetic changes in banked human autopsy brains from DS cases and controls [Citation59,Citation63,64]. Since the brain is composed of multiple cell types, steps to separate neurons from non-neuronal cells are expected to improve the yield and accuracy of molecular profiling. Therefore, in our recent work [Citation59] we purified neurons from a series of DS and control brains using the FANS procedure [Citation65], which entails NeuN staining of cell nuclei, followed by fluorescence-activated nuclear sorting. Data from Illumina 450K methylation Beadchips, with validations of DS-DM by bisulfite sequencing (Bis-Seq), revealed a gene-specific and cell-type-specific epigenetic response to Ts21 in neurons and glial cells, with substantial but only partial overlap of DS-DM loci between these cell types [Citation59]. Oxidative Bis-Seq for the ‘sixth base’, 5hydroxymethyl-C (5hmC), showed that changes in this epigenetic mark often, though not always, paralleled those in 5mC [Citation59].
In we show results of our meta-analysis combining multiple independent studies that have utilized Illumina 450K Beadchips for methylation profiling of Ts21 versus control samples and made their full data publicly available [Citation66]. The results confirm the tissue specificity of DS-DM, and also reveal a small but interesting set of genes with pan-tissue or multitissue DS-DM. This group of multitissue DS-DM loci may be special in some way – perhaps they are affected by the presence of the Hsa21 aneuploidy particularly early in development, hence propagating the DS-DM to multiple tissue types, and/or they may contain a specific type of DNA sequence motif that attracts DS-DM very strongly in all tissues. Potentially relevant to the latter possibility, as summarized in , the pan-tissue DS-DM loci are enriched in several types of sequence motifs, notably including binding sites for the insulator protein CTCF [Citation54,Citation59]. Since CTCF binds to a CpG-containing class of recognition sites in a methylation-sensitive manner, the patterns of CTCF binding, and thus presumably the 3D conformation of chromatin in the nucleus, may be particularly sensitive to epigenetic perturbations, including those due to chromosomal aneuploidies. However, the mechanisms by which Ts21 leads to altered methylation of CTCF sites remain to be determined.
While altered DNA methylation is not a mechanism for widespread dosage compensation on the triplicated Hsa21, a few genes on this chromosome do show DS-DM, an important example being RUNX1, which codes for a transcription factor that is essential for lymphocyte development. As shown in , gains of methylation in the RUNX1 promoter/enhancer region are seen both in DS versus control T cells [Citation59] and in DS versus control whole blood leukocytes [Citation57]. This CpG hypermethylation, which based on bis-seq data, is heterogeneous from cell to cell, may be acting as a type of dosage compensation at the single cell level (which would be interesting to test in future experiments), but nonetheless there is net overexpression of RUNX1 mRNA in total DS T cells. Importantly for mechanisms that we discuss in the next section, RUNX recognition motifs, associated with RUNX1 ChIP-Seq peaks in ENCODE data, are significantly enriched among hypomethylated DS-DM loci in DS T cells (). In the section dealing with mechanisms we consider a generalization of this situation, in which altered occupancy of various transcription factor-binding sites (TFBS) may play a role in shaping the cell-type-specific patterns of DS-DM. In this regard, purification of specific cell types can lead to more informative results. Similar to the situation in T cells, many RUNX1 binding sites show DS-DM in whole blood samples. However, in contrast to the findings in the purified T cells, a specific enrichment for such sites is not seen in the DS-DM data from the unfractionated whole blood leukocytes ().
Also likely of mechanistic importance, based on multiple studies DS-DM has a definite overall bias toward hypermethylation, which is particularly striking in brain samples [Citation59,Citation63,64]. In brains from individuals with DS, >80% of the DS-DM CpGs are hypermethylated relative to controls, while in both unfractionated PBL and purified T lymphocytes the bias toward hypermethylation is present but less strong (). This trend toward hypermethylation suggests a hypothesis for the mechanism of DS-DM involving increased dosage and overexpression of methylation pathway genes, which we discuss in the next section. The general predominance of hypermethylation among the DS-DM loci, including in fetal tissues, has also raised interesting questions about a possible connection between DS-DM and aging in DS. Putative accelerated aging in this syndrome has been of general interest because it might contribute to the important phenotype of early onset AD [Citation67]. Both historically, with investigations of possible early senescence in fibroblasts [Citation68], and at present with studies on epigenetics, this area is best described as controversial. Regarding the earlier onset of AD in adults with DS, the debate can be phrased as – is the accelerated onset of the disease due exclusively to the increased dosage of APP and other directly acting genes on Hsa21, or is the onset of dementia also partly attributable to a general increase in the rate of biological aging? To address this issue using epigenomics, Horvath et al. examined DS-DM and overall methylation using both previously published and original 450K methylation profiling data and concluded that there is an accelerated aging of methylation patterns in blood and brains of individuals with DS [Citation63]. In contrast, our analysis of the same results, plus additional 450K data from brains and T cells, showed faster acquisition of CpG methylation patterns during fetal and early postnatal development in DS, leading to a higher initial ‘set point’, but no evidence of accelerated aging of these patterns in adult life [Citation59].
Another important characteristic of DS-DM is that, in addition to the significant intergroup differences in methylation (i.e., DS cases vs controls), most of the DS-DM loci also show a somewhat broad range of intragroup methylation values, both among the DS cases and among the control individuals. This intragroup variation is seen in every published dataset from both blood cells and brain cells and tissues, and it is well illustrated by the examples in . In future work it will be interesting to ask whether this variation in DS-DM among individuals (which may reflect genetic background effects and/or environmental effects) might correlate with quantitative differences in DS-associated phenotypes such as immune functions and intellectual disability.
Parent-of-origin effects are well known in mammalian methylomes, specifically in the context of genomic imprinting. Therefore, one group recently searched for such effects in DS-DM, and found that among three genes analyzed (TMEM131, RUNX1 and WRB), the first two showed DS-DM independent of parental origin of the alleles, while the third, WRB, which maps to Hsa21 and is known to be maternally imprinted, showed a preserved pattern of imprinting, with maternal alleles consistently methylated, in the DS cases [Citation58]. Thus, for some loci DS-DM can be superimposed on a normal pattern of parental imprinting.
In addition to cytosine methylation and hydroxymethylation in CpG dinucleotides, the second major type of epigenetic marks are histone modifications, with post-translational modifications of lysine residues in histone H3 being the best studied. A complete description of epigenomic alterations thus should include information on both types of epigenetic marks. Letourneau et al. assessed perturbations of gene expression in Ts21 versus normal fetal fibroblasts and found differential expression organized in broad domains along all chromosomes, with the H3K4me3 profile (a mark for transcriptionally active promoter regions) of the trisomic fibroblasts following the broad domain patterns [Citation69]. How this and other types of histone marks might correlate with and interact mechanistically with DS-DM is an interesting question for ongoing research. It is already clear that the relationship between these two types of epigenetic marks will not be simple, since the H3K4me3 alterations were seen in broad chromatin domains spanning multiple genes, while in every study so far DS-DM has been found to be localized to discrete regulatory regions of single genes, without obvious domain-like effects. A partial exception is DS-DM in the protocadherin gene cluster on chromosome band 5q, which we discuss below. However, even in this gene cluster the DS-DM is focal, affecting only specific types of regulatory sequences. Future analyses using available genomic technologies should be able to address whether other types of epigenetic parameters might be recurrently altered in cells with Ts21.
Mechanisms underlying the trans-acting effects: insights from human Ts21 & mouse models
Two different scenarios for trans-acting molecular mechanisms, namely a role for increased dosage of Hsa21-linked methylation pathway genes and a role for increased dosage of Hsa21-linked transcription factor genes, have been postulated to underlie DS-DM. Regarding the ‘methylation pathway hypothesis’, as shown in , Hsa21 contains a group of genes that have known or predicted roles in the biochemistry of DNA methylation [Citation70]. The pathway leading to methylation of cytosines in genomic DNA requires donation of methyl groups from S-adenosylmethionine (SAM) which, in turn, depends on folate metabolism in the cell. The SCL19A1 gene codes for the reduced folate carrier protein that acts as a transporter at the plasma membrane, while the products of the SOD1 GART and CBS genes act in metabolic pathways involving folic acid and homocysteine in the cytoplasm. The N6AMT1 and DNMT3L genes encode enzymes or components of enzymes that act as methyltransferases, either for DNA (DNMT3L) or for other cellular components that could compete for methyl groups from SAM (N6AMT1). Last, the protein coded by the Hsa21 MIS18A gene interacts with DNMT3A/3B and has been shown to be critical for maintaining DNA methylation at centromeres [Citation71]. The increased dosage of any or all of these genes could in principle affect DNA methylation in cells with Ts21.
How can the methylation pathway hypothesis be tested and the roles of the individual candidate effector genes assessed? Here mouse models can be quite useful because the various mouse lines carry different duplicated chromosomal regions, each with only a subset of the methylation pathway genes. For example, as shown in & , the DNMT3L gene maps to the distal q-arm of Hsa21 and its ortholog is, therefore, present in three copies in the Dp(10)1Yey mice but only in the normal two copies in the Dp(16)1Yey and Ts65Dn lines. Testing the role of this gene in future experiments, by increasing and decreasing its copy number using genetic methods in the mouse models and testing for phenocopying and rescue of the DS-DM, respectively, is a high priority since the DNMT3L protein, while lacking intrinsic methyltransferase activity, is an essential heterodimerization partner for the DNMT3A or DNMT3B DNA methyltransferases that are responsible for de novo CpG methylation in mammalian development [Citation72,Citation73]. This idea is particularly compelling since expression of DNMT3B was found to be coordinately increased with DNMT3L in at least one type of cell (fibroblast-derived iPS cell lines) with Ts21 [Citation54].
To lay the groundwork for a genetic dissection of the Hsa21-linked ‘effector genes’ that contribute to DS-DM we carried out whole genome Bis-Seq (WGBS) in brain tissue from three of the mouse models. First, we analyzed newborn cerebral hemispheres from Dp(16)1Yey and Dp(10)1Yey mice, which are triplicated for 115 and 41 Hsa21 gene orthologs on Mmu16 and Mmu10, respectively. The WGBS data revealed that the human DS-DM signature is partly recapitulated in these models. After developing a bioinformatics pipeline to identify robust DM, we found that these two models have different patterns of DM, but that both show statistically significant similarities with human DS-DM, with a trend toward hypermethylation of the DM genes, albeit stronger in Dp(10)1Yey than in Dp(16)1Yey [Citation59]. In both models the bias toward hypermethylation, while present, was less strong than that seen in the human 450K Beadchip data, perhaps because WGBS covers the whole methylome while the Beadchips query mostly CG-rich sequence elements. The finding of greater hypermethylation in Dp(10)1Yey make sense, since both Dnmt3l and a Hsa21-linked methyl donor pathway gene, the reduced folate carrier gene Slc19a1, map to the Mmu10 duplicated region. However, since numerous DM loci, some hyper- and some hypo-methylated, were also detected in the Dp(16)1Yey brain, we concluded that multiple genes on Hsa21, mapping both to the Dp(10)1Yey and to the Dp(16)1Yey syntenic regions, play a role in the net phenomenon [Citation59]. In a more recent WGBS experiment [do c et al. 2016, manuscript in preparation] we examined cerebral cortical gray matter from Ts65Dn newborns compared with control littermates and obtained results that are again significantly similar to human DS and, as expected based on the duplicated regions (), are more similar to the findings in Dp(16)1Yey than Dp(10)1Yey ().
Regarding the second mechanistic hypothesis, which postulates a role for TFBS occupancies in shaping the tissue-specific patterns of DS-DM, our bioinformatic enrichment analyses in the human data support this mechanism, with strong TFBS enrichment seen for the Runx1 motif in our data from DS versus control T lymphocytes ([Citation59] and ). The RUNX1 gene maps to Hsa21 and, as noted above, despite gains of CpG methylation in its promoter/enhancer region, it is on average overexpressed in Ts21 lymphocytes. So, the simplest interpretation for the bioinformatic enrichment data is that the RUNX1 protein is occupying its cognate DNA binding sites on various chromosomes to a greater extent than normal, thereby blocking CpG methylation in and around these sites. The observation that the ‘core set’ of genes with strong DS-DM in multiple human tissues are significantly enriched in CTCF insulator binding sites is potentially relevant to both hypotheses. Since CTCF is not encoded on Hsa21, the connection between Ts21 and altered methylation at specific CTCF sites might be indirect, possibly mediated through changes in the amounts of specific DNA-binding proteins encoded on Hsa21, or through CTCF binding sites being hypersensitive to altered activity of methylation pathway enzymes and cofactors, some of which are encoded on Hsa21.
Although bioinformatic analyses of the human data have been informative, results from experimentally manipulable models will again be essential for allowing active testing of candidate molecular mechanisms and phenotypic consequences. In the WGBS data from the Ts65Dn and Dp(16)1Yey mouse brains the DM CpGs show a significant enrichment of binding sites for the Mmu16-associated transcription factors Olig2, Gabpa, Erg and Ets2 (). An example of the human-mouse overlap of DM for a biologically relevant locus with DS-DM, the PCDHGA protocadherin gene cluster, is shown in for both the human brains (450K Methylation Beadchip data) and the brains from the mouse models (WGBS data). The profiles show that there are similarities between the overall mouse and human data, with the DM CpGs concentrated at CTCF binding. In addition, the results in Dp(10)1Yey differ from those in Dp(16)1Yey and Ts65Dn, which are in turn more similar, though not identical, to each other. Since Dp(16)1Yey and Ts65Dn both have portions of Mmu16 duplicated, but no duplication of Mmu10, these findings bode well for continuing to use a genetic approach to dissect the groups of Hsa21-orthologous genes that contribute mechanistically to specific components of DS-DM.
While the TFBS occupancy hypothesis can be particularly cleanly tested in DS and mouse models, it is also more broadly relevant to how methylation patterns are shaped in mammalian cells. Work on this topic in multiple systems increasingly points to an important role for sequence-specific DNA binding proteins. Proteins for which occupancy of their binding sites on the DNA can lead to protection of those sites from CpG methylation includes not only CTCF [Citation75–77], but also zinc finger CxxC domain-containing proteins, such as CFP1 and KDM2A, and important transcription factors including SP1 and ETS-family DNA-binding proteins, which recognize sequence motifs that contain CpG dinucleotides. Conversely, a group of zinc-finger TFs, including the BTB/POZ family proteins KAISO, ZTB4 and ZBTB38, and ZFP57, which belongs to the Kruppel-associated box domain family, recognize methylated CpGs within specific DNA sequence motifs and can act as repressors and perpetuate CpG hypermethylation [Citation78]. In fact, the number of methylation-sensitive transcription factors may be much larger: experiments using protein microarrays revealed numerous examples of purified human transcription factors that showed methylated CpG-dependent DNA-binding activities [Citation79]. Epigenome-wide correlative studies have also been informative – for example, in a survey involving multiple cell lines, TFs and methylation types, Xu et al. found strong relationships between occupancies of TFBS’ and methylation levels in and around these sites [Citation80]. Thus, results from studying the role of TFBS occupancies in shaping the patterns of DS-DM may help to consolidate our understanding of the creation of DNA methylation patterns in many other biological situations.
How do the epigenetic changes contribute to DS phenotypes?
In considering the important question of phenotypic effects of DS-DM, it is useful to break the problem down into the roles of the upstream effector genes, located on Hsa21, and the downstream target genes that are affected by DS-DM, which are distributed on various chromosomes (). Based on the hypotheses discussed above, the triplicated Hsa21 gene orthologs that encode transcription factors and methylation pathway genes are likely to be the upstream effector genes that cause the DS-DM in the downstream target genes. This scenario explains, for example, the similarities of DS-DM in Ts65Dn and Dp(16)1Yey mice because the same 100 Hsa21 gene orthologs are triplicated in both models. So, by assessing and comparing key phenotypes, such as lymphocyte development and learning and memory, in mouse lines carrying DNA deletions that normalize the dosage of single or multiple methylation pathway genes in the aneuploid background, it should be possible to draw some initial conclusions as to the importance of the hypermethylation component of DS-DM for DS-associated phenotypes.
Regarding the TFBS hypothesis for DS-DM and its implications for DS phenotypes, the TFs whose binding sites were found to be enriched among DM loci in WGBS data from the mouse models have been implicated previously in both hematopoietic and brain-related phenotypes of these mice [Citation81,Citation82]. However, for these and other Hsa21-linked transcription factor genes that are putatively involved in shaping the specific patterns of DS-DM, gene dosage normalization experiments in mice, while informative for testing the mechanisms, will be less definitive for addressing the role of the epigenetic changes in producing phenotypic effects. This problem arises because transcription factors have acute effects on gene expression in cells, in addition to their putative roles in shaping the methylome. Therefore, for understanding the phenotypic contributions from DS-DM it will be necessary to study not only the upstream effector genes but also the roles of the downstream target genes in DS-associated phenotypes.
In fact, results from PBL and T lymphocytes indicate that DS-DM affects target genes encoding signaling proteins and transcription factors that are known or suspected to be necessary for lymphocyte and NK cell development and function [Citation53,Citation57–59]. Examples shown in include TMEM131, encoding a transmembrane protein that marks lymphocyte precursor cells, SH3BP2, coding for a signaling adaptor protein that has been best studied in B lymphocytes, ZDHHC14, which codes for a palmitoyl transferase that can regulate receptor tyrosine kinases, and RUNX1, which is crucial for hematopoietic stem/progenitor cell, NKT and T-cell development. DS-DM in these genes may play roles in the immunodeficiency and autoimmune components of DS. Likewise, a high percentage of the genes with DS-DM in neural cells, such as those encoding multiple protocadherin-family proteins, neuroligin-2, cytohesin-2, the signaling receptor amigo-3, the brsk-2/sad-a kinase and others, have known or predicted roles in brain development or function [Citation59]. Thus, changes in expression of these genes affecting lymphocyte development and function and neurodevelopment could lead to DS-associated phenotypes in the immune system and brain – a hypothesis that can be actively tested in the future using genetic approached in mouse models. These types of experiments will ultimately clarify whether the consequences of DS-DM at specific loci are adverse, neutral, or protective against the medically relevant DS-associated phenotypes. In fact, however, DS-DM results in transcriptional changes of only a subset of the affected genes: in our studies we assayed expression of a limited number of genes using Q-PCR and found that only about half of the genes with DS-DM showed differential mRNA expression in DS versus control brains and T cells. Since DS-DM is highly tissue specific, future studies of the relative expression levels of genes with DS-DM using purified cell populations from human individuals with and without DS, and parallel studies in the mouse models, will be a crucial next step.
Since correlative studies in epigenetics are susceptible to the classical ‘chicken and egg problem’, to close the logical loop on a causal link between DS-DM and target gene expression, future experiments using methyltransferase knockouts in mice as well as CRISPR-Cas9-mediated deletions of methyltransferases in human cells, followed by RNA-Seq, can be designed to test the general methylation dependence of the expression of genes with DS-DM. Already arguing for a functional link, we found that the expression levels of several genes with DS-DM changed in cell culture with exposure to the hypomethylating drug 5aza-dC [Citation53,Citation59]. So again, results from studying the role of DS-DM in DS phenotypes may well generalize more broadly, as similar experimental challenges arise in all studies that seeks to link epigenetic changes to biological phenotypes.
Questions for future work: implications for therapies & epigenetic effects of chromosomal aneuploidies in other developmental disorders & cancers
First, given the potential phenotypic impacts of DS-DM it is worth considering whether treatment approaches could benefit from taking epigenetics into account. Hypomethylating drugs such as 5-azadC, while useful as anticancer agents, can be mutagenic and are, therefore, not appropriate for use outside of oncology. However, dietary manipulations that are known to be nontoxic and that seem to be safe in the postnatal period can also affect DNA methylation patterns, the classical example being methyl–donor supplementation with folic acid, betaine and vitamin B12. Such diets, which can help to maintain methylation levels genome-wide, have been administered to individuals with DS in a few controlled studies, with variable but partly promising effects on measures of overall health ([Citation70]; references therein). Such diets would not be expected to reduce the methylation of the DS-DM loci that are hypermethylated (most DS-DM loci in brain cells), but might prevent losses of methylation at DS-DM loci that are hypomethylated, of which there are good number in whole blood leukocytes and lymphocytes. In a more targeted approach, the identification of DS-DM target genes and effector genes that play roles in DS-associated phenotypes such as immune dysregulation and intellectual disability may provide clues to future therapies, both in people with DS and in the wider population.
In addition to DS (Ts21), the more severe and largely nonviable syndromes due to Ts13 (Patau’s syndrome) and Ts18 (Edward’s syndrome) are also starting to be studied for alterations in DNA methylation [Citation61]. More generally, a well-known group of viable human developmental syndromes are caused by subchromosomal duplications or deletions that range from hundreds of Kb up to several Mb in size. For some of these disorders, such a Charcot-Marie Tooth syndrome (CMT1A), the phenotype can be completely explained by increased dosage of a single gene, PMP22 for CMT1A, in the duplicated region in chromosome band 17p12 [Citation83], while for others, such as Smith-Magenis syndrome caused by 3.5 Mb hemizygous deletions in band 17p11.2, Potocki-Lupski syndrome due to duplications of this same chromosomal region, Williams-Beuren syndrome caused by 1.5 Mb hemizygous deletions in band 7q11.23, DiGeorge-velocardiofacial syndrome caused by 1.5–3.0 Mb hemizygous deletions in band 22q11.2, rare syndromes such as that due to chromosome 8p23.1 duplications, and even for the classical imprinted disorder Prader–Willi syndrome, due to paternally transmitted deletions spanning about 4 Mb in band 15q11–13, the phenotypes are dominated by single genes but also have contributions from multiple genes in the deleted regions [Citation84–91]. Could there be trans-acting effects of these subchromosomal anomalies on epigenetic patterning elsewhere in the genome, and if so, could such effects be contributing to the phenotypes? To our knowledge there have been few, if any, reports so far on genome-wide epigenetic profiling in these conditions. Future studies of possible epigenetic effects due to these subchromosomal disorders will also benefit from the availability of chromosomally engineered mouse models designed specifically for these duplications and deletions [Citation92–97].
Likewise, large subchromosomal DNA deletions with probable pathogenic roles have been found in nonsyndromic cases of neurodevelopmental and psychiatric disorders including autism and schizophrenia ([Citation98–101]; references therein). In a representative study, Christian et al. used array comparative genomic hybridization to examine 397 cases of autism spectrum disorder (ASD) and, after discarding common copy number variants that were also seen in controls, found a total of 51 novel and presumably pathogenic copy number variants in 46 of the ASD subjects. These deletions ranged in size from 189 kb to 5.5 Mb and many of them contained multiple genes. Here too, although there are genes within the deleted regions that could directly account for the phenotypes (e.g. the neurexin gene NRXN1 in a recurrent class of autism- and schizophrenia-associated deletions), it will be interesting to ask whether trans-effects on epigenetic patterning might also be occurring and contributing to phenotypic severity, particularly in the individuals that carry the larger deletions.
An intriguing variation on these themes is the situation in which chromosomal trisomies, such as Ts16, are detected as mosaicism (i.e., a subpopulation of aneuploid cells) in placentas from pregnancies that have abnormal outcomes such as intrauterine growth restriction, pre-eclampsia and birth defects. Typically in these cases the mosaicism is confined to the placenta and is not found in the affected fetus or child [Citation102], and the developmental effects have been postulated to be either indirect, through altered placental function, and/or due to the presence of cells with Ts16 in the embryo early in development which are then counter-selected, and are thus undetectable later in gestation. In fact, recurrent epigenetic abnormalities have been reported in human placentas with Ts16 [Citation103], so it is possible that the types of trans-acting effects covered in this review may contribute to the adverse fetal outcomes, via epigenetic mechanisms in the placenta and in the early embryo.
Last but not least, the disease category in which chromosomal and subchromosomal aneuploidies are most abundant is neoplasia. Research on the role of chromosomal aneuploidies in human cancers has a long history, starting as early as 1890 with observations by von Hansemann, who noted abnormal mitoses in cancer cells and postulated that the resulting abnormal chromosomal segregation might be leading to altered DNA content [Citation104], followed by cytological observations of chromosomal abnormalities in cancer cells by Boveri at the turn of the century (reviewed in [Citation5]), and progressing with the development of classical cytogenetics and the construction of useful catalogs of chromosomal abnormalities in every tumor type [Citation105]. Such catalogs have laid the groundwork for identifying tumor suppressor genes in the recurrently lost chromosomal regions and proto-oncogenes in the amplified ones. More generally, direct chromosome transfers into cancer cell lines have shown that, regardless of chromosome or cancer type, chromosomal trisomies result in a significant increase in the average transcriptional activity of the trisomic chromosome [Citation106]. But at the same time there has also been a long-running debate on the relative importance of single-gene mutations versus chromosomal aneuploidies in cancer biology [Citation107]. Relevant to this debate, while recent large-scale genome sequencing efforts such as The Cancer Genome Anatomy project have uncovered numerous examples of pathogenic somatic mutations in human cancers, these data have at the same time revealed two general types of tumors: one type (M-Class, e.g. melanoma) with frequent somatic mutations and another type (C-Class, e.g. high-grade ovarian carcinomas) with frequent chromosomal aneuploidies but relatively few point mutations [Citation108,Citation109]. While the C-Class tumors do have mutations in the TP53 tumor suppressor that allow genomic instability they have very few mutations in other genes and it is thought that they acquire the full malignant phenotype via the effects of the chromosomal aneuploidies. In future work it may be productive to mine epigenome-wide data to ask whether C-Class tumors have recurrent epigenetic abnormalities that correlate with aneuploidies of specific chromosomes, although the complex karyotypes of many of these tumors will likely make this a daunting task. Analogous to our hypotheses for DS-DM, in C-class cancers it will also be interesting to test whether altered dosage of epigenetic ‘reader’ and ‘writer’ genes correlates with the observed epigenetic profiles, and to ask whether the genome wide patterns of CpG methylation in these tumors can be explained by altered occupancy of TFBS by transcription factors that are over- or under-expressed because they are encoded within the aneuploid chromosomal regions.
Understanding the role of genetics in establishing epigenetic patterns has become a high priority in the postgenomic era. Here we have reviewed an important type of genetic–epigenetic interaction – trans-effects of chromosomal aneuploidies on CpG methylation and chromatin states throughout the genome.
Down syndrome (DS), caused by trisomy 21 (Ts21), includes medically important phenotypes in blood cells, heart and brain. While the etiology of DS (the extra copy of Hsa21) has been known for many years, how the extra chromosome leads to the DS-associated phenotypes is still not well understood. A long-standing hypothesis, supported by gene expression profiling data, invokes gene dosage effects, in which increased expression of Hsa21 genes leads to direct biological effects and perturbations of downstream transcriptional networks.
Going beyond simple transcriptional effects, recent data support an additional hypothesis that involves epigenetics. The extra copy of Hsa21 can act in trans to produce tissue-specific changes in DNA methylation (DS-DM) and chromatin states at loci distributed throughout the genome. In a given tissue or cell type DS-DM affects 100–1000 genes, and the epigenetic changes are highly recurrent and predictable, with independent studies revealing DS-DM affecting many of the same genes.
Studies of human DS and mouse models carrying subchromosomal aneuploidies of Hsa21-syntenic regions, Ts65Dn, Dp(10)1Yey and Dp(16)1Yey, are starting to support two trans-acting mechanisms that may underlie DS-DM. First, there could be a role for increased dosage of Hsa21-linked methylation pathway genes (SLC19A1, FTCD, GART, CBS, PRMT2, N6AMT1, MIS18A and DNMT3L) that have known or predicted roles in SAM-dependent DNA methylation. Increased dosage of these genes, most of which map to the distal q-arm of Hsa21, might underlie the bias toward hypermethylation at DS-DM CpGs in human DS brain and in other tissues, and the greater hypermethylation that is found in Dp(10)1Yey compared with Dp(16)1Yey mouse brains. The second hypothesis postulates a role for transcription factor binding site (TFBS) occupancies in shaping the patterns of DS-DM. Bioinformatic enrichment analyses support this mechanism, with CTCF motifs enriched among pan-tissue hypermethylated DS-DM loci, and Runx1 binding motifs enriched among hypomethylated DS-DM loci in T lymphocytes. The RUNX1 gene maps to Hsa21 and is overexpressed in DS lymphocytes, so increased occupancy of Runx1 sites may be protecting these sites from CpG methylation. Enrichment analysis of WGBS data from the mouse models further supports this type of mechanism, implicating other TFs (Olig2, Gabpa, Erg, Ets2) that are encoded on Hsa21-orthologous chromosomal segments.
The phenotypic effects of DS-DM are under investigation. These studies will need to test the biological roles of upstream effector genes on Hsa21, including methylation pathway genes and transcription factor genes, and the groups of downstream target genes that are affected by DS-DM. In the blood DS-DM affects genes that encode signaling proteins and transcription factors with known or suspected roles in lymphocyte development and function, including TMEM131, SH3BP2, ZDHHC14 and RUNX1. Likewise, DS-DM in brain cells affects genes coding for multiple protocadherin-family members and the neural proteins NLGN2, CYTH2, AMIGO3 and BRSK2, all of which have known or predicted roles in the brain.
We suggest that future work should explore epigenetic effects of aneuploidies in other developmental, obstetrical and neuropsychiatric disorders, and in human cancers. Individuals with autism and schizophrenia often carry large subchromosomal deletions encompassing multiple genes, which could be studied for effects on epigenetic patterning. Human cancers can be classified as ‘M-class’, carrying multiple point mutations, and ‘C-class’, which have fewer mutations but frequent chromosomal aneuploidies. It will be important to ask whether C-class cancers might have epigenetic changes that track with specific chromosomal or subchromosomal gains and losses.
Financial & competing interests disclosure
This work was supported by grants from the NIH to B Tycko (R01AG036040, R01AG035020, P01-HD035897), by a grant from the Down Syndrome Research Foundation-UK to B Tycko, by a grant to the Herbert Irving Comprehensive Cancer Center at Columbia University (P30CA013696), by grants from the NIH to E Yu (R01NS66072 and R01HL91519) and to Roswell Park Cancer Institute (P3016056), and by a grant from the Children’s Guild Foundation to E Yu. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Tycko B . Allele-specific DNA methylation: beyond imprinting . Hum. Mol. Genet.19 ( R2 ), R210 – R220 ( 2010 ).
- Revill K , TyckoB . Epigenomic insights into common disease . Genome Med.23 ( 3 ), 71 ( 2016 ).
- Weksberg R , ButcherDT , TyckoB . Epigenetics . In : Emery and Rimoin’s Principles and Practice of Medical Genetics (6th Edition) . RimoinDL , PyeritzRE , KorfBR ( Eds ). Elsevier , NY, USA ( 2006 ).
- Arrowsmith CH , BountraC , FishPV , LeeK , SchapiraM . Epigenetic protein families: a new frontier for drug discovery . Nat. Rev. Drug Discov.11 ( 5 ), 384 – 400 ( 2012 ).
- Torres EM , WilliamsBR , AmonA . Aneuploidy: cells losing their balance . Genetics179 ( 2 ), 737 – 746 ( 2008 ).
- Savva GM , MorrisJK , MuttonDE , AlbermanE . Maternal age-specific fetal loss rates in Down syndrome pregnancies . Prenat. Diagn.26 ( 6 ), 499 – 504 ( 2006 ).
- Bray IC , WrightDE . Estimating the spontaneous loss of Down syndrome fetuses between the times of chorionic villus sampling, amniocentesis and livebirth . Prenat. Diagn.18 ( 10 ), 1045 – 1054 ( 1998 ).
- Lana-Elola E , Watson-ScalesSD , FisherEM , TybulewiczVL . Down syndrome: searching for the genetic culprits . Dis. Model Mech.4 ( 5 ), 586 – 595 ( 2011 ).
- Antonarakis SE , EpsteinCJ . The challenge of Down syndrome . Trends Mol. Med.12 ( 10 ), 473 – 479 ( 2006 ).
- Curry CJ . Autosomal trisomies . In : Emery & Rimoin’s Principles and Practice of Medical Genetics (6th Edition) . RimoinDL , PyeritzRE , KorfBR ( Eds ). Elsevier Science, NY, USA ( 2013 ).
- Bratman SV , HorstKC , CarlsonRW , KappDS . Solid malignancies in individuals with Down syndrome: a case presentation and literature review . J. Natl Compr. Canc. Netw.12 ( 11 ), 1537 – 1545 ( 2014 ).
- Yang A , ReevesRH . Increased survival following tumorigenesis in Ts65Dn mice that model Down syndrome . Cancer Res.71 ( 10 ), 3573 – 3581 ( 2011 ).
- Sussan TE , YangA , LiF , OstrowskiMC , ReevesRH . Trisomy represses Apc(Min)-mediated tumours in mouse models of Down’s syndrome . Nature451 ( 7174 ), 73 – 75 ( 2008 ).
- Schupf N , KapellD , NightingaleB , RodriguezA , TyckoB , MayeuxR . Earlier onset of Alzheimer’s disease in men with Down syndrome . Neurology50 ( 4 ), 991 – 995 ( 1998 ).
- Zigman WB , JenkinsEC , TyckoB , SchupfN , SilvermanW . Mortality is associated with apolipoprotein E epsilon4 in nondemented adults with Down syndrome . Neurosci. Lett.390 ( 2 ), 93 – 97 ( 2005 ).
- Lee JH , GurneyS , PangDet al. Polymorphisms in HSD17B1: early onset and increased risk of Alzheimer’s disease in women with Down syndrome . Curr. Gerontol. Geriatr. Res. 2012 , 361218 ( 2012 ).
- Schupf N , LeeA , ParkNet al. Candidate genes for Alzheimer’s disease are associated with individual differences in plasma levels of beta amyloid peptides in adults with Down syndrome . Neurobiol. Aging36 ( 10 ), 2907.e2901 – 2910.e2901 ( 2015 ).
- Hartley D , BlumenthalT , CarrilloMet al. Down syndrome and Alzheimer’s disease: common pathways, common goals . Alzheimers Dement.11 ( 6 ), 700 – 709 ( 2015 ).
- Zhang L , FuD , BelichenkoPVet al. Genetic analysis of Down syndrome facilitated by mouse chromosome engineering . Bioeng. Bugs3 ( 1 ), 8 – 12 ( 2012 ).
- Jiang X , LiuC , YuTet al. Genetic dissection of the Down syndrome critical region . Hum. Mol. Genet.24 ( 22 ), 6540 – 6551 ( 2015 ).
- Xing Z , LiY , PaoAet al. Mouse-based genetic modeling and analysis of Down syndrome . Br. Med. Bull. ( 2016 ) ( Epub ahead of print ).
- Xing L , SalasM , ZhangHet al. Creation and characterization of BAC-transgenic mice with physiological overexpression of epitope-tagged RCAN1 (DSCR1) . Mamm. Genome24 ( 1–2 ), 30 – 43 ( 2013 ).
- Smith DJ , StevensME , SudanaguntaSPet al. Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome . Nat. Genet.16 ( 1 ), 28 – 36 ( 1997 ).
- Reeves RH , IrvingNG , MoranTHet al. A mouse model for Down syndrome exhibits learning and behaviour deficits . Nat. Genet.11 ( 2 ), 177 – 184 ( 1995 ).
- Ruparelia A , WisemanF , SheppardO , TybulewiczVL , FisherEM . Down syndrome and the molecular pathogenesis resulting from trisomy of human chromosome 21 . J. Biomed. Res.24 ( 2 ), 87 – 99 ( 2010 ).
- O’doherty A , RufS , MulliganCet al. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes . Science309 ( 5743 ), 2033 – 2037 ( 2005 ).
- Liu C , MorishimaM , JiangXet al. Engineered chromosome-based genetic mapping establishes a 3.7 Mb critical genomic region for Down syndrome-associated heart defects in mice . Hum. Genet.133 ( 6 ), 743 – 753 ( 2014 ).
- Yu T , LiZ , JiaZet al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions . Hum. Mol. Genet.19 ( 14 ), 2780 – 2791 ( 2010 ).
- Yu T , LiuC , BelichenkoPet al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice . Brain Res.1366 , 162 – 171 ( 2010 ).
- Lange AW , RothermelBA , YutzeyKE . Restoration of DSCR1 to disomy in the trisomy 16 mouse model of Down syndrome does not correct cardiac or craniofacial development anomalies . Dev. Dyn.233 ( 3 ), 954 – 963 ( 2005 ).
- Blazek JD , AbeysekeraI , LiJ , RoperRJ . Rescue of the abnormal skeletal phenotype in Ts65Dn Down syndrome mice using genetic and therapeutic modulation of trisomic Dyrk1a . Hum. Mol. Genet.24 ( 20 ), 5687 – 5696 ( 2015 ).
- Liu C , MorishimaM , YuTet al. Genetic analysis of Down syndrome-associated heart defects in mice . Hum. Genet.130 ( 5 ), 623 – 632 ( 2011 ).
- Xing L , SalasM , LinCSet al. Faithful tissue-specific expression of the human chromosome 21-linked COL6A1 gene in BAC-transgenic mice . Mamm. Genome18 ( 2 ), 113 – 122 ( 2007 ).
- Alford KA , SlenderA , VanesLet al. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome . Blood115 ( 14 ), 2928 – 2937 ( 2010 ).
- Kirsammer G , JilaniS , LiuHet al. Highly penetrant myeloproliferative disease in the Ts65Dn mouse model of Down syndrome . Blood111 ( 2 ), 767 – 775 ( 2008 ).
- Jablonska B , FordD , TrislerD , PessacB . The growth capacity of bone marrow CD34 positive cells in culture is drastically reduced in a murine model of Down syndrome . C R Biol329 ( 9 ), 726 – 732 ( 2006 ).
- Belichenko PV , KleschevnikovAM , BeckerAet al. Down syndrome cognitive phenotypes modeled in mice trisomic for all HSA 21 homologues . PLoS ONE10 ( 7 ), e0134861 ( 2015 ).
- Ahmed MM , DhanasekaranAR , TongSet al. Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain . Hum. Mol. Genet.22 ( 9 ), 1709 – 1724 ( 2013 ).
- Seo H , IsacsonO . Abnormal APP, cholinergic and cognitive function in Ts65Dn Down’s model mice . Exp. Neurol.193 ( 2 ), 469 – 480 ( 2005 ).
- Park J , OhY , YooLet al. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells . J. Biol. Chem.285 ( 41 ), 31895 – 31906 ( 2010 ).
- Wolvetang EW , BradfieldOM , TymmsMet al. The chromosome 21 transcription factor ETS2 transactivates the beta-APP promoter: implications for Down syndrome . Biochim. Biophys. Acta1628 ( 2 ), 105 – 110 ( 2003 ).
- Hoeffer CA , DeyA , SachanNet al. The Down syndrome critical region protein RCAN1 regulates long-term potentiation and memory via inhibition of phosphatase signaling . J. Neurosci.27 ( 48 ), 13161 – 13172 ( 2007 ).
- Torres EM , SokolskyT , TuckerCMet al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast . Science317 ( 5840 ), 916 – 924 ( 2007 ).
- Torres EM , SpringerM , AmonA . No current evidence for widespread dosage compensation in S. cerevisiae . Elife5 , e10996 ( 2016 ).
- Kahlem P , SultanM , HerwigRet al. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of Down syndrome . Genome Res.14 ( 7 ), 1258 – 1267 ( 2004 ).
- Lyle R , GehrigC , Neergaard-HenrichsenC , DeutschS , AntonarakisSE . Gene expression from the aneuploid chromosome in a trisomy mouse model of Down syndrome . Genome Res.14 ( 7 ), 1268 – 1274 ( 2004 ).
- Vacik T , OrtM , GregorovaSet al. Segmental trisomy of chromosome 17: a mouse model of human aneuploidy syndromes . Proc. Natl Acad. Sci. USA102 ( 12 ), 4500 – 4505 ( 2005 ).
- Mao R , ZielkeCL , ZielkeHR , PevsnerJ . Global up-regulation of chromosome 21 gene expression in the developing Down syndrome brain . Genomics81 ( 5 ), 457 – 467 ( 2003 ).
- Saran NG , PletcherMT , NataleJE , ChengY , ReevesRH . Global disruption of the cerebellar transcriptome in a Down syndrome mouse model . Hum. Mol. Genet.12 ( 16 ), 2013 – 2019 ( 2003 ).
- Li CM , GuoM , SalasMet al. Cell type-specific over-expression of chromosome 21 genes in fibroblasts and fetal hearts with trisomy 21 . BMC Med. Genet.7 , 24 ( 2006 ).
- Prandini P , DeutschS , LyleRet al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance . Am. J. Hum. Genet.81 ( 2 ), 252 – 263 ( 2007 ).
- Costa V , AngeliniC , D’apiceLet al. Massive-scale RNA-Seq analysis of non ribosomal transcriptome in human trisomy 21 . PLoS ONE6 ( 4 ), e18493 ( 2011 ).
- Kerkel K , SchupfN , HattaKet al. Altered DNA methylation in leukocytes with trisomy 21 . PLoS Genet.6 ( 11 ), e1001212 ( 2010 ).
- Sailani MR , SantoniFA , LetourneauAet al. DNA-methylation patterns in trisomy 21 using cells from monozygotic twins . PLoS ONE10 ( 8 ), e0135555 ( 2015 ).
- Jones MJ , FarreP , McewenLMet al. Distinct DNA methylation patterns of cognitive impairment and trisomy 21 in Down syndrome . BMC Med. Genomics6 , 58 ( 2013 ).
- Eckmann-Scholz C , BensS , KolarovaJet al. DNA-methylation profiling of fetal tissues reveals marked epigenetic differences between chorionic and amniotic samples . PLoS ONE7 ( 6 ), e39014 ( 2012 ).
- Bacalini MG , GentiliniD , BoattiniAet al. Identification of a DNA methylation signature in blood cells from persons with Down syndrome . Aging (Albany NY)7 ( 2 ), 82 – 96 ( 2015 ).
- Alves Da Silva AF , MachadoFB , PavarinoECet al. Trisomy 21 Alters DNA methylation in parent-of-origin-dependent and -independent manners . PLoS ONE11 ( 4 ), e0154108 ( 2016 ).
- Mendioroz M , DoC , JiangXet al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models . Genome Biol.16 ( 1 ), 263 ( 2015 ).
- Lim JH , KimSY , HanJY , KimMY , ParkSY , RyuHM . Comprehensive investigation of DNA methylation and gene expression in trisomy 21 placenta . Placenta42 , 17 – 24 ( 2016 ).
- Hatt L , AagaardMM , BachCet al. Microarray-based analysis of methylation of 1st trimester trisomic placentas from Down syndrome, Edwards syndrome and Patau syndrome . PLoS ONE11 ( 8 ), e0160319 ( 2016 ).
- Yuen RK , NeumannSM , FokAKet al. Extensive epigenetic reprogramming in human somatic tissues between fetus and adult . Epigenetics Chromatin4 , 7 ( 2011 ).
- Horvath S , GaragnaniP , BacaliniMGet al. Accelerated epigenetic aging in Down syndrome . Aging Cell14 ( 3 ), 491 – 495 ( 2015 ).
- El Hajj N , DittrichM , BockJet al. Epigenetic dysregulation in the developing Down syndrome cortex . Epigenetics11 ( 8 ), 563 – 578 ( 2016 ).
- Matevossian A , AkbarianS . Neuronal nuclei isolation from human postmortem brain tissue . J. Vis. Exp. ( 20 ), pii: 914 ( 2008 ).
- PubMed . Gene expression omnibus . www.ncbi.nlm.nih.gov/geo/ .
- Devenny DA , WegielJ , SchupfNet al. Dementia of the Alzheimer’s type and accelerated aging in Down syndrome . Sci. Aging Knowledge Environ.2005 ( 14 ), dn1 ( 2005 ).
- Weirich-Schwaiger H , WeirichHG , GruberB , SchweigerM , Hirsch-KauffmannM . Correlation between senescence and DNA repair in cells from young and old individuals and in premature aging syndromes . Mutat. Res.316 ( 1 ), 37 – 48 ( 1994 ).
- Letourneau A , SantoniFA , BonillaXet al. Domains of genome-wide gene expression dysregulation in Down’s syndrome . Nature508 ( 7496 ), 345 – 350 ( 2014 ).
- Blehaut H , MircherC , RavelAet al. Effect of leucovorin (folinic acid) on the developmental quotient of children with Down’s syndrome (trisomy 21) and influence of thyroid status . PLoS ONE5 ( 1 ), e8394 ( 2010 ).
- Kim IS , LeeM , ParkKCet al. Roles of Mis18alpha in epigenetic regulation of centromeric chromatin and CENP-A loading . Mol. Cell46 ( 3 ), 260 – 273 ( 2012 ).
- Ooi SK , QiuC , BernsteinEet al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA . Nature448 ( 7154 ), 714 – 717 ( 2007 ).
- Bourc’his D , XuGL , LinCS , BollmanB , BestorTH . Dnmt3L and the establishment of maternal genomic imprints . Science294 ( 5551 ), 2536 – 2539 ( 2001 ).
- Suzuki M , YamadaT , Kihara-NegishiFet al. Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b . Oncogene25 ( 17 ), 2477 – 2488 ( 2006 ).
- Williams A , FlavellRA . The role of CTCF in regulating nuclear organization . J. Exp. Med.205 ( 4 ), 747 – 750 ( 2008 ).
- Ong CT , CorcesVG . CTCF: an architectural protein bridging genome topology and function . Nat. Rev. Genet.15 ( 4 ), 234 – 246 ( 2014 ).
- Do C , LangCF , LinJet al. Mechanisms and disease associations of haplotype-dependent allele-specific DNA methylation . Am. J. Hum. Genet.98 ( 5 ), 934 – 955 ( 2016 ).
- Anvar Z , CammisaM , RisoVet al. ZFP57 recognizes multiple and closely spaced sequence motif variants to maintain repressive epigenetic marks in mouse embryonic stem cells . Nucleic Acids Res.44 ( 3 ), 1118 – 1132 ( 2016 ).
- Hu S , WanJ , SuYet al. DNA methylation presents distinct binding sites for human transcription factors . Elife2 , e00726 ( 2013 ).
- Xu T , LiB , ZhaoMet al. Base-resolution methylation patterns accurately predict transcription factor bindings in vivo . Nucleic Acids Res.43 ( 5 ), 2757 – 2766 ( 2015 ).
- Hill CA , SussanTE , ReevesRH , RichtsmeierJT . Complex contributions of Ets2 to craniofacial and thymus phenotypes of trisomic ‘Down syndrome’ mice . Am. J. Med. Genet A149A ( 10 ), 2158 – 2165 ( 2009 ).
- Chakrabarti L , BestTK , CramerNPet al. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome . Nat. Neurosci.13 ( 8 ), 927 – 934 ( 2010 ).
- Warner LE , GarciaCA , LupskiJR . Hereditary peripheral neuropathies: clinical forms, genetics, and molecular mechanisms . Annu. Rev. Med.50 , 263 – 275 ( 1999 ).
- Gropman AL , ElseaS , DuncanWCJr , SmithAC . New developments in Smith-Magenis syndrome (del 17p11.2) . Curr. Opin. Neurol.20 ( 2 ), 125 – 134 ( 2007 ).
- Vilboux T , CicconeC , BlancatoJKet al. Molecular analysis of the retinoic acid induced 1 gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion . PLoS ONE6 ( 8 ), e22861 ( 2011 ).
- Schubert C . The genomic basis of the Williams-Beuren syndrome . Cell. Mol. Life Sci.66 ( 7 ), 1178 – 1197 ( 2009 ).
- Fusco C , MicaleL , AugelloBet al. Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits . Eur. J. Hum. Genet.22 ( 1 ), 64 – 70 ( 2014 ).
- Bittel DC , YuS , NewkirkHet al. Refining the 22q11.2 deletion breakpoints in DiGeorge syndrome by aCGH . Cytogenet. Genome Res.124 ( 2 ), 113 – 120 ( 2009 ).
- Merscher S , FunkeB , EpsteinJAet al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome . Cell104 ( 4 ), 619 – 629 ( 2001 ).
- Magoulas PL , LiuP , GelowaniVet al. Inherited dup(17)(p11.2p11.2): expanding the phenotype of the Potocki-Lupski syndrome . Am. J. Med. Genet A164A ( 2 ), 500 – 504 ( 2014 ).
- Barber JC , RosenfeldJA , FouldsNet al. 8p23.1 duplication syndrome; common, confirmed, and novel features in six further patients . Am. J. Med. Genet A161A ( 3 ), 487 – 500 ( 2013 ).
- Lindsay EA , BottaA , JurecicVet al. Congenital heart disease in mice deficient for the DiGeorge syndrome region . Nature401 ( 6751 ), 379 – 383 ( 1999 ).
- Carmona-Mora P , MolinaJ , EncinaCA , WalzK . Mouse models of genomic syndromes as tools for understanding the basis of complex traits: an example with the smith-magenis and the potocki-lupski syndromes . Curr. Genomics10 ( 4 ), 259 – 268 ( 2009 ).
- Molina J , Carmona-MoraP , ChrastJet al. Abnormal social behaviors and altered gene expression rates in a mouse model for Potocki-Lupski syndrome . Hum. Mol. Genet.17 ( 16 ), 2486 – 2495 ( 2008 ).
- Segura-Puimedon M , SahunI , VelotEet al. Heterozygous deletion of the Williams-Beuren syndrome critical interval in mice recapitulates most features of the human disorder . Hum. Mol. Genet.23 ( 24 ), 6481 – 6494 ( 2014 ).
- Tsai TF , JiangYH , BresslerJ , ArmstrongD , Beaudet al. Paternal deletion from SNRPN to UBE3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome . Hum. Mol. Genet.8 ( 8 ), 1357 – 1364 ( 1999 ).
- Walz K , Caratini-RiveraS , BiWet al. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance . Mol. Cell Biol.23 ( 10 ), 3646 – 3655 ( 2003 ).
- Christian SL , BruneCW , SudiJet al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder . Biol. Psychiatry63 ( 12 ), 1111 – 1117 ( 2008 ).
- Ye T , LipskaBK , TaoRet al. Analysis of copy number variations in brain DNA from patients with schizophrenia and other psychiatric disorders . Biol. Psychiatry72 ( 8 ), 651 – 654 ( 2012 ).
- Ahn K , GotayN , AndersenTMet al. High rate of disease-related copy number variations in childhood onset schizophrenia . Mol. Psychiatry19 ( 5 ), 568 – 572 ( 2014 ).
- Girirajan S , DennisMY , BakerCet al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder . Am. J. Hum. Genet.92 ( 2 ), 221 – 237 ( 2013 ).
- Benn P . Trisomy 16 and trisomy 16 Mosaicism: a review . Am. J. Med. Genet.79 ( 2 ), 121 – 133 ( 1998 ).
- Blair JD , LangloisS , McfaddenDE , RobinsonWP . Overlapping DNA methylation profile between placentas with trisomy 16 and early-onset preeclampsia . Placenta35 ( 3 ), 216 – 222 ( 2014 ).
- Von Hansemann DP . Über asymmetrische Zelltheilung in Epithelkrebsen und deren biologische Bedeutung . Virchow’s Archiv für pathologische Anatomie und Physiologie und für klinische Medicin779 , 299 – 326 ( 1890 ).
- Mitelman F , JohanssonB , MertensF . Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer . http://cgap.nci.nih.gov/Chromosomes/Mitelman .
- Upender MB , HabermannJK , McshaneLMet al. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells . Cancer Res.64 ( 19 ), 6941 – 6949 ( 2004 ).
- Duesberg P . Does aneuploidy or mutation start cancer?Science307 ( 5706 ), 41 ( 2005 ).
- Ciriello G , MillerML , AksoyBA , SenbabaogluY , SchultzN , SanderC . Emerging landscape of oncogenic signatures across human cancers . Nat. Genet.45 ( 10 ), 1127 – 1133 ( 2013 ).
- Liu Z , ZhangS . Tumor characterization and stratification by integrated molecular profiles reveals essential pan-cancer features . BMC Genomics16 , 503 ( 2015 ).