
Abstract
Aim: The expression of human SLFN11 was reported to sensitize cancer cells to DNA damaging agents. This study is to explore the epigenetic change and the function of SLFN11 in human colorectal cancer (CRC). Materials & methods: Six CRC cell lines and 128 primary CRC samples were used. Results:SLFN11 was methylated in 55.47% (71/128) of primary CRC. The expression of SLFN11 was regulated by promoter region methylation. Methylation of SLFN11 was significantly associated with age, poor 5-year overall survival and 5-year relapse-free survival (all p < 0.05). SLFN11 suppressed CRC cell growth both in vitro and in vivo and sensitized CRC cells to cisplatin. Conclusion:SLFN11 is frequently methylated in human CRC, and the expression of SLFN11 is regulated by promoter region methylation. Methylation of SLFN11 reduced the sensitivity of CRC cells to cisplatin.
First draft submitted: 1 February 2017; Accepted for publication: 27 March 2017; Published online: 13 April 2017
Colorectal cancer (CRC) is the third most common cancer and the fourth most common cause of cancer-related death worldwide [Citation1]. Incidence is low at ages younger than 50 years but increases significantly with age. Median age at diagnosis is approximately 70 years in developed countries. However, rapid increases of CRC incidence have been noted with the changes in dietary patterns in China [Citation2]. Most sporadic cases have chromosomal instability, an allelic imbalance at several chromosomal loci, chromosomal amplification and translocation [Citation3]. Defects in DNA damage repair and checkpoint control can result in gene mutations and chromosomal instability to promote tumorigenesis [Citation4,Citation5]. Familial adenomatous polyposis and Lynch syndrome, which are caused by gene mutations in the DNA mismatch repair system, account for less than 5% of all CRC [Citation6]. Aberrant methylation changes in DNA damage repair genes were reported to be an important mechanism of CRC development [Citation7].
The Schlafen (SLFN) gene family was first identified by Schwarz and co-workers in 1998 [Citation8]. To date, five human SLFN genes have been identified [Citation9,Citation10]. SLFN gene products have been demonstrated to be involved in cell biological processes including proliferation, differentiation and immune function [Citation11]. From the Cancer Genome Atlas database, SLFN11 has a wide expression range [Citation12]. The expression of human SLFN11 was reported to sensitize cancer cells to DNA-damaging agents [Citation13,Citation14]. High SLFN11 expression independently predicts overall survival (OS) in a group of ovarian cancer patients treated with cisplatin-containing regiments [Citation13]. The role and epigenetic regulation of SLFN11 in human CRC remain to be elucidated.
Materials & methods
Human tissue samples & cell lines
Six CRC cell lines (RKO, DLD1, SW620, LOVO, Ls180 and DKO) were examined in this study and maintained in 90% Roswell Park Memorial Institute (RPMI) 1640 media supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin. 293T cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. All cell lines were cultured in an atmosphere of 5% carbon dioxide at 37°C.
A total of 128 cases of primary CRC and five cases of noncancerous colorectal mucosa were collected from the Chinese PLA General Hospital in Beijing between May 2009 and November 2013. All cancer samples were classified according to WHO Classification of Digestive Tumors: the Fourth Edition [Citation15]. This study was approved by the Chinese PLA General Hospital’s institutional review board.
5-aza-2′-deoxycytidine treatment
CRC cell lines (RKO, DLD1, SW620, LOVO and Ls180) were split to a low density (30% confluence) 12 h before treatment with 2 μM 5-aza-2′-deoxycytidine (5-AZA, Sigma, MO, USA). Growth medium conditioned with 5-AZA at 2 μM was exchanged every 24 h for a total of 96 h. At the end of the treatment course, RNA was extracted from the cells as described below.
RNA isolation & semiquantitative reverse transcription PCR
Total RNA was extracted using Trizol reagent; cDNA was synthesized according to the manufacturer’s instructions (Invitrogen, CA, USA) and glyceraldehyde-3-phosphatedehydrogenase (GAPDH) was used as control. The SLFN11 PCR primer sequences were as follows: 5′-AACGCCCGATAACCTTCACA-3′ (forward) and 5′-CTAAGGGGAGGCCCACTAGA-3′ (reverse). Thermal cycling parameters were as follows: 95°C 5 min; (95°C 30 s, 60°C 30 s and 72°C 30 s) 35 cycles; 72°C 5 min. The GAPDH PCR primer sequences were as follows: 5′-GACCACAGTCCATGCCATCAC-3′ (forward) and 5′-GTCCACCACCCTGTTGCTGTA-3′ (reverse). Thermal cycling parameters were as follows: 95°C 5 min; (95°C 30 s, 63°C 30 s and 72°C 40 s) 25 cycles; 72°C 5 min.
KRAS & BRAF mutation detection, bisulfite modification, methylation-specific PCR & bisulfite sequencing
Genomic DNA from colorectal cell lines and CRC tissue samples were prepared by the proteinase-K method. KRAS codons 12 and 13 and BRAF codon 600 were amplified by PCR and sequenced according to previously reports [Citation16,Citation17]. The primer sequences are listed in . SLFN11 methylation-specfic PCR (MSP) and bisulfite sequencing (BSSQ) were performed as described previously [Citation18]. DNA samples were then purified with the Wizard DNA Clean-Up System (Promega, CA, USA). MSP primers were designed as follows: 5′-ATTATTAGTAGCGTGACGGTTATC-3′ (M-forward), 5′-CGACAAATATACAAATTAAACCGCG-3′ (M-reverse), 5′-TATATTATTAGTAGTGTGATGGTTATT-3′ (U-forward) and 5′-ATACAACAAATATACAAATTAAACCACA-3′ (U-reverse). Thermal cycling parameters were as follows: 95°C 5 min; (95°C 30 s, 58°C 30 s and 72°C 40 s) 35 cycles; 72°C 5 min. BSSQ primers were designed including the MSP region in a 225-bp region upstream of the SLFN11 transcription start site (-319 to -544 bp). BSSQ primers were designed as follows: 5′-TAGAAAAGTAGAAYGTTGGTAG-3′ (forward) and 5′-CAAAAAATAAATCTTAAAAAC-3′ (reverse). Thermal cycling parameters were as follows: 95° 5 min; (95° 30 s, 55° 30 s and 72° 40 s) 35 cycles; 72° 5 min. Methylation status was detected by MSP in four genes (RUNX3, CACNA1G, IGF2 and MLH1) to represent CpG island methylator phenotype (CIMP) as described previously [Citation19]. The primer sequences are listed in .
Construction of human full length SLFN11 cDNA lentiviral expression vector & transient full length SLFN11 cDNA pcDNA3.1(+) expression vectors
The human full length SLFN11 cDNA (NM_001104587) was cloned into the PCDH (pCDH-CMV-MCS-EF1-Puro) lentiviral plasmid (Genetimes ExCell Technology, Shanghai, China). Transfection was performed at a ratio of 1:3 (DNA mass:lipo mass) following the manufacturer’s instructions for Lipofectamine 3000 Reagent (Invitrogen, CA, USA). Packaged recombinant lentiviruses were harvested from the supernatant of cell cultures at 48 h post-transfection. RKO and DLD1 cells were then infected with viral supernatant. Cells stably expressing SLFN11 were selected with puromycin at a concentration of 2 μg/ml for 2 weeks.
For transient transfection, the human full length SLFN11 cDNA (NM_001104587) was cloned into the pcDNA3.1(+) plasmid (Era Biotech, Shanghai, China). Transfection was performed according to the manufacturer’s instructions by using Lipofectamine 2000 in SW620 cell (Invitrogen, CA, USA).
siRNA knockdown technique
Selected siRNAs targeting SLFN11 and RNAi negative control duplex (Gene Pharma, Shanghai, China) were used in this study. The sequences were as follows: siRNA-1726 duplex (sense: 5′-CCAGGAUAUUUGCGAUAUATT-3′; antisense: 5′-UAUAUCGCAAAUAUCCUGGTT-3′); RNAi negative control duplex (sense: 5′-UUC UCC GAA CGU GUC ACG UTT-3′; and antisense: 5′-ACG UGA CAC GUU CGG AGA ATT-3′). RNAi oligonucleotide or RNAi negative control duplex was transfected into SLFN11 highly expressed DKO cells using Lipofectamine RNAiMAX (Invitrogen, CA, USA).
Cell viability assay
Cells were plated into 96-well plates at a density of 2 × 103 cells per well, and cell viability was measured by the methyl thiazolyl tetrazolium assay (KeyGEN Biotech, Nanjing, China) at 0, 24, 48 and 72 h. Absorbance was measured on a microplate reader of Thermo Multiskan MK3 (Termo Fisher Scientific, MA, USA) at a wavelength of 490 nm.
The IC50 was also detected by the MTT assay. Cells were plated at a density of 3000 cells per well into 96-well plates. After overnight incubation, cells were treated with cisplatin at concentrations of 0, 1, 2.5, 5, 10, 25 and 50 µM for 48 h.
Colony formation assay
SLFN11 stably expressed and unexpressed RKO and DLD1 cells were plated onto 6-well plates in a total of 200 cells per well. After 2 weeks, cells were fixed with 75% ethanol for 30 min. Colonies were then stained with 0.5% crystal violet solution and counted. The experiment was performed in triplicate. Another method was employed in this study to further validate the results. The detailed protocol was described as previously [Citation20]. In brief, SLFN11 stably expressed and unexpressed RKO and DLD1 cells were plated at a confluency of 6 × 104 cells per 6-cm dish. Media were replenished every 2 days. After 7–10 days, cells were stained with crystal violet in formalin for 30 min. After excess stain was washed away, colonies were imaged and the percent plate coverage is indicated as determined from five independent areas per plate using ImageJ software.
After 48 h of transfection, the SLFN11 expressing DKO cells and DKO cells with knockdown of SLFN11 were treated as mentioned above.
Colony formation assay treated with cisplatin
This was performed as previously reported [Citation21]. For the colony-forming assay, SLFN11 stably expressed and unexpressed RKO and DLD1 cell lines were plated onto 6-well plates in a total of 1000 cells per well. After overnight incubation, cells were treated with cisplatin (2 μM) for 24 h and maintained in a drug-free medium for an additional 2 weeks until visible colonies were evident. After staining with the 0.5% crystal violet, the number of colonies of >0.1 mm in diameter was counted. After 48 h post transfection, DKO cells and DKO cells with knockdown of SLFN11 were treated as mentioned above.
Flow cytometry for cell cycle & apoptosis
SLFN11 stably expressed and unexpressed RKO and DLD1 cell lines were plated into 6-well plates at 1 × 106 cells per well. The cells were harvested at 48 h, washed with phosphate-buffered saline twice and stained according to manufacturer’s instructions of Cycletest™ Plus DNA Reagent Kit (Becton, Dickinson and company, CA, USA). Samples were analyzed with an FACS Caliber flow cytometer (Becton, Dickinson and company).
Apoptosis was detected and quantified by Annexin V-FITC and propidium iodide staining. SLFN11 stably expressed and unexpressed RKO and DLD1 cell lines were seeded in 6-well plates at a density of 1.5 × 106 cells/ml. Cells were treated with a concentration of 10 µM cisplatin for 36 h. The cells were then harvested using 0.2% trypsin (no ethylenediaminetetraacetic acid), stained according to manufacturer’s instructions of FITC Annexin V apoptosis Detection kit I (Becton, Dickinson and company), and analyzed by flow cytometry. After 48 h of transfection, DKO cells with and without knockdown of SLFN11 were treated with a concentration of 10 µM cisplatin for 36 h; apoptosis was detected.
Protein preparation & western blotting
Cells were lysed in icecold Tris buffer (20 mmol/l Tris; pH 7.5) containing 137 mmol/l NaCl, 2 mmol/l ethylenediaminetetraacetic acid, 1% Triton X, 10% glycerol, 50 mmol/l NaF, 1 mmol/l DTT and a protease inhibitor cocktail (Roche Applied Science, IN, USA). Protein concentrations were quantified using the BCA protein assay kit (CWBIO, Beijing, China). The protein lysates were then separated by SDS-PAGE and electroblotted onto polyvinylidene fluoride (PVDF) membranes (Hybond-P). After blocking with 5% nonfat milk and 0.1% Tween-20 in tris buffered saline (TBS), the membranes were incubated with antibodies.
The antibodies for immunoblot analysis were as follows: SLFN11 (1:200, Santa Cruz Biotechnology, CA, USA), cyclin D1 (1:500, Proteintech, IL, USA), cyclin E1 (1:500, Proteintech, IL, USA) and β-actin (1:1000 Beyotime Biotech, Jiangsu, China). β-actin antibody was used as a loading control. The blots were visualized using enhanced chemiluminescence (Pierce Bioscience, IL, USA).
SLFN11 unexpressed & re-expressed DLD1 cell xenograft mouse model
Animal experiments were reviewed and approved by the Chinese PLA General Hospital Experimental Animal Committee. SLFN11 unexpressed and SLFN11 stably expressed DLD1 cells were injected subcutaneously into the dorsal left side of 4-week-old male Balb/c nude mice (n = 10) weighing 12–18 g. Tumor volumes were measured every 4 days for 20 days starting 4 days after implantation. Tumor volumes were calculated according to the formula: V = L × W2/2, where V represents the volume (mm3), L represents largest length (mm) and W represents width (mm), respectively. Mice were sacrificed after 20 days.
Statistical analysis
Statistical analysis was performed using SPSS 17.0 software (SPSS, IL, USA). Either χ2 or Fisher’s exact tests were used to evaluate the relationship between methylation status and clinicopathological characteristics. The two-tailed independent samples’ t-test was applied to determine the statistical significance of the differences between the two experimental groups. Survival rates were calculated by the Kaplan–Meier method, and differences in survival curve were evaluated using the log-rank test. Cox proportional hazards models were fit to determine independent associations of SLFN11 methylation with OS and relapse-free survival (RFS) outcome. Two-sided tests were used to determine significance, and p < 0.05 was considered statistically significant.
Results
The expression of SLFN11 is regulated by promoter region methylation in CRC
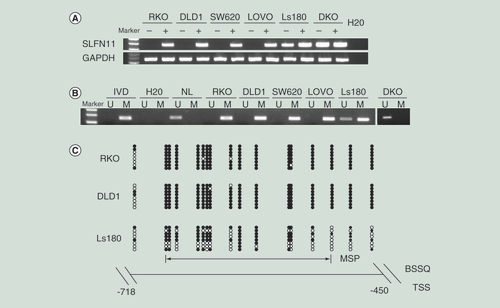
The expression levels of SLFN11 were examined by semiquantitative reverse transcription PCR (RT-PCR) to explore the regulation of SLFN11 in CRC. SLFN11 was expressed in Ls180 and DKO cells and unexpressed in RKO, DLD1, SW620 and LOVO cells (A). Complete methylation was observed in RKO, DLD1, SW620 and LOVO cells; partial methylation was detected in Ls180; and unmethylation was found in DKO cells (B). MSP results were validated by BSSQ in RKO, DLD1 and Ls180 cells (C). Loss of/reduced expression of SLFN11 was correlated with promoter region methylation. Upon treatment with 5-AZA, a demethylating agent, restoration of SLFN11 expression was observed in RKO, DLD1, SW620 and LOVO cells, and increased expression was detected in Ls180 (A). No expression changes were detected in DKO cells. These results suggest that the expression of SLFN11 is regulated by promoter region methylation.
(A) RT-PCR shows the levels of SLFN11 expression – (-) untreated; (+) 5-AZA treated; H2O: double distilled water. GAPDH was used as internal control. (B) MSP results of SLFN11 in colorectal cancer cell lines. (C) Bisulfite sequencing results: the double-headed arrow indicates the region of the MSP product.
5-AZA: 5-Aza-2′-deoxycytidine; Filled circles: Methylated CpG sites; IVD: In vitro methylated DNA; M: Methylated alleles; NL: Normal lymphocyte DNA; Open circles: Unmethylated CpG sites; RT: Reverse transcription; TSS: transcription start site; U: Unmethylated alleles.
SLFN11 is frequently methylated in human CRC
The methylation status of SLFN11 was detected by MSP (A). SLFN11 was methylated in 55.47% (71/128) of CRC patient samples. No methylation was found in five cases of noncancerous colorectal mucosa. Methylation of SLFN11 was significantly associated with age, poor 5-year OS and 5-year RFS (all p < 0.05; B & ), while no association was found between SLFN11 methylation and gender, tumor differentiation, lymphatic metastasis, location, stage, size (all p > 0.05, ). Among 128 cases of CRC, KRAS mutation was found in 29 cases (22.66%), and BRAF mutation in 3 cases (2.34%). No association was found between SLFN11 methylation and KRAS or BRAF mutation (all p > 0.05, ). Among 128 cases of CRC, CIMP (+) was detected in 38 cases (29.69%) and CIMP (-) in 90 cases (70.31%). No association was found between SLFN11 methylation and CIMP (+) in CRC (p > 0.05, ).
(A) Representative MSP results of SLFN11 in normal colorectal tissues (N1, N2, N3, N4 and N5) and primary CRC tissues. (B)SLFN11 methylation status is associated with RFS and OS of CRC patients.
CRC: Colorectal cancer; OS: Overall survival; RFS: Relapse-free survival.
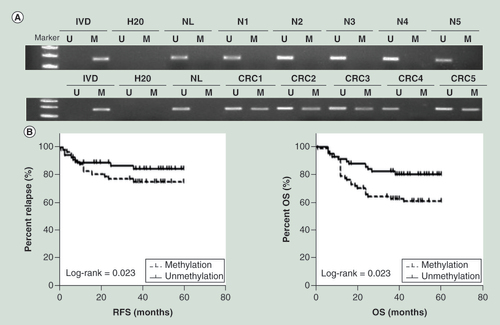
Cox proportional hazards model analysis indicated that SLFN11 methylation was an independent prognostic factor for OS and RFS after adjusting for tumor differentiation and N stage (both p < 0.05, ).
SLFN11 suppresses CRC cell proliferation
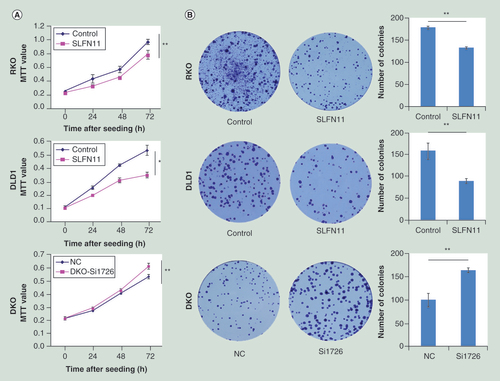
SLFN11 stably expressing cells were established by transfecting PCDH-SLFN11 expression vector into SLFN11 unexpressed RKO and DLD1 cells. MTT and colony formation assays were employed to evaluate the effects of SLFN11 on cell proliferation. The optical delnsity (OD) values were 0.533 ± 0.037 versus 0.349 ± 0.019 (p < 0.05) in DLD1 cells and 0.968 ± 0.037 versus 0.782 ± 0.065 (p < 0.01) in RKO cells before and after restoration of SLFN11 expression (A). Re-expression of SLFN11 significantly suppressed colony formation in RKO and DLD1 cells (B; Supplementary Figure 1). The effects of SLFN11 on cell cycle were analyzed by flow cytometry. The cell phase distributions before and after re-expression of SLFN11 in DLD1 cells were as follows: G0/G1 phase: 45.93 ± 2.05% versus 50.23 ± 0.95%, S phase 17.51 ± 0.24% versus 14.72 ± 0.59% and G2/M phase: 36.55 ± 1.84% versus 35.05 ± 0.57%. In RKO cells, the cell phase distributions before and after re-expression of SLFN11 were as follows: G0/G1 phase: 35.57 ± 0.82% versus 38.66 ± 0.85%, S phase 14.77 ± 0.98% versus 13.62 ± 1.12; and G2/M phase: 49.66 ± 0.75% versus 47.72 ± 0.29%. The number of cells in G1/G0 phase increased significantly after re-expression of SLFN11 in DLD1 and RKO cells (both p < 0.05, C). To further validate these results, siRNA knockdown technique was employed. The OD values were 0.591 ± 0.015 versus 0.677 ± 0.022 before and after knockdown of SLFN11 in DKO cells (p < 0.05, A). Knockdown of SLFN11 significantly promotes colony formation in DKO cells (B; Supplementary Figure 1). The cell phase distributions before and after knockdown of SLFN11 were as follows: G0/G1 phase 37.36 ± 0.98% versus 34.36 ± 0.43%, S phase 33.9 ± 0.61% versus 31.41 ± 0.05%, and G2/M phase 28.74 ± 0.91% versus 34.23 ± 0.44%. The number of cells in G0/G1 phase was significantly reduced after knockdown of SLFN11 in DKO cells (p < 0.05, C).
(A) The effect of SLFN11 on cell proliferation was measured by the MTT assay for 72 h – SLFN11: SFLN11 re-expressed CRC cells. Control: empty vector. *p < 0.05, **p < 0.01. (B) The effects of SLFN11 on colony formation in colorectal cells; **p < 0.01. (C) Cell phase distribution in SLFN11 re-expressed cells (RKO and DLD1) and the effects of knockdown of SLFN11 (DKO) on cell cycle. *p < 0.05, **p < 0.01. (D) SLFN11, cyclin D1 and cyclin E1 were detected by western blot in SLFN11 re-expressed (RKO and DLD1) cells and control cells. (E) SLFN11, cyclin D1 and cyclin E1 were detected by western blot in SLFN11 knockdown DKO cells and control cells.
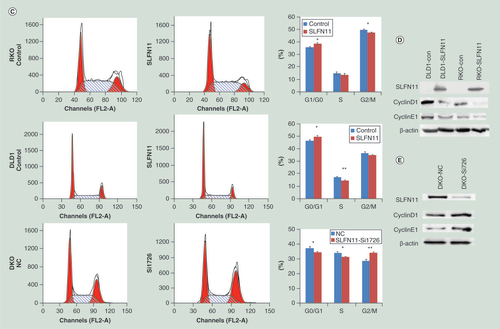
To further validate the effects of SLFN11 on cell cycle, the levels of cyclin D1 and cyclin E1 were examined by western blot in DLD1 and RKO cells before and after re-expression of SLFN11. The expression levels of cyclin D1 and cyclin E1 were reduced after re-expression of SLFN11 in RKO and DLD1 (D), while the expression levels of cyclin D1 and cyclin E1 were increased after knockdown of SLFN11 in DKO (E). These results further suggest that SLFN11 inhibits cell proliferation in CRC.
SLFN11 sensitized CRC cells to cisplatin
The expression of SLFN11 has been reported to sensitize ovarian cancer to cisplatin-containing regimens [Citation13]. To explore the sensitizing effects of SLFN11 on CRC cells, the MTT assay was employed. The IC50 values of cisplatin were 21.77 ± 0.46 versus 9.99 ± 1.12 µM, 6.23 ± 0.77 versus 2.59 ± 0.28 µM and 17.56 ± 0.30 versus 13.19 ± 1.48 µM before and after re-expression of SLFN11 in RKO, DLD1 and SW620 cells, respectively. The IC50 values were significantly reduced after re-expression of SLFN11 (all p < 0.05, A). The IC50 values were 14.19 ± 0.21 versus 24.20 ± 2.51 µM in DKO cell before and after knockdown of SLFN11. The IC50 value was significantly increased after knockdown of SLFN11 (all p < 0.05, A). These results suggest that SLFN11 sensitizes CRC cells to cisplatin.
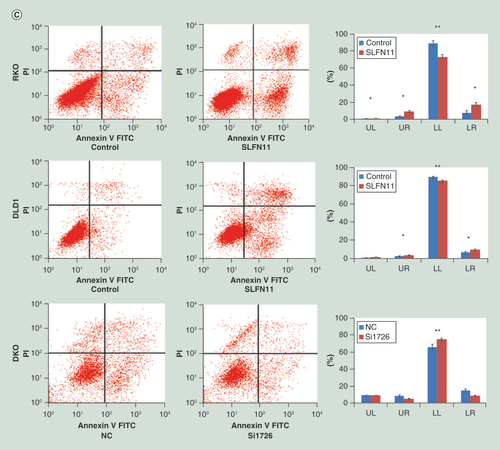
(A) Cell viability determined by MTT assays following exposure to cisplatin. (B) Colony formation under treatment with 2 µM of cisplatin in RKO, DLD1 and SW620 cells before and after re-expression of SLFN11, and in DKO cells before and after knockdown of SLFN11. (C) Apoptosis was detected and quantified by annexin V-FITC and propidium iodide staining.
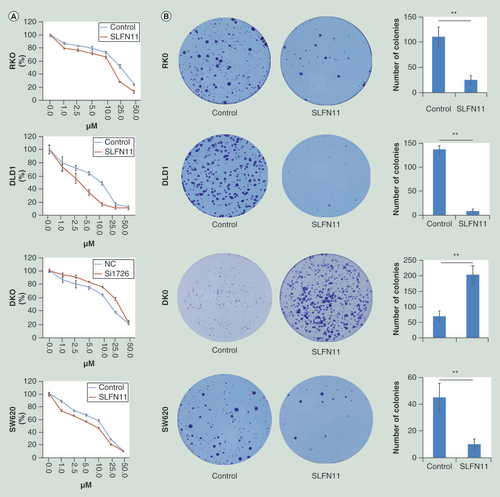
Under treatment with 2 µM of cisplatin, the number of colonies was 111 ± 20 versus 26.33 ± 8.14, 137.67 ± 8.02 versus 9.33 ± 3.51 and 47.50 ± 10.61 versus 10.00 ± 4.00 in RKO, DLD1 and SW620 cells, respectively, before and after re-expression of SLFN11. The colony numbers were reduced significantly after re-expression of SLFN11 in RKO, DLD1 and SW620 cells (all p < 0.01, B). Upon treatment with 2 µM of cisplatin, the number of colonies was 71.33 ± 15.95 versus 204.66 ± 28.15 in DKO cells before and after knockdown of SLFN11. The colony numbers were increased significantly after knockdown of SLFN11 in DKO cells (p < 0.01, B). These results further demonstrate that SLFN11 sensitizes CRC cells to cisplatin.
To further validate these results, the effects of SLFN11 on apoptosis were analyzed upon cisplatin treatment. Before and after re-expression of SLFN11 in RKO and DLD1 cells upon treatment with 10 µM of cisplatin, the percentages of early apoptosis in lower right quadrant (LR) cells were 7.26 ± 2.2% versus 16.82 ± 1.81% (p < 0.01) and 6.85 ± 0.87% versus 9.68 ± 0.64%; the percentages of necrosis/late apoptosis in upper left quadrant (UL) and upper right quadrant (UR) cells were 3.97 ± 0.96% versus 10.15 ± 1.61% and 3.89 ± 0.05% versus 5.17 ± 0.63%, respectively (p < 0.05) (C). In DKO cells before and knockdown of SLFN11, the percentages of early apoptotic cells (LR) were 15.22 ± 2.36% versus 9.37 ± 0.49% (p < 0.05), and necrosis/late apoptosis (UL + UR) cells were 18.76 ± 1.25% versus 15.09 ± 0.69% (p > 0.05), respectively (C).
These results demonstrate that the amount of apoptosis induced by cisplatin treatment significantly increased after re-expression of SLFN11 and significantly decreased after knockdown of SLFN11. These results further suggest that SLFN11 sensitizes CRC cells to cisplatin.
SLFN11 suppresses human CRC cell xenograft growth in mice
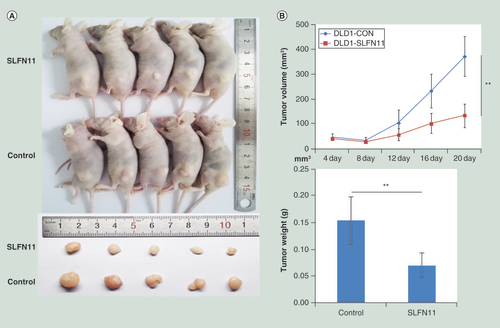
To further investigate the effects of SLFN11 on CRC, SLFN11 unexpressed and re-expressed DLD1 cell xenograft mouse models were employed (A). The tumor volumes were 372.83 ± 79.59 versus 133.11 ± 46.41 mm3 in SLFN11 unexpressed and re-expressed DLD1 cell xenografts (p < 0.01; B). The tumor weights were 0.15 ± 0.04 versus 0.07 ± 0.02 g in SLFN11 unexpressed and re-expressed DLD1 cell xenografts (p < 0.01; B).
(A) Results of SLFN11 re-expressed and unexpressed DLD1 cell xenografts in mice – top: SLFN11 re-expressed colorectal cells group; Bottom: control group. (B) Tumor growth curves and average weights of SLFN11 re-expressed and unexpressed DLD1 cell xenografts.
**p < 0.01.
Discussion
In humans, there are only five SLFN members, SLFN12 is located in cytoplasm and the remaining members (SLFN5, SLFN11, SLFN13 and SLFN14) are localized to the nucleus [Citation10]. The function of human SLFN has not been extensively studied. Previous proteomic studies pointed to SLFN11 as a putative interactor of several key proteins of the DNA damage cell response system, such as replication proteins RPA1, RPA2 and RPA3 or BRCA1-associated Ring Domain protein [Citation14]. Nogales et al. found that SLFN11 methylation is a predictive biomarker of platinum resistance in ovarian and non-small-cell lung cancer by analyzing the DNA methylome database [Citation14]. In another study, the authors found that SLFN11 sensitizes CRC cells to irinotecan [Citation22]. Our study demonstrated that SLFN11 is methylated in 55.47% of human CRC samples, and the expression of SLFN11 is regulated by promoter region methylation. The results suggest that methylation of SLFN11 is involved in human CRC development and is a potential CRC detection marker. SLFN11 suppressed CRC growth both in vitro and in vivo. Methylation of SLFN11 may serve as a potential therapeutic target in CRC. Methylation of SLFN11 was significantly associated with age, poor 5-year OS and 5-year RFS, suggesting that SLFN11 methylation is a poor prognostic marker in CRC.
Defects of the DNA damage repair system can contribute to tumorigenesis and progression [Citation23]. On the other hand, defects of DNA damage repair pathways may sensitize cancer cells to chemotherapy [Citation24,Citation25]. Human SLFN11 interacts with RPA and negatively regulates the DNA damage response by inhibiting checkpoint maintenance and homologous recombination repair [Citation26]. Our study demonstrated that SLFN11 suppresses CRC cell proliferation by inducing G1/S arrest. In addition, SLFN11 sensitizes CRC cells to cisplatin. Thus, SLFN11 methylation may serve as a cisplatin insensitive marker. However, the chemotherapeutic regimens and other therapies were different in this population, it limited further analysis the association between SLFN11 methylation and each chemical agent. Because there is no reliable antibody available for SLFN11, we were not able to perform immunohistochemistry staining in primary CRC.
In conclusion, SLFN11 is frequently methylated in human CRC, and the expression of SLFN11 is regulated by promoter region methylation. Methylation of SLFN11 is associated with age, poor 5-year OS and 5-year RFS. SLFN11 suppresses CRC growth both in vitro and in vivo and SLFN11 sensitizes CRC cells to cisplatin.
Future perspective
SLFN11 is a candidate tumor suppressor in CRC. Methylation of SLFN11 may be a frequent event in CRC and is a potential detection and prognostic marker in CRC. Methylation of SLFN11 may serve as a cisplatin-resistant marker in CRC.
Table 1. Primer sequences and PCR conditions.
Table 2. Clinic-pathological features and SLFN11 methylation status in colorectal cancer patients.
Table 3. Univariate and multivariate analysis of SLFN11 methylation status with relapse-free survival or overall survival in colorectal cancer patients.
SLFN11 is frequently methylated in human colorectal cancer (CRC).
The expression of SLFN11 is regulated by promoter region methylation.
Methylation of SLFN11 is associated with age, poor 5-year overall survival and 5-year relapse-free survival.
SLFN11 suppresses CRC growth both in vitro and in vivo.
Methylation of SLFN11 reduced the sensitivity of CRC cells to cisplatin.
Supplemental Figure 1
Download TIFF Image (2 MB)Supplementary data
Financial & competing interests disclosure
This work was supported by the following grants: National Basic Research Program of China (973 Program No. 2012CB934002); National Key research and development Programme (Programme No. 2016YFC1303600); National Key Scientific Instrument Special Programme of China (Grant No. 2011YQ03013405); National Science Foundation of China (NSFC Nos. 81672318 81402345, U1604281, 8167100001, 81230047 and 81490753); Beijing Science Foundation of China (BJSFC No. 7171008). JG Herman is a consultant to MDxHealth. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Ferlay J , SoerjomataramI , DikshitRet al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012 . Int. J. Cancer136 ( 5 ), E359 – E386 ( 2015 ).
- Center MM , JemalA , SmithRA , WardE . Worldwide variations in colorectal cancer . CA Cancer J. Clin.59 ( 6 ), 366 – 378 ( 2009 ).
- Armaghany T , WilsonJD . ChuQet al. Geneticalterations in colorectal cancer . Gastrointestinal cancer research.5 ( 1 ), 19 – 27 ( 2012 ).
- Grady WM , CarethersJM . Genomic and epigenetic instability in colorectal cancer pathogenesis . Gastroenterology135 ( 4 ), 1079 – 1099 ( 2008 ).
- Nyberg KA , MichelsonRJ , CWP , WeinertTA . Toward maintaining the genome: DNA damage and replication checkpoints . Annu. Rev. Genet.36 ( 1 ), 617 ( 2002 ).
- Jasperson KW , TuohyTM , NeklasonDW , BurtRW . Hereditary and familial colon cancer . Gastroenterology138 ( 6 ), 2044 – 2058 ( 2010 ).
- Gao D , HermanJG , GuoM . The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer . Oncotarget7 ( 24 ), 37331 – 37346 ( 2016 ).
- Schwarz DA , KatayamaCD , HedrickSM . Schlafen, a new family of growth regulatory genes that affect thymocyte development . Immunity9 ( 5 ), 657 – 668 ( 1998 ).
- Bustos O , NaikS , AyersGet al. Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence . Gene447 ( 1 ), 1 – 11 ( 2009 ).
- Mavrommatis E , FishEN , PlataniasLC . The Schlafen family of proteins and their regulation by interferons . J. Interferon Cytokine Res.33 ( 4 ), 206 – 210 ( 2013 ).
- De La Casa-Esperon E . From mammals to viruses: the Schlafen genes in developmental, proliferative and immune processes . Biomol. Concepts2 ( 3 ), 159 – 169 ( 2011 ).
- The Cancer Genome ATLAS . http://cancergenome.nih.gov .
- Zoppoli G , RegairazM , LeoEet al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents . Proc. Natl Acad. Sci. USA109 ( 37 ), 15030 – 15035 ( 2012 ).
- Nogales V , ReinholdWC , VarmaSet al. Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs . Oncotarget7 ( 3 ), 3084 – 3097 ( 2016 ).
- Flejou JF . [WHO Classification of digestive tumors: the fourth edition] . Ann. Pathol.31 ( Suppl. 5 ), S27 – S31 ( 2011 ).
- Morandi L . WO2011104695 A2 ( 2011 ).
- Samowitz WS , SweeneyC , HerrickJet al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers . Cancer Res.65 ( 14 ), 6063 ( 2005 ).
- Herman JG , GraffJR , MyöhänenS , NelkinBD , BaylinSB . Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands . Proc. Natl Acad. Sci. USA93 ( 18 ), 9821 – 9826 ( 1996 ).
- Ogino S , KawasakiT , KirknerGJ , KraftP , LodaM , FuchsCS . Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample . J. Mol. Diagn.9 ( 3 ), 305 – 314 ( 2007 ).
- Basile KJ , AbelEV , DadpeyN , HartsoughEJ , FortinaP , AplinAE . In vivo MAPK reporting reveals the heterogeneity in tumoral selection of resistance to RAF inhibitors . Cancer Res.73 ( 23 ), 7101 – 7110 ( 2013 ).
- Zhang X , YueP , FletcherS , ZhaoW , GunningPT , TurksonJ . A novel small-molecule disrupts Stat3 SH2 domain–phosphotyrosine interactions and Stat3-dependent tumor processes . Biochem. Pharmacol.79 ( 10 ), 1398 ( 2010 ).
- Tian L , SongS , LiuXet al. Schlafen-11 platinum drugssensitizes colorectal carcinoma cells toirinotecan . Anticancer Drugs25 ( 10 ), 1175 – 1181 ( 2014 ).
- Bartkova J , HorejsíZ , KoedKet al. DNA irinotecan damage response as a candidate anti-cancer barrier in early human tumorigenesis . Nature434 ( 7035 ), 864 – 870 ( 2005 ).
- Rottenberg S , JaspersJE , KersbergenAet al. High sensitivity tumorigenesis of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs . Proc. Natl Acad. Sci.105 ( 44 ), 17079 – 17084 ( 2008 ).
- Zaanan A , Cuilliere-DartiguesP , GuillouxAet al. Impactof p53 expression and microsatellite instability on stage III colon cancerdisease-free survival in patients treated by 5-fluorouracil and leucovorin withor without oxaliplatin . Annals of oncology: official journal of the European Society for Medical Oncology/ESMO21 ( 4 ), 772 – 780 ( 2010 ).
- Mu Y , LouJ , SrivastavaMet al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair . EMBO Rep.17 ( 1 ), 94 – 109 ( 2016 ).