
Abstract
Aim: We hypothesized that cross-generational effects of alcohol exposure could alter DNA methylation and expression of the HRAS oncogene and TP53 tumor suppressor gene that drive cancer development. Methods: DNA methylation of the HRAS and TP53 genes was tested in samples from young participants (Mean age of 13.4 years). Results: Controlling for both personal use and maternal use of substances during pregnancy, familial alcohol dependence was associated with hypomethylation of CpG sites in the HRAS promoter region and hypermethylation of the TP53 gene. Conclusion: The results suggest that ancestral exposure to alcohol can have enduring effects that impact epigenetic processes such as DNA methylation that controls expression of genes that drive cancer development such as HRAS and TP53.
Keywords::
First draft submitted: 11 April 2017; Accepted for publication: 19 June 2017; Published online: 11 August 2017
Alcohol use is responsible for about 4% of all deaths worldwide [Citation1]. A portion of this increased incidence of all-cause mortality is due to head and neck cancers, particularly of the pharynx, larynx, oral cavity and esophagus as well as the liver [Citation2]. Alcohol abuse and dependence are frequently seen in individuals diagnosed with Head and Neck Squamous Cell Carcinoma [Citation3]. Moreover, prospective epidemiological data show that alcohol use has a significant dose–response relationship with increased hazard ratios for developing cancer starting at three drinks per day [Citation4]. Consumption of alcohol appears to have a direct effect on gastrointestinal tissue promoting the development of malignancy. The incidence of the head and neck cancers is in direct proportion to the dilution of alcohol in the gut so that mouth cancers are most common followed by esophageal cancers with the least incidence occurring in stomach and intestinal cancers [Citation5].
Integrated approaches to understanding cancer risk appear to require analysis of genetic and epigenetic factors along with gene expression profiles to address the complexity of cancer etiology [Citation6]. In addition to the direct effect of alcohol on tissues exposed to alcohol, the potential exists for alcohol to change the methylation of oncogenes and tumor suppressor genes involved in tumor development. Defining the factors that contribute to this increased risk for these cancers is critical to the ultimate goal of reducing cancer development.
A diverse set of environmental conditions affect DNA methylation [Citation7–9] with the potential for altering gene expression [Citation10]. Alcohol use and smoking can produce altered methylation in humans [Citation11,Citation12]. Cross generational effects have been reported with famine experienced by parents affecting medical conditions and longevity in offspring [Citation13,Citation14]. Although not previously reported in humans, animal studies have shown that ethanol administration can influence DNA methylation through the germline in offspring exposed either through fetal exposure [Citation15] or as a result of paternal exposure [Citation16,Citation17]; changes with the potential to alter every somatic cell in the body across generations. Determining if alcohol consumption in the preconception period in parents influences genes involved in cancer risk in offspring is critical to improvement in prevention initiatives.
DNA methylation of several genes involved in cell cycle control has been associated with the risk for developing head and neck squamous cell cancer [Citation18], in breast cancer where changes in peripheral blood have been reported years before breast cancer is diagnosed [Citation19], in urocystic tumorgenesis [Citation20], in non-polyposis colorectal cancer [Citation21] and in germline epimutation of MLH1 in individuals with multiple cancers [Citation22].
In addition to personal exposure to alcohol, parental or even grandparental behaviors may influence the methylation of genes which can influence the expression of genes involved in tumor promotion (oncogenes) or suppression. Establishing a connection between DNA methylation and its potential for development of a malignancy would ideally require an in depth history of alcohol and tobacco exposure in those in whom methylation effects are studied, and knowledge of alcohol and tobacco use in their first and second degree relatives in order to examine potential germ line effects. Obtaining histories for their mothers’ use of alcohol and tobacco during pregnancy is also necessary to rule out direct effects on the fetus and to establish that cross-generation methylation effects occur.
Two of the most important genes in the development of cancer are the Ras family of oncogenes and the TP53 tumor suppressor gene. HRAS is one of the oncogenes belonging to the Ras family that is involved in cell proliferation and survival which can lead to neoplastic formation. Abnormal function of HRAS has been shown to cause tumor growth [Citation23]. Moreover, mutated HRAS genes are found in a wide variety of cancers [Citation24]. Because HRAS has been an important marker for diagnosis and prognosis in human cancers, it was chosen as a prototypic oncogene for study of the potential relationship between alcohol dependence diagnoses in parents and grandparents and methylation effects in offspring. TP53 is a transcription factor that responds to stress signals leading to multiple cellular responses including cell cycle arrest, apoptosis and DNA repair [Citation25]. Through signal transduction the HRAS protein relays signals to the nucleus of the cell providing instruction to divide giving the protein the potential to turn normal cells into cancerous ones. Because alteration in the CpG methylation of the oncogene promoter region can induce cancers, the factors promoting hypomethylation of this oncogene is of considerable importance. Additionally, DNA hypermethylation at CpG islands in or near gene promoter regions of the TP53 tumor suppressor gene is associated with reduced expression and gene silencing [Citation25].
Materials & methods
Participants
Participants were from pedigrees acquired through ongoing family studies in which two types of families were recruited, those with a high density of alcohol dependence cases (High-Risk) and those with minimal alcohol dependence within the extended pedigree, as previously described [Citation26]. Third generation members of these families were seen multiple times as part of a longitudinal follow-up that extended from childhood to young-adulthood. Extensive alcohol and other drug use histories were obtained at approximately yearly intervals in childhood/adolescence enabling the determination of lifetime exposure up to the time of the DNA collection for the third generation participants. A total of 503 participants were included; 62 first and second generation individuals () and 441 third generation offspring () for whom DNA samples had been banked at the time of their first clinical visit. All adult participants signed consent forms indicating their willingness to participate in interviews concerning their lifetime history of alcohol and drug use and to provide a blood sample for DNA extraction. Children were asked to give their assent with their parents providing written consent for the child’s participation.
Table 1. First- and second-generation participant characteristics and recruitment source (n = 62).
Table 2. Alcohol dependence and smoking status of second-generation parents of third generation.
Table 3. Number of relatives with alcohol dependence across generations.
Table 4. Number of parents and grandparents with alcohol dependence.
Table 5. Third generation participant characteristics and recruitment source (n = 441).
Table 6. Personal use of substances prior to DNA collection for third generation participants (n = 441).
The focus of the present report is on the third generation high and low-risk (control) offspring first identified in childhood for whom a blood sample for DNA extraction was obtained at an average age of 13.4 years and banked. First and second generation DNA samples were obtained in adulthood and analyzed to determine the effects of long-term use of alcohol in older participants some of whom were alcohol dependent. A typical High-Risk family is illustrated in .
Black squares or circles indicate the presence of an alcohol dependence diagnosis. Gray indicates phenotype unknown. Squares indicate males; circles females.
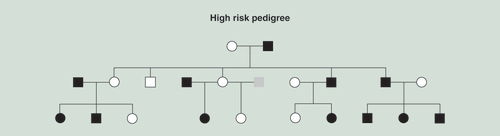
High-risk multiplex families
The high-risk families had been identified through a proband pair of same sex alcohol dependent siblings while one member of the pair was in a substance abuse treatment facility in the Pittsburgh area at the time of recruitment. Probands and all willing first-degree relatives (first generation parents of the probands) and (second generation siblings of the probands), were screened for the presence or absence of alcohol dependence (AD) as previously described [Citation26].
Low-risk control families
Low-risk community control families consisting of either two adult brothers or two adult sisters and their parents were identified through an index case who responded to a newspaper advertisement requesting participants who were interested in a study of heritable aspects of personality and lifestyle [Citation26]. Low-risk families were included if all first- and second-degree relatives of the index case were free of alcohol and other drug dependence. The third generation offspring from these control families along with third generation offspring from multiplex alcohol dependence families were included in the current analyses.
DNA isolation & genotyping
Genomic DNA was utilized from a resource extracted from whole blood or from EBV transformation and cryopreservation.
HRAS & TP53 methylation studies
Using EpiTect Bisulfite kits, DNA was converted and cleaned. HRAS PCR amplification included 45 cycles of thermal cycling following Qiagen guidelines for predesigned primers. For TP53 PCR amplification, 50 cycles of thermal cycling was completed at 95°C for 60 min, 57°C for 60 min, and 72°C for 60 min. The methylation assays were completed using a Biotage PSQ 96MA Pyrosequencer (Biotage AB, Uppsala, Sweden) using PyroMark CpG software (1.0.11) with the assay-specific assay set up file from Qiagen.
SNP genotyping
Genotyping was completed on the Biotage PSQ 96MA Pyrosequencer. Each polymorphism was analyzed by PCR amplification incorporating a biotinylated primer. Thermal cycling included 45 cycles at an annealing temperature of 60°C. The Biotage workstation was used to isolate the biotinylated single strand from the double strand PCR products. The isolated product was then sequenced using the complementary sequencing primer.
HRAS
Amplification was performed on the converted DNA using PyroMark PCR kits and the predesigned HRAS-05_PM PyroMark CpG assay for the HRAS sequence located on Chromosome 11 (BP 534,850–534,831) in the promoter region/transcriptional start site. This assay utilizes the following forward sequence (CpG sites are highlighted in bold): 5′ – TCGGCGGCGCCTAGTACGCA – 3′.
TP53
For the TP53 analysis, a custom primer assay based on published sequences [Citation27,Citation28] was generated by Qiagen. The assay utilized the following forward sequence 5′-CGGGGACACTTTGCGTTCGGGCTGGGAGCGTGCTTTCCA-3′. The biotinylated PCR product was captured on Streptavidin-coated Sepharose beads and the bound single-strand product prepared on a Qiagen workstation. The prepared biotinylated PCR product was released to plates containing the predesigned sequenced primer provided with the Qiagen Pyromark CpG assay. The software provides three suggested levels of confidence in the resulting percent methylation using a color coded system to allow for rejection of samples not passing quality control. All samples tested met the top level of control. In addition, pyrograms of the data were visually inspected to insure that data was of high quality.
RNA isolation & reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR)
Random selection from among lymphocyte samples with methylation data was used to determine the levels of HRAS expression. Extraction was completed using 1 ml Trizol (Ambion, NY, USA) added to lymphocyte pellets and cells which were lysed with a dounce homogenizer prior to phenol-chloroform separation. Samples were further processed using RNA Clean and Concentrator with DNAse I treatment (Zymo Research, CA, USA) and elution into 14 μl nuclease free water. Reverse transcription of RNA was performed with the iScript cDNA Synthesis Kit (BioRad Laboratories, CA, USA) according to manufacturer’s protocol. The cDNA product was diluted 1:10 before qPCR using BioRad SYBR Green Fluorescent Master Mix and a BioRad iCycler. Oligo sequences were: HRAS Forward (F): 5′-TTTGAGGACATCCACCAGTACA-3′ and reverse (R): 5′-GCCGAGATTCCACAGTGC, β-actin (F): 5′-GAGCACAGAGCCTCGCCTTT-3′ and (R): 5′-TCATCATCCATGGTGAGCTGG-3′, and GAPDH (F): 5′-AGGGCTGCTTTTAACTCTGGT-3′ and (R): 5′- ACATGTAAACCATGTAGTTGAGGT. Threshold cycle values for HRAS were normalized within sample to the average of housekeeping genes β-actin and GAPDH (HRAS cycle value – housekeeping gene average cycle value).
Leukocyte HRAS expression was obtained along with two housekeeping genes, Actin and GAPDH. HRAS expression was determined by averaging the count values of the two housekeeping genes, subtracting this value from the counts obtained from the HRAS gene so that lower count values signify increased expression of the HRAS gene.
Quality control
Quality control of the methylation assays included ongoing monitoring provided by Qiagen software. The software provides sequence data for the assay of interest along with a pyrogram showing the peak height of each nucleotide. Signals are flagged by the software as problematic if the signal to noise ratio is too low, dispensation errors occur, or inadequate DNA is present for analysis. Data analysis was performed for only those signals meeting our ‘pass’ criterion determined by the PyroMark CpG software (1.0.11) algorithm and by visual inspection of the pyrograms. In addition, a reliability check for the HRAS primers was performed using samples in duplicate run on the same plate. This analysis of 87 pairs of samples showed a Pearson correlation of 0.77, p < 0.0001.
SNP variation
Two SNPs were genotyped rs11246176 and rs12628. The SNPs were selected on the basis of their proximity to the HRAS methylation sequence tested. The HRAS Qiagen primer set is located on Chromosome 11 (534,795–535,020). Within this interval are four CpG islands between 534,831 and 534,850. SNP rs12628 is located at 534, 242 with rs112461 at 534,916.
Statistical analyses
The percent methylation observed for each subject was transformed using the formula: log (% methylation + 1). Mixed model analyses of variance (SPSS 20) was the primary method of analysis. Where the distribution of methylation values indicated a need to do so, STATA (version 14) tobit regression analysis with censoring was used. All analyses included covariates that could potentially affect DNA methylation including socioeconomic status, age at blood draw, prenatal exposures, and familial relatedness. Each family was assigned a family identification code to identify related individuals. If the effect of each of these variables proved to be non-significant, they were dropped from further analyses. The rationale for including SES was to control for possible effects that lower SES might have on methylation such as poorer nutrition. Familial relatedness was statistically controlled because of its expected influence on allelic variation that may be related to DNA methylation.
Results
Effect of alcohol dependence on HRAS methylation
A total of 62 individuals from generations 1 and 2 () were tested to determine the effect of alcohol dependence diagnosis on HRAS methylation controlling for age at blood draw, sex, familial alcohol dependence risk, and family relatedness. The effect of having an alcohol dependence diagnosis was significant (F = 9.3, df = 1, 58, p = 0.003), as was familial risk variable (high risk vs low risk) (F = 5.90, df = 1, 58, p = 0.018). The effect of sex and age at DNA collection was not significant.
Parental effects on HRAS methylation
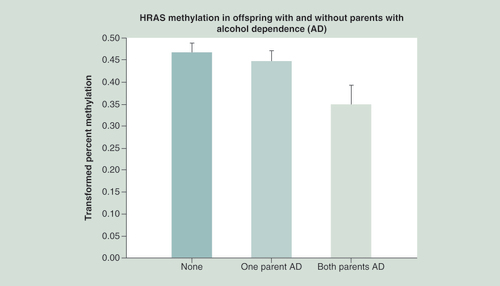
Using methylation data from the third generation subjects, a regression model was constructed that included the number of parents with alcohol dependence (none, one or both), with covariates that included age at blood draw, total days during pregnancy on which the mother used any street drugs, the amount of alcohol consumed during pregnancy and the total number of cigarettes smoked during pregnancy. The model also incorporated the cumulative amount of alcohol, cigarettes and marihuana used by the third generation offspring up to the time of the blood draw at a mean age of 13.4 years.
With 309 observations that included the covariates of interest, the statistical model provided an overall χ2 of 25.21, p = 0.0027 (). In comparison with having no alcohol dependent parent, those with two affected parents were significantly more likely to show a reduction in mean HRAS methylation (t = 2.30, df = 275, p = 0.022), with a significant difference also seen for those having a single parent with alcohol dependence (father or mother) in comparison to none (t = 1.98, df = 386, p = 0.048).
Statistical analysis controlled for the age that DNA was obtained from offspring, prenatal use of substances by mothers (alcohol, drugs and cigarettes), and the personal use by the offspring up to the time that DNA was obtained. Hypomethylation of HRAS in offspring is seen in association with increased loading of parental AD (p = 0.022). This hypomethylation suggests increased expression of the oncogene.
AD: Alcohol dependence.
The sex of the parent (mother or father) with alcohol dependence was tested. In a regression model that included only mothers (294 observations), covarying the mothers prenatal use of substances and the offspring’s personal use up to the time of DNA collection, having a mother with AD in comparison with not having one resulted in a significant effect on methylation (t = 2.43, df = 294, p = 0.016). The mothers’ prenatal use of cigarettes (t = 2.00, df = 294, p = 0.046) and the number of days on which drugs were used during her pregnancy were significant (t = 1.93, df = 294, p = 0.054). Presence of an AD father alone did not influence the offspring’s level of HRAS methylation.
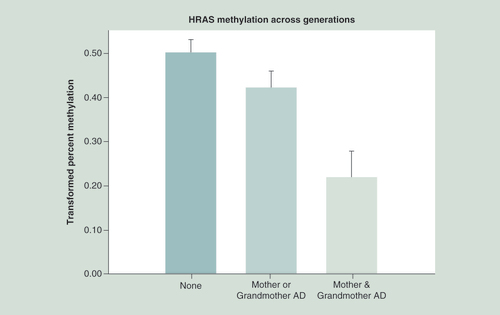
HRAS across two generations effect
A regression model was constructed that included whether the mother was alcohol dependent and whether her mother was also alcohol dependent. The model included prenatal covariates (total alcohol, cigarettes and drug days) and personal use covariates (cumulative alcohol, cigarettes and marihuana prior to DNA collection). This analysis with a total of 188 observations was highly significant (X2 = 27.69, p = 0.0003). In comparison with having neither a mother nor maternal grandmother with alcohol dependence (AD), having both with AD showed a highly significant effect on the offspring’s HRAS mean methylation (t = 4.15, df = 153, p < 0.0001).
Additionally, a graded effect was seen such that having both a mother and grandmother with AD was significantly different than having one AD relative (mother or grandmother) (t = 3.05, df = 102, p = 0.003) (). A similar analysis of 87 fathers and grandfathers found that having both was significantly different from having only a father or grandfather (X2 = 15.93, p = 0.01). The mean methylation values observed for those having no relative (0.462) versus those with either one or both relatives with AD were 0.489 and 0.486, respectively.
Statistical analysis controlled for age at DNA collection, the mothers’ prenatal use of substances and the offspring’s personal use up to the time of DNA collection. Hypomethylation of HRAS CpG sites is seen in association with increased familial AD across generations (p = 0.003).
AD: Alcohol dependence.
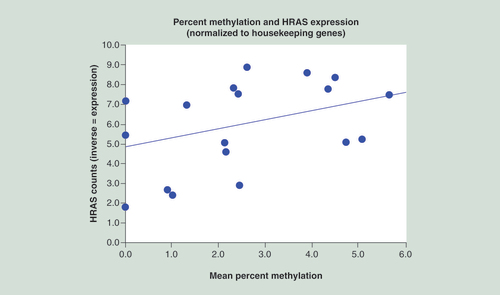
HRAS expression & methylation
A parametric analysis was completed to test the relationship between mean methylation and mRNA levels, a measure of HRAS expression. A trend was seen with a Pearson r of 0.380 (p = 0.109) based on 19 cases (). (A positive correlation is seen because HRAS count values are the inverse of HRAS expression.) A nonparametric Sign test relating to the ordinal values of methylation and expression showed an exact significance of 0.0001.
The parametric relationship showed an r of 0.38 with a marginally significant value, with the ordinal relationship showing good significance (p = 0.001).
Parental effects on TP53 methylation
A regression model was tested that contrasted the number of parents with alcohol dependence (none, one or both), which included all relevant covariates (age at DNA collection, mothers’ use of street drugs, alcohol and cigarettes during pregnancy). The model also incorporated the cumulative amount of alcohol, cigarettes and marihuana used by the offspring up to the time of the DNA collection. The percent methylation observed for each subject was transformed using log (% methylation + 1). Examination of the distribution of the transformed scores indicated that left censoring of the data for statistical analysis was not required.
Analysis of the full model indicated that all of the tested covariates did not significantly influence TP53 methylation. Based on 416 observations, the overall effect of having 0, 1 or 2 parents was significant (F = 6.95, df = 1, 414, p = 0.0087). Additionally, comparison of having one parent with AD versus having no parent with AD was significant (t = 3.15, df = 369, p = 0.002). Based on this result, further analysis was done to determine if both the father and the mother’s AD diagnosis was significantly associated with hypermethylation of the CpG sites tested within the TP53 gene.
Mother versus father
A model was tested in which AD mothers versus mothers without AD were contrasted. A significant effect of the mothers’ AD was seen (F = 6.85, df = 1, 352, p = 0.0092). A significant effect was also found in a model that tested the effect of having an AD father versus the absence of an AD father (F = 6.62, df = 1, 342, p = 0.0105).
TP53 cross-generational effects
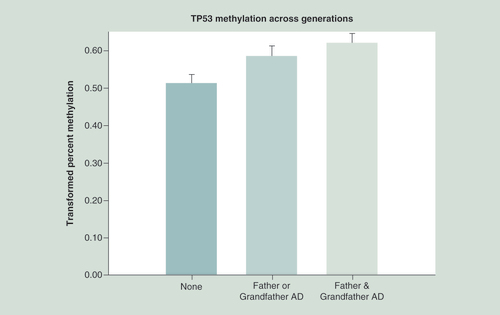
The effect of having a mother, grandmother or both on TP53 methylation of the third generation was tested using a regression model that included the same prenatal and personal use covariates used in the analysis completed to determine the effect of having 0, 1 or 2 parents with alcohol dependence. This multigenerational model with 213 observations was highly significant (F = 4.80, df = 2, 210, p = 0.0091). In comparison with having either a mother or maternal grandmother with AD, versus none we find a highly significant effect on the offspring’s TP53 mean methylation of the CpG sites tested (t = 2.65, df = 197, p = 0.009). A similar model with 110 observations tested whether TP53 methylation of third generation offspring differed between those having either a father or grandfather with AD, having both with AD, or neither with AD. This analysis also revealed a highly significant difference (F = 5.23, df = 2,107, p = 0.0068) ().
TP53 methylation in offspring was compared with those without a father or grandfather with alcohol dependence, with those with either a father or grandfather and those having both. Statistical analysis controlled for age at DNA collection, the mothers’ prenatal use of substances and the offspring’s personal use up to the time of DNA collection. Hypermethylation of CpG sites is seen in association with increased familial AD across generations (p = 0.0068). This hypermethylation suggests lesser expression of the tumor suppressor gene.
AD: Alcohol dependence.
Allele specific methylation (ASM) & HRAS methylation
Allele specific methylation (ASM) is present in varying degrees throughout the genome with complete association between methylation and genotype in some cases or intermediate with either allele associated with the methylation mark in others [Citation29]. In order to test for ASM in this dataset, a mixed model analysis was first performed for all 509 subjects from the three generations to test for the association between rs12628 genotype and percent methylation, covarying personal diagnosis of alcohol dependence, the age of the participant when DNA was obtained, and a family identification variable to correct for within-family relatedness. The same analysis was done using rs11246176. The results showed a significant association for rs12628 with HRAS methylation (F = 3.88, df = 253, p = 0.027). Mean methylation effects were 0.431 for the 11 genotype, 0.600 for the heterozygote 12 group, and 0.438 for those with 22. Results for rs11246176 were not significant. Importantly, analysis of parental AD effects on offspring methylation remained significant when rs12628 was entered into a statistical model that included all relevant covariates (t = 2.17, df = 306, p = 0.30). Similarly, the methylation effect seen in offspring in which those without AD mothers were compared with those with either an AD mother or grandmother versus those with both remained significant when rs12628 was included as a covariate (t = 4.26, df = 185, p < 0.001).
Discussion
Tumorigenesis involves accumulation of mutations in both proto-oncogenes and in tumor-suppressor genes. Human cancers are most frequently impacted by the proto-oncogene Ras [Citation30,Citation31] and the TP53 tumor-suppressor gene (TP53). DNA methylation in the promoter region of the HRAS gene provides one mechanism for gene expression. In general, lower methylation translates into increased gene expression and greater risk for tumor development. Similarly, increased methylation of the TP53 tumor suppressor gene is associated with reduced expression and greater risk of tumor development.
Mutated HRAS genes are seen in a wide variety of cancers [Citation24,Citation32] and are a factor in the expression of cell cycle regulatory proteins that affect disease prognosis in oral cancers [Citation33]. Methylation levels of the HRAS gene have been shown to have clinical relevance in urocystic cancers with lesser methylation associated with presence of tumor in many of the CpG sites tested [Citation20]. The TP53 tumor suppressor protein functions as a transcription factor coordinating key cellular responses to stress signals resulting in DNA damage and hyperproliferation through cell cycle arrest, apoptosis and senescence. TP53 has demonstrated clinical relevance to development of breast tumors [Citation34,Citation35].
In the present study, hypomethylation was seen for CpG sites within the HRAS oncogene in association with increasing numbers of affected parents. The cross-generational parental effect was cumulative; having two parents with AD resulted in greater reduction of methylation than did having one or none. Considering two generations, we find that when both the mother and the maternal grandmother were alcohol dependent greater hypomethylation was seen than with one or none. Similarly, increased hypomethylation was seen when both fathers and grandfathers were alcohol dependent in comparison with either one having AD. These effects were seen even when prenatal exposure to alcohol, cigarettes and drugs used by the mothers and personal use of substances by the third generation offspring were statistically controlled. Our results suggest that cross-generational effects can occur that may render offspring more vulnerable to developing a variety of cancers for which the HRAS is involved. Hypermethylation of the TP53 CpG sites though not enhanced by having two parents versus one, importantly, exhibited cross-generational effects extending to two generations.
Parental behaviors have now been shown to affect methylation of genes with wide-spread effects on the offspring’s health and well-being [Citation7]. Evidence that alcohol and other substance use in parents can affect lifetime risk for cancer development in offspring is currently quite limited. However, the effects of parental alcohol consumption and cigarette smoking have been studied in children with pediatric cancers. In a review of 33 studies of parental behaviors among offspring diagnosed with some form of childhood cancer, ten studies found a statistically significant effect. In seven of these the association was with maternal consumption, either preconceptual or prenatal, and in three it was related to paternal use [Citation36]. Results obtained for the effects of maternal use of alcohol during pregnancy and pediatric cancers have been mixed [Citation37–40]. A meta-analysis of childhood acute myeloid leukemia (AML) and acute lymphocytic leukemia (ALL) reported significantly increased odds for AML (odds ratio = 1.56) but not for ALL in association with maternal use of alcohol during pregnancy [Citation41]. Two large-scale studies [Citation37,Citation38] assessed the effects of maternal drinking during pregnancy on ALL risk finding that alcohol consumption greater than one drink per day was related to incidence of ALL in offspring [Citation38], though the other study did not find a relationship [Citation37]. An association between mothers’ use of alcohol during either the preconception or prenatal period was not seen in another study [Citation40].
All of the studies were observational and did not include the study of epigenetic effects or gene expression. Moreover, all of the studies linking parental behaviors and offspring outcome have assessed pediatric cancers. Due to the length of follow-up needed to make the association with adult onset cancers, there are currently no studies that can shed light on this question. The first step in evaluating such a connection would be to determine if familial alcohol dependence or prenatal use of alcohol or tobacco is associated with changes in DNA methylation of oncogenes or tumor suppressor genes.
Differential methylation of the TP53 has now become a topic of considerable interest [Citation42]. Because TP53 is the most frequently inactivated gene in human cancers, the potential for environmental factors to inactivate this gene is of considerable importance. Current evidence suggests that diet, alcohol consumption and cigarette smoking are associated with TP53 mutations and in turn the development of breast tumors [Citation34,Citation35]. Although personal use of alcohol and cigarettes has been studied with respect to TP53 mutations, methylation studies examining cross-generational effects have not previously been done.
The present results showing hypermethylation of the TP53 gene have implications for risk for developing cancers due to the potential for hypermethylation to decrease expression of this gene. The TP53 gene has been shown to influence the development of cancers through its role in tumor suppression. DNA hypermethylation of the TP53 gene in peripheral blood is a significant independent predictor of worse survival [Citation27]. Germline sequence mutations in tumor suppressor genes are now recognized to cause cancer predisposition syndromes including hereditary nonpolyposis colorectal cancer [Citation43]. The influence of environmental carcinogens has been demonstrated to occur through hypermethylation of the TP53 gene [Citation28].
Two positive features of the present report suggest the importance of our findings. First, the present study utilized a wide variety of covariates in an attempt to control for factors that might explain differences in methylation other than the hypothesized role of parental behaviors on HRAS and TP53 methylation. These included assessment of methylation in young offspring who had very limited exposure to alcohol and drugs so that the effects of these substances on methylation could be largely ruled out. Additionally, use of a family study data in which personal interviews of first and second degree relatives had been conducted to reliably determine presence or absence of alcohol dependence is an important aspect of the study.
Second, the DNA was analyzed using a bisulfite reaction that is a known dependable and sensitive method for studying CpG methylation that can detect the methylation status of a single molecule [Citation44]. Sodium bisulfite treatment of samples allows the unmethylated cytosines to be converted to uracil, leaving the methylated cytosines intact. The percent methylation can then be assessed for the targeted CpG sites of interest.
Although there are many positive aspects of our analysis, there are limitations. These include the fact that only a single HRAS region was analyzed for epigenetic change. Other studies have taken a more comprehensive approach to epigenetic analysis of the HRAS gene with respect to specific cancers. Analysis of 28 CpG sites revealed particular sites to be statistically related to the presence of bladder cancer [Citation20]. Two sites (3 and 28) were significant though sites 7–10 were not [Citation20]. Sites 7 through 10 overlap the sequence tested in the present analysis. However, the present sample was not chosen based on presence of any particular cancer but rather considered to be at higher risk for cancers due to their familial background.
A second potential concern is that the percent methylation obtained was within a narrow range of values with methylation of the HRAS CpG sites showing a range of 0 to 20.97% (90th percentile = 4.71%). Similarly, the TP53 methylation data showed a rather restricted range of 0 to 8.44% (90th percentile = 4.93%). As a result, the values obtained for comparisons were based on small percent methylation. The multigenerational effect of mothers and grandmothers with AD varied from 2.11 to 1.74 to 0.76%. This may be viewed as rather small differences of questionable impact. Although methylation of large change is seen in the case of imprinted genes and in malignant tissue, more subtle processes involving changes of <10% are now being recognized as typical in complex disorders [Citation45]. Several studies now illustrate this point with environmental effects having significant health-related consequences being seen that are accompanied by methylation changes that are subtle, in the range of 1–5% [Citation46–49]. Methylation of the NPSR1 gene, a gene that is associated with severe adult asthma, has shown hypomethylation in the range of 1.4 to 3.29% [Citation46]. Similarly, the Dutch famine of 1944–45 has been shown to effect DNA methylation of the IGF2 gene six decades later at about 5% methylation change [Citation47]. Health consequences of this famine have been documented to include impaired insulin secretion, changes in glucose tolerance, obesity and blood pressure [Citation50–53]. In short, small changes in percent methylation appear to be related to significant clinical effects.
Another possible limitation is the use of peripheral blood derived DNA as a source for determining DNA methylation. Most of the literature that associates methylation changes with development of specific cancers has been based on analysis of tumor samples where a clear relationship to disease can be established. Use of peripheral blood sources for determining active disease is less common. Recent studies suggest that DNA methylation of peripheral blood can be reliably associated with head and neck squamous cell carcinoma [Citation54]. Using receiver operator curves for analysis of the data, these investigators established that after controlling for age, gender, alcohol consumption, smoking and HPV16 serostatus, cases and controls could be distinguished based on methylation of six CpG sites with 85% of samples captured in the area under the curve.
Assessment of a greater number of SNPs within the HRAS gene could provide additional information concerning those who might be at highest risk for developing cancers based on the observation that CpG methylation is highly correlated with DNA sequences [Citation55]. However, we did genotype two SNPs that bracketed the methylation site studied, finding that one of these SNPs showed a significant association with HRAS methylation and suggesting an ASM effect. Importantly, the relationship between number of parents and number of generations with AD on methylation seen in the third generation remained significant when the rs12628 SNP was statistically controlled.
Finally, we recognize that the third generation subject sample that is currently still in their 20′s and 30′s has not entered the period of highest risk for developing cancers and was not assessed for the presence of cancers. Therefore, the link between methylation effects and increased cancer risk could not be demonstrated within this study. Nevertheless, the present results demonstrate that preconception parental behaviors can have significant effects on methylation of genes involved in the development of cancers with significant decreases in methylation of CpG sites within the HRAS oncogene along with significant increases in methylation of CpG sites within the TP53 tumor suppressor gene being observed in offspring of parents and grandparents with alcohol dependence. Identification of cross-generational effects of alcohol dependence on methylation of these genes in offspring is particularly noteworthy.
The present results suggest that individuals with a family history of alcohol dependence may be at higher risk of developing cancers involving the HRAS and TP53 genes. This is especially important in view of the tendency for individuals with a family history of alcohol dependence to develop alcohol dependence at higher rates than those without such a history [Citation26]. With heavier alcohol use having an association with increased risk for developing head and neck cancers, the cross generational effects on HRAS and TP53 methylation may put these individuals at a compounded risk due to their familial background. Future follow-up will be required to determine if these offsprings with hypomethylation of HRAS and or hypermethylation of TP53 will experience a greater incidence of cancers.
The significance of the present work is also underscored by the demonstrated relationship between TP53 mutant proteins and the Ras gene in inducing cancers, first established in early cancer research [Citation56,Citation57] with continuing reports in contemporary research [Citation58]. The importance of the present findings for public health initiatives designed to provide early screening for cancers, particularly those associated with HRAS and TP53, is clear.
Future perspective
Parental behaviors before conception appear to have cross-generational effects on the methylation of genes involved in the development of a variety of cancers. The present study found significant decreases in methylation of CpG sites within the HRAS oncogene along with significant increases in methylation of CpG sites within the TP53 tumor suppressor gene in offspring of parents with alcohol dependence. Public health awareness of the influence that parental preconception behaviors have on offspring needs to be added to those targeting prenatal behaviors of the mother.
Having an alcohol dependence diagnosis is associated with hypomethylation of the HRAS gene, suggesting increased expression of this oncogene.
Prenatal use of cigarettes and drugs by mothers was associated with hypomethylation of the HRAS oncogene.
Cross-generational effects were observed; mothers who were alcohol dependent by lifetime history had offspring who showed hypomethylation of the HRAS oncogene controlling for prenatal exposures.
Having a mother and maternal grandmother with a lifetime diagnosis of alcohol dependence was associated with greater HRAS hypomethylation than having neither or having one only.
Having a father and paternal grandfather with a lifetime diagnosis of alcohol dependence was associated with greater HRAS hypomethylation than having neither or having one only.
The observed effects on HRAS methylation were significantly associated with mRNA levels, a measure of HRAS expression.
Having either no alcohol dependent parent, one parent or both with alcohol dependence was significantly associated with TP53 methylation; with alcohol dependence was associated with hypermethylation of the tumor suppressor gene suggesting reduced expression of TP53 and impaired functioning of tumor suppression.
The number of generations with alcohol dependence by lifetime history was associated with the degree of hypermethylation of the TP53 gene.
Ethical conduct of research
The University of Pittsburgh Institutional Review Board approved this protocol.
Acknowledgements
The authors are grateful for the long-term support of the families who come to their lab multiple times in the course of the longitudinal study of the third generation offspring that made this research possible.
Financial & competing interests disclosure
Funding for this study was provided by NIAAA grants AA018289, AA005909, AA008082, AA015168 and AA021746 to SY Hill. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Rehm J , MathersC , PopovaS , ThavorncharoensapM , TeerawattananonY , PatraJ . Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders . Lancet373 ( 9682 ), 2223 – 2233 ( 2009 ).
- Nelson DE , JarmanDW , RehmJet al. Alcohol-attributable cancer deaths and years of potential life lost in the United States . Am. J. Public Health103 ( 4 ), 641 – 648 ( 2013 ).
- McCaffrey JC , WeitznerM , KamboukasD , HaselhuhnG , LamondeL , Booth-JonesM . Alcoholism, depression, and abnormal cognition in head and neck cancer: a pilot study . Otolaryngol. Head Neck Surg.136 ( 1 ), 92 – 97 ( 2007 ).
- Freedman ND , SchatzkinA , LeitzmannMF , HollenbeckAR , AbnetCC . Alcohol and head and neck cancer risk in a prospective study . Br. J. Cancer96 ( 9 ), 1469 – 1474 ( 2007 ).
- Williams RR , HornJW . Association of cancer sites with tobacco and alcohol consumption and socioeconomic status of patients: interview study from the Third National Cancer Survey . J. Natl Cancer Inst.58 ( 3 ), 525 – 547 ( 1977 ).
- Thingholm LB , AndersenL , MakalicE , SoutheyMC , ThomassenM , HansenLL . Strategies for integrated analysis of genetic, epigenetic, and gene expression variation in cancer: addressing the challenges . Front. Genet.7 , 2 ( 2016 ).
- Jaenisch R , BirdA . Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals . Nat. Genet.33 ( Suppl. ), 245 – 254 ( 2003 ).
- Bird A . Perceptions of epigenetics . Nature447 ( 7143 ), 396 – 398 ( 2007 ).
- Champagne FA . Epigenetic mechanisms and the transgenerational effects of maternal care . Front. Neuroendocrinol.29 ( 3 ), 386 – 397 ( 2008 ).
- van Eijk KR , de JongS , BoksMPet al. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects . BMC Genomics13 , 636 ( 2012 ).
- Philibert RA , PlumeJM , GibbonsFX , BrodyGH , BeachSR . The impact of recent alcohol use on genome wide DNA methylation signatures . Front. Genet.3 , 54 ( 2012 ).
- Monick MM , BeachSR , PlumeJet al. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers . Am. J. Med. Genet. B. Neuropsychiatr. Genet.159B ( 2 ), 141 – 151 ( 2012 ).
- Kaati G , BygrenLO , PembreyM , SjostromM . Transgenerational response to nutrition, early life circumstances and longevity . Eur. J. Hum. Genet.15 ( 7 ), 784 – 790 ( 2007 ).
- Pembrey M , SafferyR , BygrenLO . Network in epigenetic epidemiology: human transgenerational responses to early-life experience: potential impact on development, health and biomedical research . J. Med. Genet.51 ( 9 ), 563 – 572 ( 2014 ).
- Govorko D , BekdashRA , ZhangC , SarkarDK . Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations . Biol. Psychiatry72 ( 5 ), 378 – 388 ( 2012 ).
- Knezovich JG , RamsayM . The effect of preconception paternal alcohol exposure on epigenetic remodeling of the h19 and rasgrf1 imprinting control regions in mouse offspring . Front. Genet.3 , 10 ( 2012 ).
- Finegersh A , HomanicsG . Paternal alcohol exposure reduces alcohol drinking and increases behavioral sensitivity to alcohol selectively in male offspring . PLoS ONE9 ( 6 ), e99078 ( 2014 ).
- Magic Z , SupicG , Brankovic-MagicM , JovicN . DNA methylation in the pathogenesis of head and neck cancer . In : Methylation - From DNA, RNA and Histones to Diseases and Treatment . DricuA ( Ed. ). InTech , Rijeka, HR , 185 – 216 ( 2013 ).
- van Veldhoven K , PolidoroS , BagliettoLet al. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis . Clin. Epigenetics7 , 67 ( 2015 ).
- Sun XF , LiL , LiXJ , ShenW . Methylation pattern of oncogene HRAS gene promoter region and its clinical relevance to urocystic tumorigenesis . Mol. Biol. Rep.39 ( 8 ), 8431 – 8437 ( 2012 ).
- Hitchins MP , WardRL . Constitutional (germline) MLH1 epimutations as an aetiological mechanism for hereditary non-polyposis colorectal cancer . J. Med. Genet.46 ( 12 ), 793 – 802 ( 2009 ).
- Suter CM , MartinDIK , WardRL . Germline epimutation of MLH1 in individuals with multiple cancers . Nat. Genet.36 ( 5 ), 497 – 501 ( 2004 ).
- Goodsell DS . The molecular perspective: the ras oncogene . Oncologist4 ( 3 ), 263 – 264 ( 1999 ).
- Campbell SL , Khosravi-FarR , RossmanKL , ClarkGJ , DerCJ . Increasing complexity of Ras signaling . Oncogene17 ( 11 ), 1395 – 1413 ( 1998 ).
- Vousden KH , LuX . Live or let die: the cell’s response to p53 . Nat. Rev. Cancer2 ( 8 ), 594 – 604 ( 2002 ).
- Hill SY , ShenS , Locke-WellmanJ , MatthewsAG , McDermottM . Psychopathology in offspring from multiplex alcohol dependence families: a prospective study during childhood and adolescence . Psychiatry Res.160 ( 2 ), 155 – 166 ( 2008 ).
- Al-Moundhri MS , Al-NabhaniM , TarantiniL , BaccarelliA , RusieckiJA . The prognostic significance of whole blood global and specific DNA methylation levels in gastric adenocarcinoma . PLoS ONE5 ( 12 ), e15585 ( 2010 ).
- Pavanello S , BollatiV , PesatoriACet al. Global and gene-specific promoter methylation changes are related to anti-B[a]PDE-DNA adduct levels and influence micronuclei levels in polycyclic aromatic hydrocarbon-exposed individuals . Int. J. Cancer125 ( 7 ), 1692 – 1697 ( 2009 ).
- Hutchinson JN , RajT , FagernessJet al. Allele-specific methylation occurs at genetic variants associated with complex disease . PLoS ONE9 ( 6 ), e98464 ( 2014 ).
- Bos JL . ras oncogenes in human cancer: a review . Cancer Res.49 ( 17 ), 4682 – 4689 ( 1989 ).
- Hollstein M , SidranskyD , VogelsteinB , HarrisCC . p53 mutations in human cancers . Science253 ( 5015 ), 49 – 53 ( 1991 ).
- Downward J . Targeting RAS signalling pathways in cancer therapy . Nat. Rev. Cancer3 ( 1 ), 11 – 22 ( 2003 ).
- Sathyan KM , NalinakumariKR , KannanS . H-Ras mutation modulates the expression of major cell cycle regulatory proteins and disease prognosis in oral carcinoma . Mod. Pathol.20 ( 11 ), 1141 – 1148 ( 2007 ).
- Freudenheim JL , BonnerM , KrishnanSet al. Diet and alcohol consumption in relation to p53 mutations in breast tumors . Carcinogenesis25 ( 6 ), 931 – 939 ( 2004 ).
- Conway K , EdmistonSN , CuiLet al. Prevalence and spectrum of p53 mutations associated with smoking in breast cancer . Cancer Res.62 ( 7 ), 1987 – 1995 ( 2002 ).
- Infante-Rivard C , El-ZeinM . Parental alcohol consumption and childhood cancers: a review . J. Toxicol. Environ. Health B. Crit. Rev.10 ( 1–2 ), 101 – 129 ( 2007 ).
- Shu XO , RossJA , PendergrassTW , ReamanGH , LampkinB , RobisonLL . Parental alcohol consumption, cigarette smoking, and risk of infant leukemia: a Childrens Cancer Group study . J. Natl Cancer Inst.88 ( 1 ), 24 – 31 ( 1996 ).
- Menegaux F , RipertM , HémonD , ClavelJ . Maternal alcohol and coffee drinking, parental smoking and childhood leukaemia: a French population-based case-control study . Paediatr. Perinat. Epidemiol.21 ( 4 ), 293 – 299 ( 2007 ).
- Rudant J , MenegauxF , LevergerGet al. Childhood hematopoietic malignancies and parental use of tobacco and alcohol: the ESCALE study (SFCE) . Cancer Causes Control19 ( 10 ), 1277 – 1290 ( 2008 ).
- Milne E , GreenopKR , ScottRJet al. Parental alcohol consumption and risk of childhood acute lymphoblastic leukemia and brain tumors . Cancer Causes Control24 ( 2 ), 391 – 402 ( 2013 ).
- Latino-Martel P , ChanDS , Druesne-PecolloN , BarrandonE , HercbergS , NoratT . Maternal alcohol consumption during pregnancy and risk of childhood leukemia: systematic review and meta-analysis . Cancer Epidemiol. Biomarkers Prev.19 ( 5 ), 1238 – 1260 ( 2010 ).
- Scoumanne A , ChenX . Protein methylation: a new regulator of the p53 tumor suppressor . Histol. Histopathol.23 ( 9 ), 1143 – 1149 ( 2008 ).
- Hesson LB , HitchinsMP , WardRL . Epimutations and cancer predisposition: importance and mechanisms . Curr. Opin. Genet. Dev.20 ( 3 ), 290 – 298 ( 2010 ).
- Varley KE , MitraRD . Bisulfite Patch PCR enables multiplexed sequencing of promoter methylation across cancer samples . Genome Res.20 ( 9 ), 1279 – 1287 ( 2010 ).
- Leenen FA , MullerCP , TurnerJD . DNA methylation: conducting the orchestra from exposure to phenotype?Clin. Epigenetics8 , 92 ( 2016 ).
- Reinius LE , GrefA , SääfAet al. DNA methylation in the Neuropeptide S Receptor 1 (NPSR1) promoter in relation to asthma and environmental factors . PLoS ONE8 ( 1 ), e53877 ( 2013 ).
- Heijmans BT , TobiEW , SteinADet al. Persistent epigenetic differences associated with prenatal exposure to famine in humans . Proc. Natl Acad. Sci. USA105 ( 44 ), 17046 – 17049 ( 2008 ).
- Tobi EW , LumeyLH , TalensRPet al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific . Hum. Mol. Genet.18 ( 21 ), 4046 – 4053 ( 2009 ).
- Liu Y , MurphySK , MurthaAPet al. Depression in pregnancy, infant birth weight and DNA methylation of imprint regulatory elements . Epigenetics7 ( 7 ), 735 – 746 ( 2012 ).
- de Rooij SR , PainterRC , PhillipsDIet al. Impaired insulin secretion after prenatal exposure to the Dutch famine . Diabetes Care29 ( 8 ), 1897 – 1901 ( 2006 ).
- de Rooij SR , PainterRC , RoseboomTJet al. Glucose tolerance at age 58 and the decline of glucose tolerance in comparison with age 50 in people prenatally exposed to the Dutch famine . Diabetologia49 ( 4 ), 637 – 643 ( 2006 ).
- Stein AD , ZybertPA , van der Pal-de BruinK , LumeyLH . Exposure to famine during gestation, size at birth, and blood pressure at age 59 y: evidence from the Dutch Famine . Eur. J. Epidemiol.21 ( 10 ), 759 – 765 ( 2006 ).
- Ravelli AC , van Der MeulenJH , OsmondC , BarkerDJ , BlekerOP . Obesity at the age of 50 y in men and women exposed to famine prenatally . Am. J. Clin. Nutr.70 ( 5 ), 811 – 816 ( 1999 ).
- Langevin SM , KoestlerDC , ChristensenBCet al. Peripheral blood DNA methylation profiles are indicative of head and neck squamous cell carcinoma: an epigenome-wide association study . Epigenetics7 ( 3 ), 291 – 299 ( 2012 ).
- Bock C , PaulsenM , TierlingS , MikeskaT , LengauerT , WalterJ . CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure . PLoS Genet.2 ( 3 ), e26 ( 2006 ).
- Eliyahu D , RazA , GrussP , GivolD , OrenM . Participation of p53 cellular tumour antigen in transformation of normal embryonic cells . Nature312 ( 5995 ), 646 – 649 ( 1984 ).
- Parada LF , LandH , WeinbergRA , WolfD , RotterV . Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation . Nature312 ( 5995 ), 649 – 651 ( 1984 ).
- Solomon H , BuganimY , Kogan-SakinIet al. Various p53 mutant proteins differently regulate the Ras circuit to induce a cancer-related gene signature . J. Cell. Sci.125 ( pt 13 ), 3144 – 3152 ( 2012 ).