
Abstract
Aim: To identify differentially methylated probes (DMPs) and regions (DMRs) in relation to chronic obstructive pulmonary disease (COPD) and lung function traits. Methods: We performed an epigenome-wide association study of COPD and spirometric parameters, including forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC) and FEV1/FVC, in blood DNA using the Infinium HumanMethylation450 (n = 100, a Korean COPD cohort). Results: We found one significant DMP (cg03559389, DIP2C) and 104 significant DMRs after multiple-testing correction. Of these, 34 DMRs mapped to genes differential expressed with respect to the same trait. Five of the genes were associated with more than two traits: CTU2, USP36, ZNF516, KLK10 and CPT1B. Conclusion: We identified novel differential methylation loci related to COPD and lung function in blood DNA in Koreans and confirmed previous findings in non-Asians. Epigenetic modification could contribute to the etiology of these phenotypes.
First draft submitted: 3 January 2017; Accepted for publication: 3 May 2017; Published online: 15 June 2017
Reduced lung function is not only an important health indicator in respiratory illnesses, but also predicts mortality in the population as a whole [Citation1]. Lung function traits, including forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC) and FEV1/FVC, are useful in the diagnosis or monitoring of lung diseases such as chronic obstructive pulmonary disease (COPD). COPD is one of the leading causes of mortality and morbidity worldwide [Citation2], is a risk factor for lung cancer [Citation3] and impairs health-related quality of life [Citation4].
Numerous genetic loci have been associated with COPD and/or lung function traits; however, a limited proportion of variance of the phenotypes can be explained by these loci [Citation5,Citation6]. This opens the possibility that epigenetic modifications may play a role [Citation7]. DNA methylation is one of the epigenetic mechanisms that can regulate gene expression.
Five epigenome-wide association studies (EWASs) of COPD and/or lung function traits have been published [Citation8–12]. Of these, two [Citation9,Citation12] measured methylation in blood DNA and only one [Citation11] used the Illumina HumanMethylation450 (450k) array. There are few data on COPD or lung function reporting differentially methylated regions (DMRs), involving Asian populations, or connecting methylation alterations in blood to transcriptome data in lung tissue.
The earlier EWASs [Citation8–12] have focused on identification of differentially methylated probes (DMPs) in relation to COPD or lung function metrics. In other words, these studies evaluated associations with methylation levels at individual CpG sites (CpGs). Analysis of DMRs can provide stronger evidence of causality than analysis of individual DMPs [Citation13] and greater power to detect associations with phenotypic traits [Citation14].
We conducted an EWAS of COPD and pulmonary traits in 100 individuals from a Korean COPD cohort using the 450k. Both individual CpGs and regions of differential methylation were examined. For genes to which differential methylation signals are annotated, we checked whether the related trait was associated with gene expression in lung tissue from a separate cohort (n = 188, Asan Biobank). In addition, previously identified probes in non-Asians were checked for differential methylation and gene expression in relation to COPD and lung function.
Materials & methods
Study participants & lung function: the Korean COPD cohort
For methylation profiling, study participants (n = 100) were sampled from a Korean COPD cohort [Citation15]. Participants in the cohort were recruited from a rural area in Korea. Among 190 participants (135 COPD cases and 55 noncases) enrolled in 2012, 100 (60 COPD cases and 40 noncases) were chosen based on availability of clinical information, computed tomography (CT) data, blood/urine samples and survey questionnaire. We additionally applied approximate frequency matching on age in 10 years categories and smoking status. Survey questionnaires, spirometry measurements and blood/urine samples obtained from each participant were used in further analyses. One trained nurse measured height (cm) and weight (kg) twice for each participant with the body composition analyzer IOI 353 (Aarna Systems., Udaipur, India), and the average of two measurements was used in analyses. We calculated BMI by dividing the weight (kg) by the square of the height (m2). Smoking status (current, former and never smoking) was self-reported in the questionnaire. Urine cotinine levels (nmol/l) measured by immunoassay (Immulite 2000 Xpi; Siemens Healthcare Diagnostics, NY, USA) were used to confirm the current nonsmoking status. For current and former smokers, pack-years of smoking were calculated by multiplying the number of years smoked by the number of cigarette packs smoked per day.
In this study, we analyzed COPD status and spirometric parameters: FEV1, FVC and FEV1/FVC. The FEV1 and FVC were measured in liters by using the EasyOne spirometer (ndd Medical Technologies, Inc., MA, USA). To assess post-bronchodilator FEV1, spirometry is performed 15 min after inhalation of 400 mcg of salbutamol, through a metered-dose inhaler (MDI) with a spacer. We defined COPD cases based on post-bronchodilator FEV1/FVC < 0.7 [Citation16]. We used questionnaires for COPD assessment test scores and the Modified Medical Research Council dyspnea grade. Emphysema index and wall area (%) were generated based on CT data [Citation17].
DNA methylation profiling
Blood DNA samples from participants’ baseline visits were used for methylation profiling. Bisulfite conversion was done by using the EZ DNA methylation kit (Zymo Research, CA, USA). Genome-wide methylation profiles were obtained by using the Infinium HumanMethylation450 BeadChip (Illumina, Inc., CA, USA). The signal extraction and Beta Mixture Quantile dilation [Citation18] normalization to correct for probe design bias was done and followed by ComBat [Citation19] for batch effect corrections. We only included CpGs in autosomal chromosomes for our association analyses. The 450k array provides single nucleotide resolutions of methylation status on 473,864 probes across 22 autosomes. Of these, as a quality control (QC) procedure, we excluded probes that were of low quality (having a detection p < 0.01 in any sample or having a bead-count <3 in 5% or more of samples), non-CpG, non-specific [Citation20], or potentially influenced by nearby genetic variations such as single nucleotide polymorphisms (SNPs) [Citation20]. From the signal extraction to QC procedures, we used the complete pipeline in ChAMP R package [Citation21]. Additionally, we removed 31,831 nonspecific probes [Citation22] not included in prior filtering steps. The remaining 402,508 CpGs were included in association analyses. Probe filtering steps are summarized in Supplementary Table 1. To reduce potential influences of extreme values on association results, we trimmed methylation values at the outer fences [Citation23], meaning outside of three-times the interquartile range from the first and third quartiles of each probe. This procedure removed 75,549 (0.19%) β values across all participants. The methylation value (β), the proportion of methylation at a given CpG site, was used for statistical analyses. The value ranges from 0 (unmethylated) to 1 (methylated). Cell-type proportions were estimated by applying Houseman algorithm [Citation24] using the Reinius reference panel [Citation25].
Epigenome-wide association study
To identify DMPs in relation to COPD, a logistic regression model was used with the response variable of COPD status and the predictor variable of methylation values. To evaluate the association between methylation values and pulmonary traits (FEV1, FVC and FEV1/FVC), we used a robust linear regression model with each trait as the response variable and methylation values as the predictor variable. Covariates in the statistical models for FEV1, FEV1/FVC and COPD were age, sex, height, smoking status, pack-years and the estimated cell-type proportions. Weight was also included in models for FVC. For statistical significance, we set a threshold of p < 1.2 × 10-7 after Bonferroni correction (0.05/402,508 = 1.2 × 10-7) for significant DMPs and p < 1.0 × 10-5, an arbitrary threshold, for suggestive DMPs.
To detect regional differential methylation signals, we used two DMR methods: DMRcate [Citation26] and comb-p [Citation27]. These methods identify DMRs using different algorithms. DMRcate identifies DMRs from tunable kernel smoothing process of association signals, whereas comb-p identifies DMRs by regional clustering of low p-values from irregularly spaced p-values. The dmrcate function in the DMRcate R package was used. Input files for both DMR analyses were the epigenome-wide association results: regression coefficients, standard deviations, uncorrected p-values for DMRcate and uncorrected p-values and chromosomal locations for comb-p.
Significant DMRs were defined based on three criteria. First, a DMR should contain more than one probe. Second, regional information can be combined from probes within 1000 basepairs (bp). Third, the region showed multiple-testing corrected p < 0.01 in both methods (false-discovery rate [FDR] [Citation28] for DMRcate and Sidak [Citation29] for comb-p). The minimum number of CpGs in a region and the minimum length of a distance were the defaults in DMRcate [Citation30], so the same values were used for comb-p to harmonize results from two approaches. Parameters used for the DMR calling can be found in Supplementary Table 2. A significant DMR can be detected even if there is no genome-wide significant DMP in the region.
Enrichment & functional network analysis & visualization
We performed an enrichment analysis to evaluate over- or under-representation of genomic features in our differential methylation signals compared with all probes in the 450k array. The differential methylation signals contain significant (Bonferroni corrected p < 0.05) and suggestive (uncorrected p < 0.05) DMPs and nominally significant CpGs (uncorrected p < 0.05) within significant DMRs. To assess enrichment or depletion for genomic features, we used the hypergeometric test (two-sided doubling mid-p).
For biological insights of loci from our EWAS, we conducted a functional network analysis and disease mapping with genes to which our significant (corrected p < 0.05) and suggestive DMPs (p < 1.0 × 10-5) and significant DMRs (corrected p < 0.01 from both DMR methods) were annotated. A list of mapped genes was input to the network analysis and disease mapping in ‘core analysis’ of Ingenuity Pathway Analysis (IPA; Ingenuity Systems, CA, USA).
For graphical display, we used the University of California, Santa Cruz (UCSC) Genome Browser [Citation31,Citation32] and coMET [Citation33]. To check for presence of regulatory elements in loci containing significant DMPs or DMRs, we used the UCSC Genome Browser. The regulatory elements included DNaseI hypersensitivity sites (DHSs), transcription factor binding sites, chromatin state segmentation and histone modification. We added SNPs in publications containing a keyword ‘lung’ and gene expression in lung tissue. We connected three databases to the Genome Browser [Citation31,Citation32]: ‘Roadmap Epigenomics Data Complete Collection at Wash U VizHub (Assembly hg19)’ [Citation34], ‘ENCODE Analysis Hub (Assembly hg19)’ [Citation35] and ‘Genotype-Tissue Expression (GTEx) RNA-seq Signal Hub (Assembly hg19)’ [Citation36]. For top two DMRs (based on FDR from DMRcate), we used coMET to show statistical significance of association, comethylation patterns, functional annotations and regulation tracks. The functional annotations include genes/transcripts and regulation tracks from Ensembl and CpG islands, Broad Chromatin State Segmentation by hidden Markov model (ChromHMM) domains, Digital DNaseI Hypersensitivity Clusters (DNaseI clusters) and SNPs from the UCSC database.
We used the manufacturer’s annotation file [Citation37] for gene annotations in tables and for an enrichment and a functional network analysis.
Replication look-up
We checked differential methylation at DMPs in relation to COPD and lung function in three previous EWASs that used the Illumina 450k in lung tissue from African-Americans [Citation11] or the Illumina 27k in blood from Caucasians [Citation9] or African-Americans [Citation12]. Of 535 CpGs from the 450k [Citation11], 523 were ready for association after probe QC steps in our data. Of 349 CpGs from the 27k [Citation9], 332 probes were available in the 450k, 315 of which were available for association after probe filtering in our data. Of 12 CpGs from the 27k [Citation12], 11 were available for association in our data. For replicated CpGs, we examined whether the annotated genes were differentially expressed in relation to the same trait. The cutoff for statistical significance was set to uncorrected p < 0.05 in replication analyses of differential methylation and gene expression analyses.
Transcriptome analysis: Asan Biobank
Transcriptome profiles were from lung tissue of Korean male smokers (97 COPD cases and 91 noncases, Asan Biobank). Details of the RNA-seq (HiSeq 2000 system, Illumina Inc., CA, USA) transcriptome data can be found elsewhere [Citation38]. We included expression data of the genes to which significant (Bonferroni corrected p < 0.05) or suggestive (uncorrected p < 1.0 × 10-5) DMPs or significant DMRs (corrected p < 0.01 from two DMR methods) annotated. Extreme values, outside of three-times the interquartile range away from the first and third quartiles of each gene transcript, were removed to limit their potential impact on association results [Citation23]. This removed 35,607 (1.1%) gene expression values across all participants.
We lacked information on height and weight for the participants, so we analyzed FEV1 percent predicted and FVC percent predicted instead of FEV1 and FVC, respectively. We multiplied the number of cigarette smoked (packs per day) by the number of years smoked for pack-years.
To check differential gene expression in relation to COPD or lung function, a logistic regression model was used for COPD status and robust linear regression models were used for FEV1 percent predicted, FVC percent predicted and FEV1 percent predicted/FVC percent predicted. The gene expression values for each gene transcript were the predictor variables and COPD or the pulmonary traits were the response variables. Covariates were age and pack-years. The threshold of statistical significance was set to uncorrected p < 0.05.
All the methylation data processing, statistical analyses and visualizations were conducted in R (version 3.0.2) [Citation39] except for comb-p, IPA and the UCSC Genome Browser.
Results
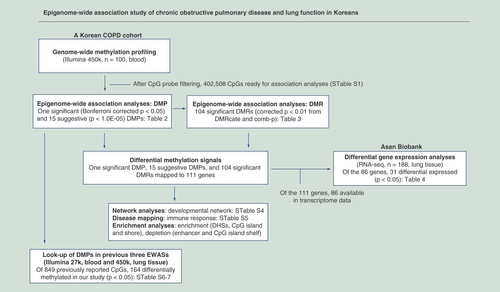
Of 100 participants in our EWAS, 60 were COPD cases and 40 were noncases. The average age was 72.8 years. There were 39 never, 30 former and 31 current smokers. Age and height were not significantly different between COPD cases and noncases, whereas weight and BMI were lower in COPD cases than noncases (p = 0.02 and 0.008, respectively). Characteristics of study participants can be found in . We provide a workflow of our study in .
Table 1. Characteristics of the study populations in our epigenome-wide association and transcriptome analysis.
From our DMP analyses, we found a significant association between cg03559389 (DIP2C) and FEV1/FVC after Bonferroni correction (p = 8.1 × 10-8) (). There was no significant DMP for FEV1, FVC nor COPD after multiple-testing correction. We found suggestive associations (p < 1. × 10-5) at 15 CpGs: four CpGs for FEV1, two for FVC and nine for FEV1/FVC. The top CpG for FEV1 was cg09440215 (C19orf18) and the top for FVC was cg19904425 (SERPINA12). There was no systematic inflation in our association results based on genomic inflation factor (λ) values: 1.00 for FEV1, 0.93 for FVC, 1.16 for FEV1/FVC and 1.37 for COPD. We provide Manhattan plots (Supplementary Figure 1) and quantile–quantile (qq) plots (Supplementary Figure 2).
Table 2. CpGs with differential methylation (p < 1.0 × 10-5) in relation to lung function in a Korean chronic obstructive pulmonary disease cohort, ordered by chromosomal location.
From our DMR analyses, we identified 104 regions of significant differential methylation (corrected p < 0.01) in relation to either the COPD or spirometric measures (Supplementary Table 3). Of the 104, ten (SRPRB, ZC3H3, HLA-C, KLK10, FAM196A, CTU2, USP36, BHMT, CPT1B and CD300A) were associated with more than one trait ().
Table 3. Differentially methylated regions in relation to more than one lung function trait or chronic obstructive pulmonary disease (multiple-testing corrected p < 0.01 from both DMRcate and comb-p) in a Korean chronic obstructive pulmonary disease cohort, ordered by chromosomal location.
For the enrichment analysis, we included one significant and 15 suggestive DMPs and among the 845 CpGs within 104 DMRs we examined 505 nominally significant ones. After taking two overlapping CpGs into account we analyzed 519 CpGs. We found enrichment for CpG islands (50 vs 31% overall from the array; p = 7.0 × 10-21), CpG island shores (29 vs 23% overall; p = 0.0004) and DHSs (25 vs 12% overall; p = 1.2 × 10-14) in Supplementary Table 8.
Our functional network analysis highlighted embryonic/organ development, cell death/survival and gene expression (Supplementary Table 4). Inflammatory response was highlighted from the disease mapping (Supplementary Table 5).
We visualized regulatory elements at 11 loci containing the genome-wide significant DMP (Supplementary Figure 3) and the ten significant DMRs associated with more than one trait (Supplementary Figure 4). Regulatory elements appear to be well-represented in the 11 loci (Supplementary Table 10). For example, a locus ± 10 kb around a DMR ‘chr22:51016501–51017432’ (Supplementary Table 4-J) contains potential functional elements including DHSs, open chromatin, active promoter, active transcription start sites, enhancers and histone modification. Of the ten DMRs, we visualized top two loci (CPT1B and SRPRB) based on the FDR values from DMRcate to show regional association results and functional annotation tracks (Supplementary Figure 5).
For 111 genes to which the 16 DMPs or 104 DMRs mapped, we had expression data at 86 genes from the Asan Biobank. Of these 86, we identified differential gene expression in relation to the same trait (uncorrected p < 0.05) at 31 genes (). Of the 31 genes, five (CTU2, USP36, ZNF516, KLK10 and CPT1B) were associated with more than one trait. Of the 31 genes, 21 showed differential gene expression in relation to COPD. Of these 21, four (CTU2, USP36, ZNF516 and CPT1B) were associated with FEV1 or FEV1/FVC, as well as COPD.
Table 4. Associations with gene expression (unadjusted p < 0.05) in the Asan Biobank for genes to which differentially methylated probes or genes annotated, ordered by chromosomal location.
From a look-up of differential methylation at 315 probes from an EWAS in blood DNA [Citation9] and 523 probes from an EWAS in lung tissue [Citation11], 77 and 87 showed differential methylation in relation to COPD or lung function (uncorrected p < 0.05) in our data, respectively. Of the 77 replicated probes from the EWAS [Citation9] in blood, 72 probes mapped to 71 genes for which we had gene expression values in our data. Of the 71 genes, differential gene expression in relation to the same trait of methylation differences was observed at 29 genes (Supplementary Table 6). Of the 87 replicated probes from the EWAS [Citation11] in lung tissue, 65 probes mapped to 62 genes with gene expression in our data. Of the 62 genes, 31 genes were differentially expressed (Supplementary Table 7). No CpG of the 11 from an EWAS in blood DNA [Citation12] was replicated in our study.
Discussion
Our study identified novel differential methylation signals associated with COPD and lung function traits in blood DNA and linked the methylation alterations to differential gene expression in lung tissue. Previous studies of COPD or lung function used DNA methylation in blood from smokers using the Infinium HumanMethylation27 BeadChip (27k) [Citation9,Citation12], in small airways epithelia from former smokers using the 27k [Citation10], in lung tissue using the Nimblegen array [Citation8], or in lung tissue from former smokers using the 450k [Citation11]. This is the first study to use the Illumina 450k in blood DNA.
We found one genome-wide significant CpG and 15 suggestive CpGs in relation to lung function traits. These DMPs are novel, meaning never reported in the previous EWASs of COPD or lung function. Cg03559389 (DIP2C) showed a significant association with FEV1/FVC after Bonferroni correction. DIP2C has a DMAP1 binding domain. Mutations in DIP2C have been identified in lung cancer samples [Citation40]. Among the 15 suggestive DMPs, cg19904425 (SERPINA12) is 127kb away from SERPINA1, an important gene for COPD [Citation41] and lung function [Citation42]. A recent exome-wide association analysis [Citation43] reported a rare variant (rs140198372) in SERPINA12 30kb away from cg19904425 was associated with airflow limitation.
Several loci from our DMR study overlap loci associated with COPD or pulmonary function in previous genome-wide association studies (GWASs), candidate gene studies or gene expression analyses. Four loci have been reported in GWASs: NCR3 [Citation44], AGER-PPT2 [Citation45], CCL18 [Citation46] and UPK3A [Citation47]. Three overlap loci from candidate gene studies: IL1RN [Citation48], F2R [Citation49] and MMP9 [Citation50]. Two were reported in gene expression studies: PSORS1C1 [Citation51] and USP36 [Citation52]. Two DMRs in our study were near HLA-DQB1/HLA-DQA2, a locus from a SNP-by-smoking interaction study of lung function [Citation53]. This is notable because we excluded 28,652 CpGs at which methylation can be influenced by neighboring SNPs [Citation20]. We found differential methylation in a locus previously associated with a putative biomarker of COPD, surfactant protein D. A GWAS [Citation54] of surfactant protein D reported five genome-wide significant SNPs in/near PSORS1C1. Our DMR resides between the SNPs. This may suggest an epigenetic contribution modulating the association between the genetic variants and pulmonary traits or COPD.
Of interest, the highlighted fundamental pathways in our study support the emerging hypothesis of the importance of fundamental developmental processes in COPD pathogenesis [Citation55,Citation56]. This study supports a possible role for epigenetic modifications in this process.
The enrichment for CpG shores (regions within 2kb from a CpG island) in differentially methylation signals related to lung function is consistent with the findings from a previous EWAS of COPD [Citation11]. To assess enrichment for gene ontology terms in the 111 genes to which significant and suggestive DMPs and significant DMRs mapped, we used WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) [Citation57] with databases for biological process, cellular process and molecular function. No gene ontology terms were significantly enriched.
Among epigenetic modifications, we examined methylation alterations in relation to COPD and lung function in this study. The well-represented regulatory elements including DNase Hypersensitivity sites/clusters, histone modification and chromatin state segmentation in our significant loci suggest that these loci may play functional roles in control of COPD or lung function.
From our replication look-up, we confirmed differential methylation in relation to COPD and lung function previously identified in non-Asian populations (Caucasians [Citation9,Citation11] and African Americans [Citation11]). Of interest, differential methylation from an EWAS [Citation11] in lung tissue was also replicated in our study in blood, supporting the possibility of blood DNA methylation biomarkers for lung function.
In our data, there were 19 mild and 41 moderate to severe COPD cases [Citation16]. To check whether there were CpGs associated with moderate to severe COPD, we performed an EWAS of COPD using 41 moderate to severe cases and 40 noncases. There were no significant DMPs in relation to moderate to severe COPD after multiple-testing correction. In our study, there was no genome-wide significant DMP in relation to COPD. However, all 14 DMPs related to FEV1 or FEV1/FVC (unadjusted p < 1 × 10-5) were also related to COPD (p < 0.05) as shown in Supplementary Table 9.
Our study has limitations. First, the data were cross-sectional for this analysis that hampers the causal inference between methylation alterations and COPD status or lung function levels. We cannot ascertain if these might have predated the COPD or are the result of the COPD. Second, we did not have an available population for replication to decrease the chance of reporting false positives at the lower significance threshold used for the DMP analysis. As strengths, we explored regional differential methylation alterations by using two different methods (DMRcate and comb-p) in addition to examining individual probes. Further, the gene expression profiles in lung tissue strengthen the biologic evidence that the DNA methylation alterations observed in blood are related to lung function and thus provide a type of functional replication. Finally, our study was conducted in Asian populations; there are few methylation and transcriptome data on these populations.
Although GWASs of COPD and lung function have identified numerous loci, these findings only explain the small proportion of variance in these traits [Citation5,Citation6]. Epigenetics may explain some of the remaining variance [Citation58]. Several EWASs reported candidate loci in relation to these phenotypes [Citation8–12]. Studies in blood or lung tissue involving Asians, Caucasians or African Americans could help understand underlying mechanisms of COPD and lung function, and provide candidate target genes for clinical purposes.
Conclusion
We report novel differential methylation alterations associated with COPD or pulmonary traits in blood DNA, and link methylation alterations to differential gene expression related to lung function in lung tissue. Our finding of differential methylation near reported loci from genetic studies of COPD and pulmonary traits supports a possible epigenetic role in control of lung function.
Future perspective
The methylation alterations in blood related to COPD and lung function traits could play a role in development of biomarkers of COPD and lung function. Future studies would be necessary to confirm these findings and understand their mechanistic basis.
This is the first epigenome-wide association study of chronic obstructive pulmonary disease and lung function traits in an Asian population.
Differentially methylated probes from earlier epigenome-wide association studies in non-Asian populations were differentially methylated in our study.
Our study revealed differentially methylated probes and differentially methylated regions in relation to chronic obstructive pulmonary disease and lung function traits in blood DNA. Candidate loci includes DIP2C, SRPRB, ZC3H3, HLA-C, KLK10, FAM196A, CTU2, USP36, BHMT, CPT1B and CD300A.
Genes including CTU2, USP36, ZNF516, KLK10 and CPT1B to which the differential methylation alterations mapped were also differentially expressed in lung tissue with respect to the same traits.
Ethical conduct of research
The Institute Review Board of the Kangwon National University Hospital approved analyses of the data (Institutional Review Board of Kangwon National University Hospital 2012-06-007-001 and KNUH-2016–05-003-001). Informed written consent was obtained from all participants. The study adhered to the tenets of the Helsinki Declaration of 1975, as revised in 2008.
Supplemental Information 1
Download MS Word (13.5 MB)Acknowledgements
The authors appreciate all of the study participants for their contribution to this research. The authors thank F Day, T Wang and S Reese of NIEHS and J Jin of Westat, Inc. for expert computational assistance.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at:www.tandfonline.com/doi/full/10.2217/epi-2017-0002
Financial & competing interest disclosure
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013R1A1A1057961, 2017R1A2B4003790), the Ministry of Education (2013R1A6A3A04059017) and grants from the Environmental Health Center funded by the Ministry of Environment, Republic of Korea. Supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (ZO1 ES043012). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Schunemann HJ , DornJ , GrantBJ , WinkelsteinW , Jr. , TrevisanM . Pulmonary function is a long-term predictor of mortality in the general population: 29-year follow-up of the Buffalo Health Study . Chest118 ( 3 ), 656 – 664 ( 2000 ).
- Mannino DM , BuistAS . Global burden of COPD: risk factors, prevalence, and future trends . Lancet370 ( 9589 ), 765 – 773 ( 2007 ).
- Koshiol J , RotunnoM , ConsonniDet al. Chronic obstructive pulmonary disease and altered risk of lung cancer in a population-based case-control study . PLoS ONE4 ( 10 ), e7380 ( 2009 ).
- Carrasco Garrido P , De Miguel DiezJ , Rejas GutierrezJet al. Negative impact of chronic obstructive pulmonary disease on the health-related quality of life of patients. Results of the EPIDEPOC study . Health Qual. Life Outcomes4 , 31 ( 2006 ).
- Loth DW , Soler ArtigasM , GharibSAet al. Genome-wide association analysis identifies six new loci associated with forced vital capacity . Nat. Genet.46 ( 7 ), 669 – 677 ( 2014 ).
- Cho MH , CastaldiPJ , WanESet al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13 . Hum. Mol. Genet.21 ( 4 ), 947 – 957 ( 2012 ).
- Boezen HM , ManninoDM . The future of nature versus nurture in understanding chronic obstructive pulmonary disease . Am. J. Respir. Crit. Care Med.188 ( 8 ), 891 – 892 ( 2013 ).
- Yoo S , TakikawaS , GeraghtyPet al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD . PLoS Genet.11 ( 1 ), e1004898 ( 2015 ).
- Qiu W , BaccarelliA , CareyVJet al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function . Am. J. Respir. Crit. Care Med.185 ( 4 ), 373 – 381 ( 2012 ).
- Vucic EA , ChariR , ThuKLet al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways . Am. J. Respir. Cell Mol. Biol.50 ( 5 ), 912 – 922 ( 2014 ).
- Morrow JD , ChoMH , HershCPet al. DNA methylation profiling in human lung tissue identifies genes associated with COPD . Epigenetics doi:10.1080/15592294.2016.1226451 ( 2016 ) ( Epub ahead of print ).
- Busch R , QiuW , Lasky-SuJ , MorrowJ , CrinerG , DemeoD . Differential DNA methylation marks and gene comethylation of COPD in African–Americans with COPD exacerbations . Respir. Res.17 ( 1 ), 143 ( 2016 ).
- Lister R , PelizzolaM , DowenRHet al. Human DNA methylomes at base resolution show widespread epigenomic differences . Nature462 ( 7271 ), 315 – 322 ( 2009 ).
- Jaffe AE , MurakamiP , LeeHet al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies . Int. J. Epidemiol.41 ( 1 ), 200 – 209 ( 2012 ).
- Kim WJ , HongY . Methodology of an observational cohort study for subjects with chronic obstructive pulmonary disease in dusty areas near cement plants . J. Pulm. Respir. Med.04 ( 01 ), 169 – 174 ( 2014 ).
- Global Initiative for Chronic Obstructive Lung Disease . Global stregaty for the diagnosis, management, and prevention of chronic obstructive pulmonary disease ( 2016 ). http://goldcopd.org/global-strategy-diagnosis-management-prevention-copd-2016/WatermarkedGlobal-Strategy-20161.pdf .
- Ji W , LimMN , BakSHet al. Differences in chronic obstructive pulmonary disease phenotypes between non-smokers and smokers . Clin. Respir. J. doi:10.1111/crj.12577 ( 2016 ) ( Epub ahead of print ).
- Teschendorff AE , MarabitaF , LechnerMet al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data . Bioinformatics29 ( 2 ), 189 – 196 ( 2013 ).
- Johnson WE , LiC , RabinovicA . Adjusting batch effects in microarray expression data using empirical Bayes methods . Biostatistics8 ( 1 ), 118 – 127 ( 2007 ).
- Nordlund J , BacklinCL , WahlbergPet al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia . Genome Biol.14 ( 9 ), r105 ( 2013 ).
- Morris TJ , ButcherLM , FeberAet al. ChAMP: 450k chip analysis methylation pipeline . Bioinformatics30 ( 3 ), 428 – 430 ( 2014 ).
- Price ME , CottonAM , LamLLet al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array . Epigenetics Chromatin6 ( 1 ), 4 ( 2013 ).
- What are outliers in the data? www.itl.nist.gov/div898/handbook/prc/section1/prc16.htm .
- Houseman EA , AccomandoWP , KoestlerDCet al. DNA methylation arrays as surrogate measures of cell mixture distribution . BMC Bioinformatics13 , 86 ( 2012 ).
- Reinius LE , AcevedoN , JoerinkMet al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility . PLoS ONE7 ( 7 ), e41361 ( 2012 ).
- Peters TJ , BuckleyMJ , StathamALet al. De novo identification of differentially methylated regions in the human genome . Epigenetics Chromatin8 , 6 ( 2015 ).
- Pedersen BS , SchwartzDA , YangIV , KechrisKJ . Comb-p: software for combining, analyzing, grouping and correcting spatially correlated p-values . Bioinformatics28 ( 22 ), 2986 – 2988 ( 2012 ).
- Benjamini Y , HochbergY . Controlling the false discovery rate – a practical and powerful approach to multiple testing . J. R. Stat. Soc. Series. B Stat. Methodol.57 ( 1 ), 289 – 300 ( 1995 ).
- Sidak Z . Rectangular confidence regions for the means of multivariate normal distributions . J. Am. Stat. Assoc.62 ( 318 ), 8 ( 1967 ).
- Peters T . Package ‘DMRcate’ ( 2016 ).
- Kent WJ , SugnetCW , FureyTSet al. The human genome browser at UCSC . Genome Res.12 ( 6 ), 996 – 1006 ( 2002 ).
- UCSC Genome Browser . http://genome.ucsc.edu .
- Martin TC , YetI , TsaiPC , BellJT . coMET: visualisation of regional epigenome-wide association scan results and DNA co-methylation patterns . BMC Bioinformatics16 , 131 ( 2015 ).
- Roadmap Epigenomics Visualization Hub (VizHub) . http://vizhub.wustl.edu/ .
- Consortium EP . An integrated encyclopedia of DNA elements in the human genome . Nature489 ( 7414 ), 57 – 74 ( 2012 ).
- Consortium GT . The Genotype-Tissue Expression (GTEx) project . Nat. Genet.45 ( 6 ), 580 – 585 ( 2013 ).
- Illumina I . Illumina Infinium HumanMethylation450 BeadChip Annotation . https://support.illumina.com/array/array_kits/infinium_humanmethylation450_beadchip_kit/downloads.html .
- Kim WJ , LimJH , LeeJS , LeeSD , KimJH , OhYM . Comprehensive analysis of transcriptome sequencing data in the lung tissues of COPD subjects . Int. J. Genomics2015 , 206937 ( 2015 ).
- R Foundation for Statistical Computing . R: A language and environment for statistical computing ( 2013 ). www.gbif.org/resource/81287 .
- Integrative analysis of complex cancer genomics and clinical profiles . www.cbioportal.org/beta/index.do .
- Kumar M , BhadoriaDP , DuttaKet al. Combinatorial effect of TIMP-1 and alpha1AT gene polymorphisms on development of chronic obstructive pulmonary disease . Clin. Biochem.44 ( 13 ), 1067 – 1073 ( 2011 ).
- Perez-Rubio G , Jimenez-ValverdeLO , Ramirez-VenegasAet al. Prevalence of alpha-1 antitrypsin high-risk variants in Mexican mestizo population and their association with lung function values . Arch. Bronconeumol.51 ( 2 ), 80 – 85 ( 2015 ).
- Jackson VE , NtallaI , SayersIet al. Exome-wide analysis of rare coding variation identifies novel associations with COPD and airflow limitation in MOCS3, IFIT3 and SERPINA12 . Thorax71 ( 6 ), 501 – 509 ( 2016 ).
- Soler Artigas M , LothDW , WainLVet al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function . Nat. Genet.43 ( 11 ), 1082 – 1090 ( 2011 ).
- Hancock DB , EijgelsheimM , WilkJBet al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function . Nat. Genet.42 ( 1 ), 45 – 52 ( 2010 ).
- Yao TC , DuG , HanLet al. Genome-wide association study of lung function phenotypes in a founder population . J. Allergy Clin. Immunol.133 ( 1 ), 248 – 255 ; e241–e210 ( 2014 ).
- Li X , HawkinsGA , AmplefordEJet al. Genome-wide association study identifies TH1 pathway genes associated with lung function in asthmatic patients . J. Allergy Clin. Immunol.132 ( 2 ), 313e315 – 320e315 ( 2013 ).
- Hegab AE , SakamotoT , SaitohWet al. Polymorphisms of TNFalpha, IL1beta, and IL1RN genes in chronic obstructive pulmonary disease . Biochem. Biophys. Res. Commun.329 ( 4 ), 1246 – 1252 ( 2005 ).
- Plate M , LawsonPJ , HillMRet al. Impact of a functional polymorphism in the PAR-1 gene promoter in COPD and COPD exacerbations . Am. J. Physiol. Lung Cell Mol. Physiol.307 ( 4 ), L311 – L316 ( 2014 ).
- Lee SY , KimMJ , KangHGet al. Polymorphisms in matrix metalloproteinase-1, -9 and -12 genes and the risk of chronic obstructive pulmonary disease in a Korean population . Respiration80 ( 2 ), 133 – 138 ( 2010 ).
- Qiu W , ChoMH , RileyJHet al. Genetics of sputum gene expression in chronic obstructive pulmonary disease . PLoS ONE6 ( 9 ), e24395 ( 2011 ).
- Sun XX , HeX , YinL , KomadaM , SearsRC , DaiMS . The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc . Proc. Natl Acad. Sci. USA112 ( 12 ), 3734 – 3739 ( 2015 ).
- Hancock DB , Soler ArtigasM , GharibSAet al. Genome-wide joint meta-analysis of SNP and SNP-by-smoking interaction identifies novel loci for pulmonary function . PLoS Genet.8 ( 12 ), e1003098 ( 2012 ).
- Kim DK , ChoMH , HershCPet al. Genome-wide association analysis of blood biomarkers in chronic obstructive pulmonary disease . Am. J. Respir. Crit. Care Med.186 ( 12 ), 1238 – 1247 ( 2012 ).
- Boucherat O , MorissetteMC , ProvencherS , BonnetS , MaltaisF . Bridging lung development with chronic obstructive pulmonary disease. Relevance of developmental pathways in chronic obstructive pulmonary disease pathogenesis . Am. J. Respir. Crit. Care Med.193 ( 4 ), 362 – 375 ( 2016 ).
- Martinez FD . Early-life origins of chronic obstructive pulmonary disease . N. Engl. J. Med.375 ( 9 ), 871 – 878 ( 2016 ).
- Wang J , DuncanD , ShiZ , ZhangB . WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013 . Nucleic Acids Res.41 , W77 – W83 ( 2013 ).
- Manolio TA , CollinsFS , CoxNJet al. Finding the missing heritability of complex diseases . Nature461 ( 7265 ), 747 – 753 ( 2009 ).