
Abstract
Cancer patients are commonly affected by cachexia, a wasting process involving muscle and fat. Specifically, loss of the muscle compartment has been associated with poor prognosis and suboptimal response to therapy. Nutritional support has been ineffective in treating this process leading to investigations into the underlying molecular processes governing muscle catabolism. In this commentary, we discuss the molecular mechanisms of cancer-associated muscle metabolism and the epigenetic processes responsible for the muscle wasting phenotype. Ultimately, we highlight how the epigenome may serve as a promising therapeutic target in reversing cancer-associated muscle catabolism.
First draft submitted: 25 April 2017; Accepted for publication: 16 May 2017; Published online: 25 September 2017
Cancer-associated cachexia is a common comorbidity in patients with advanced forms of cancer [Citation1]. Fundamentally, cachexia is an energy wasting process associated with muscle and adipose tissue loss that has significant clinical implications including increased chemotherapy-related toxicity, chemoresistance and premature mortality [Citation2]. As opposed to total body weight loss, a recent study shows that skeletal muscle mass decline is independently associated with poor prognosis and may be the most relevant clinical variable associated with this complex wasting syndrome [Citation3]. Nutritional support and orexigenic therapies have seen limited clinical efficacy, suggesting that cancer-associated muscle loss is not passively associated with caloric insufficiency, but rather the result of active catabolic processes. Targeting drivers of cancer-associated muscle loss therefore holds significant promise for improving cancer patient survival and quality of life. In this regard, a major barrier to develop novel therapies is our incomplete understanding of the mechanisms underlying cancer-related muscle catabolism. The catabolic phenotype arises from a combination of anabolic growth suppression and enhanced protein breakdown. Unlike malignant tumors, the cancer-associated catabolic state in skeletal muscle is not driven by somatic mutation, but rather arises at least in part, via epigenetic alterations. In this commentary, we discuss the current knowledge of the epigenetic regulation of muscle catabolism and propose that the epigenome represents an attractive target for attenuating or reversing muscle wasting in cancer patients.
Mechanisms of cancer-associated muscle catabolism
Mechanisms of cancer-associated muscle catabolism can be loosely broken down into processes that either affect mature myocytes/myofibers or muscle progenitor cells, termed satellite cells. Catabolism within myocytes occur in response to various extrinsic mediators including pro-inflammatory cytokines such as TNF-α or IL-1, proteolysis inducing factor, as well as TGF-β-related molecules such as myostatin. Pro-inflammatory cytokines exert their actions through their cognate receptors, initiating intracellular signaling cascades including the p38 MAPK and NF-κB pathways [Citation4–9]. Both of these signaling pathways converge to upregulate enzymes that drive muscle protein breakdown. These E3 ubiquitin ligase enzymes: atrogin-1 and MuRF-1 positively regulate protein turnover via the ubiquitin proteasome system and are key cachexic factors across diverse models of muscle atrophy () [Citation10–12].
Muscle fiber atrophy is induced by a number of stressors including tumor/host factors such as TNF-α, IL-1, IL-6, INF-γ and Angiotensin which can induce Pax7, a transcription factor that with persistent expression inhibits muscle stem cell differentiation, through NF-κB mediated regulation. Myostatin and MyoD expression promotes muscle growth and differentiation, respectively. These proteins may be inhibited with sulforophane treatment, a DNA methyltransferase resulting in reduced MyoD expression along with hypoacetylation and repression of that MSTN promoter. TGF-β signaling via SMAD transcription factors result in induction of atrogin-1 and MuRF-1 expression, both ubiquitin ligases that promote proteolysis events. Lastly, induction of DNA deacetylation via p300 or HDAC1 results in persistent enhancer binding protein beta binding to the atrogin-1 promoter resulting in upregulation of protein expression. Muscle fiber atrophy may be hindered through various cellular processes. Micro RNAs, such as miR-206, are involved in post-translational gene silencing of proteins associated with cancer-associated muscle catabolism. In addition, IGF-1/mTOR signaling pathway impedes muscle loss partly by inhibiting FoxO transcription factor, translocation/gene activation preventing atrogin-1/MuRF-1 upregulation. IGF-1 can also stimulate muscle growth/hypertrophy by activating AKT signaling targets that promote protein synthesis, such as mTORC1/p70S6K and MyoD.
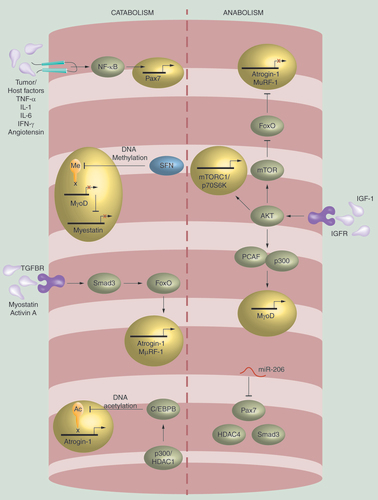
While protein catabolism contributes significantly to muscle wasting, suppression of anabolic muscle growth and regeneration is also a key feature of cancer cachexia. TGF-β family member myostatin, is a key negative regulator of muscle anabolism [Citation13–16]. Myostatin is known to act through the activin receptor of myocytes to phosphorylate and activate transcription factors Smad2 and Smad3 [Citation17]. This leads to the upregulation of atrogin-1 and MuRF-1, although there is conflicting evidence as to whether Smad proteins directly or indirectly upregulate atrogin-1 and MuRF-1 [Citation18–20]. In addition to activation of the Smad/atrogin-1/MuRF-1 signaling cascade, myostatin suppresses PI3K/AKT activity. Normally, PI3K/AKT activation results in phosphorylation of the FoxO family of transcription factors, sequestering them in the cytoplasm. Myostatin relieves this inhibition allowing FoxO to stimulate autophagy, promote expression of atrogin-1/MuRF-1 and upregulate other mediators of muscle catabolism [Citation21–23]. Alternatively, one of the most well characterized signaling molecules promoting anabolic processes is IGF-1. IGF-1 promotes protein synthesis, myofiber hypertrophy and skeletal muscle regeneration and serves to both promote muscle growth and suppress muscle atrophy. IGF-1 stimulates these processes by activating PI3K/AKT signaling resulting in activation of targets promoting protein synthesis, such as mTORC1/p70S6K () [Citation24]. Loss or blockade of IGF-1 therefore results in diminished PI3K/AKT activity promoting muscle loss partly by phosphorylating FoxO. Importantly, the IGF-1 signaling axis is inhibited by several of the aforementioned catabolic pathways including FoxO and Smad2/3.
The catabolic phenotype is also mediated by distinct processes affecting satellite cells. These are the primary ‘building blocks’ of muscle and are indispensable contributors to muscle development and regeneration. During early muscle development, myogenesis arises from primitive progenitor cells expressing Pax3 and Pax7 transcription factors [Citation25]. Late in fetal development, Pax3/Pax7 progenitors migrate to a satellite cell position along the myofiber and are the putative source of adult muscle satellite cells. Adult satellite cells are critical in maintaining muscle homeostasis by balancing self-replication and myocyte differentiation with disruption resulting in muscle wasting. Activation of satellite cells involves upregulation of muscle-specific transcription factors including Myf5, MyoD, MRF4 and myogenin [Citation25]. It is interesting to note that many of the myogenic factors required for embryonic development are also recruited during growth and regeneration of mature skeletal muscle and are dysregulated in cachexia [Citation26]. In addition, C/EBPβ is an important transcription factor expressed in satellite cells. In the context of tumor burden-associated inflammation, its expression is enhanced, conferring resistance to muscle apoptosis with experimental knockdown, abrogating appropriate muscle regeneration in response to injury [Citation27]. Pax7, itself, also contributes to muscle wasting in an NF-κB-dependent manner. While Pax7 is critical for satellite cell self-renewal, its persistent expression results in muscle atrophy by preventing myogenic pathways. In cachexia, extrinsic factors are thought to stimulate NF-κB signaling resulting in persistent Pax7 expression () [Citation29]. Ultimately, myogenic factors are subject to extensive epigenetic regulation, ranging from regulation of myogenic loci by histone modifying enzymes, to post-transcriptional mRNA stability and translational control [Citation29–31]. As such, recent attention has turned to epigenetic pathways as potential therapeutic targets aimed at reversing the wasting phenotype by augmenting muscle regeneration.
Epigenetic control of muscle catabolism
Given the lack of somatic mutations in cachexic skeletal muscle, gene expression alterations observed in wasting syndromes are likely driven by epigenetic mechanisms. Indeed, epigenetics is gaining importance as a key regulatory event above the genome; it interacts with the environment to dictate phenotype via tunable gene expression changes. The reversible nature of epigenetic modifications makes them conceptually attractive therapeutic targets [Citation30]. A deeper understanding of epigenetic modifications in cancer-induced muscle wasting could therefore lead to novel therapeutic approaches to inhibit protein catabolism and stimulate muscle regeneration. The regenerative capacity of satellite cells is heavily influenced by epigenetic factors that impact proliferation, differentiation, commitment and fusion via nucleosome positioning, histone post-translational modifications, miRNAs differential expression and DNA methylation [Citation32].
The many histone biochemical modifications that make up the histone code have been demonstrated to be important in epigenetic regulation in muscle development. Among these modifications, histone acetylation is a key regulator of satellite cell biology. Histone acetyltransferases and deacetylases are critical enzymes affecting a variety of genes involved in muscle wasting by modulating satellite cell activation and differentiation [Citation41]. A recent study of satellite cell activation identified a metabolic switch from fatty acid oxidation to glycolysis, thus decreasing histone deacetylase SIRT1 activity [Citation34]. This switch resulted in increased H4K16 acetylation and initiation of muscle gene expression. In separate studies, the formation of HDAC4/5-MEF2 complexes resulted in repression of Mef2, but upon differentiation, CaMK signaling directed phosphorylation of HDAC and dissociation of MEF2-HDAC4/5 complexes () [Citation37,Citation38,Citation42,Citation43]. Additionally, HDAC can also complex with pRb to induce disruption of MyoD HDAC complexes [Citation44]. Thus, at the initiation of stem cell differentiation, HDAC is regulated through multiple post-translational modifications to inhibit its activity. Interestingly, during exercise, phosphorylation of class II HDACs can relieve Mef2 inhibition, thus leading to transcriptional activation of myosin heavy chain genes and muscle fiber type transformation [Citation45]. In this context, a fiber type II to I switch via HDAC inhibition may result in resistance to muscle atrophy.
As skeletal muscle differentiates and matures, some satellite cells remain in an embryonic state (retaining Pax3 and Pax7 expression) with an ability to self-renew and differentiate. Some epigenetic events associated with differentiation also take place during this growth and regeneration phase. For example, IGF-1 activated AKT1 and AKT2 stimulate the recruitment of acetyltransferases p300 and PCAF to the chromatin of muscle differentiation gene loci, an event initiating transcription of these genes including MyoD in response to regeneration cues () [Citation46].
While muscle damage caused by cancer-associated muscle loss results in activation of satellite cells in an effort to counteract muscle wasting, differentiation often stalls with impaired myoblast fusion from persistent Pax7 expression [Citation33]. Sustained Pax7 expression could prevent proliferating myoblasts from exiting the cell cycle to terminally differentiate and fuse into repairing or newly formed myofibers. Other studies show that myogenic cell differentiation is inhibited due to constant expression of C/EBPβ, a transcription factor in muscle myoblasts that positively regulates atrogin-1 [Citation47,Citation48]. C/EBPβ activation is mediated through interaction with p300 or HDAC1 causing deacetylation and persistent C/EBPβ binding to the atrogin-1 promoter () [Citation48]. Stimulation of C/EBPβ expression in satellite cells and myoblasts within a muscle wasting environment blocks the normal myogenic response [Citation47]. Accordingly, as promiscuous HDAC activity interferes with myogenic differentiation, HDAC inhibitors may prevent cancer-associated atrophy. For example, valproic acid, which has HDAC inhibitor activity, attenuates myopenia by preventing C/EBPβ from regulating atrogin-1 expression, inhibiting myostatin signaling and activating AKT signaling [Citation48]. AR-42, another HDAC inhibitor, preserves skeletal muscle integrity in cachexia by inducing changes in multiple transcriptional programs and maintaining metabolic and expression profiles comparable to those in muscle from tumor-free mice [Citation49].
Another group of cells found to be important in muscle homeostasis is called FAP precursor cells. FAPs are generally quiescent within muscle tissue but, mounting evidence supports a role for FAPs in muscle fiber regeneration by: creating a scaffold that supports muscle repair and releasing cytokines that stimulate satellite cell regeneration [Citation35]. Mouse models show that FAP cells can mediate fibrotic and fat deposition, a phenotype regulated at the epigenetic level by targeting the SWI/SNF chromatin remodeling complex via HDAC activity [Citation29]. Trichostatin A, an HDAC inhibitor can alter the epigenetic landscape of FAPs, promoting muscle regeneration through repression of MyoD and follistatin expression in mouse models () [Citation32,Citation35]. In muscular dystrophy, Trichostatin A redirected FAPs from their fibro/adipogenic fate toward the myogenic lineage via MyoMiR/BAF60c/MyoD/SWI/SNF [Citation30,Citation35]. By extension, HDAC inhibitors could be repurposed to treat cancer-induced muscle loss given similarities in atrophy mechanisms [Citation50]. However, the precise role of individual HDACs need to be explored further for optimal therapeutic use.
In addition to histone modification, methylation of DNA also plays a key role in regulation of gene expression. While little is known about the mechanisms and changes in DNA methylation in cachexic skeletal muscle, several studies have examined DNA methylation during normal satellite cell differentiation. During muscle development, there is a global DNA demethylation trend in satellite cells leading to a stable and unique DNA methylation profile in mature muscle [Citation30,Citation36]. If methylation plays a role in inhibiting normal muscle structure and function, then DNA methyltransferase inhibitors currently in clinical use would be hypothesized to alter the chromatin structure responsible for inhibition of satellite cell differentiation. Indeed, satellite cell differentiation was enhanced with sulforaphane, a DNA methyltransferase inhibitor, which resulted in hypoacetylation of the MSTN promoter, causing repression of the myostatin signaling pathway () [Citation30]. Further investigation comparing DNA methylation patterns of muscle in healthy individuals versus those that have cancer-associated wasting would be informative.
Independent of direct histone and DNA biochemical modifications, noncoding miRNAs are another source of potent epigenetic gene expression regulation. There are several well-characterized miRNAs critical in the regulation of muscle homeostasis. The MyoMiR family is a class of miRNAs including miR-133, miR-208, miR-486 and miR-206 and contribute to post-transcriptional gene silencing by associating with the 3′ untranslated region of target mRNAs important for myogenesis [Citation38,Citation39]. For example, miR-206 inhibits the expression of a number of negative regulators of myogenesis including SMAD3, Pax7 and HDAC4 [Citation37,Citation38,Citation40]. Overexpression of miR-206 in myoblast cells blocks cell cycle progression and promotes differentiation into myotubes leading to regeneration and growth. The effect of miR-206 on the muscle cell phenotype may therefore be relevant to cancer-associated myopenia by virtue of miRNA-mediated targeting of wasting genes such as myostatin () [Citation39].
Conclusion & future perspective
Myopenia is a common feature of advanced tumors and is associated with poor survival. Numerous studies describe the limited therapeutic benefit of nutritional support and orexigenic agents, underscoring a need to better understand molecular underpinnings of cancer-related muscle atrophy. While targeting somatic mutations in cancer is a promising therapeutic strategy, such an approach is not viable for cancer-related myopenia as somatic mutations are not drivers of muscle wasting. The preclinical observations described in this review suggest that epigenetic changes in skeletal muscle would likely serve as effective therapeutic targets in myopenia. Despite the paucity of clinical knowledge regarding molecular and epigenetic determinants of myopenia, targeting the epigenome is a promising strategy to reverse catabolic phenotypes given the known contribution of epigenetic modifications to normal myogenesis processes.
It would be prudent, however, to note that there are several significant obstacles limiting advancement of epigenetic therapies. First, one disadvantage is the relative lack of specificity when targeting the epigenome. While epigenetic drugs have specific protein targets, they effectively alter expression of multiple genetic loci due to the widespread action of these epigenetic regulators. The identification and development of compounds or therapeutic regimens targeting particular genomic loci could therefore improve treatment options. Second, in assessing clinical efficacy, the non-specific nature of these therapeutics may have unintentional effects that are challenging to predict or measure. Unlike the cytotoxic effect chemotherapy regimens, the effect of epigenetic treatments is aimed to modify phenotypes by reprogramming gene expression. Thus, understanding epigenetic regulation of cancer-associated muscle catabolism will aid development of therapeutic strategies designed to prevent or reverse muscle wasting, thereby improving overall survival and quality of life for cancer cachexia patients [Citation30,Citation34,Citation42,Citation46].
Acknowledgements
We would like to apologize to all the investigators whose work could not be cited because of the strict limit on the number of allowed references.
Financial & competing interests disclosure
Our work was supported by NCI CA195473 and NIAMS AR066696. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Walsh D , RybickiL . Symptom clustering in advanced cancer . Support. Care Cancer14 ( 8 ), 831 – 836 ( 2006 ).
- Dewys WD , BeggC , LavinPTet al. Prognostic effect of weight loss prior to chemotherapy in cancer patients . Am. J. Med.69 ( 4 ), 491 – 497 ( 1980 ).
- Martin L , BirdsellL , MacdonaldNet al. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index . J. Clin. Oncol.31 , 1539 – 1547 ( 2013 ).
- Cai D , FrantzJD , TawaNEJret al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice . Cell199 , 285 – 298 ( 2004 ).
- Moore-Carrasco R , BusquetsS , AlmendroVet al. The AP-1/NfkB double inhibitor SP100030 can revert muscle wasting during experimental cancer cachexia . Int. J. Oncol.30 , 1239 – 1245 ( 2007 ).
- Clarke BA , DrujanD , WillisMSet al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle . Cell Metab.6 , 376 – 385 ( 2007 ).
- Cohen S , BraultJJ , GygiSPet al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation . J. Cell Biol.185 , 1083 – 1095 ( 2009 ).
- Csibi A , LeibovitchMP , CornilleKet al. MAFbx/Atrogin-1 controls the activity of the initiation factor eIF3-f in skeletal muscle atrophy by targeting multiple C-terminal lysines . J. Biol. Chem.284 , 4413 – 4421 ( 2009 ).
- Mittal A , BhatnagarS , KumarAet al. The TWEAK, Fa14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice . J. Cell Biol.188 , 833 – 849 ( 2010 ).
- Gomes MD , LeckerSH , JagoeRTet al. Atrogin-1, a muscle specific F-box protein highly expressed during muscle atrophy . Proc. Natl Acad. Sci. USA98 , 14440 – 14445 ( 2001 ).
- Bodine SC , LatresE , BaumhueterSet al. Identification of ubiquitin ligases required for skeletal muscle atrophy . Science294 , 1704 – 1708 ( 2001 ).
- Argiles JM , OrpiM , BusquetsS , Lopez-SorianoFJ . Myostatin, more than just a regulator of muscle mass . Drug Discov. Today17 , 702 – 709 ( 2012 ).
- Clop A , MarcqF , TakedaHet al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep . Nat. Genet.28 , 813 – 818 ( 2006 ).
- Lee SJ , McPherronAC . Reulation of myostatin activity and muscle growth . Proc. Natl Acad. Sci. USA98 , 9306 – 9311 ( 2001 ).
- McPherron AC , LeeSJ . Double muscling in cattle due to mutations in the myostatin gene . Proc. Natl Acad. Sci. USA94 , 12457 – 12461 ( 1997 ).
- Schuelke M , WagnerKR , StolzLEet al. Myostatin mutation associated with gross muscle hypertrophy in a child . N. Engl. J. Med.350 , 2682 – 2688 ( 2004 ).
- Lee SJ , ReedLA , DaviesMVet al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors . Proc. Natl Acad. Sci. USA102 , 18117 – 18122 ( 2005 ).
- Goodman CA , McNallyRM , HoffmannFMet al. Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo . Mol. Endocrinol.27 ( 11 ), 1946 – 1957 ( 2013 ).
- Bollinger LM , WitczakCA , HoumardJAet al. SMAD3 augments FoxO3-induced MuRF-1 promoter activity in a DNA-binding-dependent manner . Am. J. Physiol. Cell Physiol.307 ( 3 ), C278 – C287 ( 2014 ).
- Elkina Y , von HaehlingS , AnkerSDet al. The role of myostatin in muscle wasting: an overview . J. Cachexia Sarcopenia Muscle2 ( 3 ), 143 – 151 ( 2011 ).
- Sandri M , SandriC , GilbertAet al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy . Cell117 , 399 – 412 ( 2004 ).
- Sartori R , MilanG , PatronMet al. Smad2 and 3 transcription factors control muscle mass in adulthood . Am. J. Physiol. Cell Physiol.296 , C1248 – C1257 ( 2009 ).
- Trendelenburg AU , MeyerA , RohnerDet al. Myostatin reduces Akt/TORC1/p70SK signaling, inhibiting myoblast differentiation and myotube size . Am. J. Physiol. Cell Physiol.296 , C1258 – C1270 ( 2009 ).
- Rommel C , BodineSC , ClarkeBAet al. Mediation of IGF-1 induced skeletal myotube hypertrophy by PI(3)K/AKT/mTOR and PI(3)K/AKT/GSK3 pathways . Nat. Cell Biol.3 , 1009 – 1013 ( 2001 ).
- Ryall JG , Dell’OrsoS , DerfoulAet al. The NAD+-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells . Cell Stem Cell16 ( 2 ), 171 – 183 ( 2015 ).
- Faralli H , DilworthFJ . Dystrophic muscle environment induces changes in cell plasticity . Genes Dev.28 ( 8 ), 809 – 811 ( 2014 ).
- Marchildon F , FuD , Lala-TabbertN , Wiper-BergeronN . CCAAT/enhancer binding protein beta protects muscle satellite cells from apoptosis after injury and in cancer cachexia . Cell Death Dis.7 , e2109 ( 2016 ).
- He WA , BerandiE , CardilloVMet al. NF-kB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia . J. Clin. Invest.123 ( 11 ), 4821 – 4835 ( 2013 ).
- Carrió E , MagliA , MuñozMet al. Muscle cell identity requires Pax7-mediated lineage-specific DNA demethylation . BMC Biol.14 , 30 ( 2016 ).
- Fan H , ZhangR , TesfayeDet al. Sulforaphane causes a major epigenetic repression of myostatin in porcine satellite cells . Epigenetics7 ( 12 ), 1379 – 1390 ( 2012 ).
- Moresi V , MarroncelliN , ColettiDet al. Regulation of skeletal muscle development and homeostasis by gene imprinting, histone acetylation and microRNA . Biochim. Biophys. Acta1849 ( 3 ), 309 – 316 ( 2015 ).
- Ma G , WangY , LiYet al. MiR-206, a key modulator of skeletal muscle development and disease . Int. J. Biol. Sci.11 ( 3 ), 345 – 352 ( 2015 ).
- Marchildon F , FuD , Lala-TabbertNet al. CCAAT/enhancer binding protein beta protects muscle satellite cells from apoptosis after injury and in cancer cachexia . Cell Death Dis.7 , e2109 ( 2016 ).
- Liu N , WilliamsAH , MaxeinerJMet al. MicroRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice . J. Clin. Invest.122 ( 6 ), 2054 – 2065 ( 2012 ).
- McKinsey TA , ZhangCL , LuJet al. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation . Nature408 ( 6808 ), 106 – 111 ( 2000 ).
- Puri PL , IezziS , StieglerPet al. Class I histone deacetylases sequentially interact with MyoD and pRB during skeletal myogenesis . Mol. Cell8 ( 4 ), 885 – 897 ( 2000 ).
- Sun R , ZhangS , HuWet al. Valproic acid attenuates skeletal muscle wasting by inhibiting C/EBPβ-regulated Atrogin1 expression in cancer cachexia . Am. J. Physiol. Cell Physiol.311 ( 1 ), C101 – C115 ( 2016 ).
- Tseng YC , KulpDK , LaiILet al. Preclinical investigation of the novel histone deacetylase inhibitor AR-42 in the treatment of cancer-induced cachexia . J. Natl Cancer Inst.107 ( 12 ), doi:10.1093/jnci/djv274. ( 2015 ) ( Epub ahead of print ).
- Acharyya S , SharmaSM , ChengASet al. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy . PLoS ONE5 ( 8 ), e12479 ( 2010 ).
- Kim MS , FielitzJ , McAnallyJet al. Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle performance . Mol. Cell Biol.28 ( 11 ), 3600 – 3609 ( 2008 ).
- Serra C , PalaciosD , MozzettaCet al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation . Mol. Cell28 ( 2 ), 200 – 213 ( 2007 ).
- Sincennes MC , BrunCE , RudnickiMA . Concise review: epigenetic regulation of myogenesis in health and disease . Stem Cells Transl. Med.5 ( 3 ), 282 – 290 ( 2016 ).
- Gonnella P , AlamdariN , TizioSet al. C/EBPbeta regulates dexamethasone-induced muscle cell atrophy and expression of atrogin-1 and MuRF1 . J. Cell Biochem.112 ( 7 ), 1737 – 1748 ( 2011 ).
- Chamberlain W , GonellaP , AlamdariNet al. Multiple muscle wasting-related transcription factors are acetylated in dexamethasone-treated muscle cells . Biochem. Cell Biol.90 ( 2 ), 200 – 208 ( 2012 ).
- Tseng YC , KulpSK , LaiILet al. Preclinical investigation of the novel histone deacetylase inhibitor AR-42 in the treatment of cancer-induced cachexia . J. Natl Cancer Inst.107 ( 12 ), doi:10.1093/jnci/djv274. ( 2015 ) ( Epub ahead of print ).
- Alamdari N , AversaZ , CastilleroEet al. Acetylation and deacetylation – novel factors in muscle wasting . Metabolism62 ( 1 ), 1 – 11 ( 2013 ).
- Guasconi V , PuriPL . Epigenetic drugs in the treatment of skeletal muscle atrophy . Curr. Opin. Clin. Nutr. Metab. Care11 ( 3 ), 233 – 241 ( 2008 ).
- Lu J , McKinseyTA , NicolRLet al. Signal-dependent activation of the MEF2 transcription factor by disassociation from histone deacetylases . Proc. Natl. Acad. Sci. USA97 ( 8 ), 4070 – 4075 ( 2000 ).
- Bassel-Duby R , OlsonEN . Signaling pathways in skeletal muscle remodeling . Annu. Rev. Biochem.75 , 19 – 37 ( 2006 ).
- Serra C , PalaciosD , MozzettaCet al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation . Mol. Cell28 ( 2 ), 200 – 213 ( 2007 ).