
Abstract
Long noncoding RNAs (lncRNAs) have been implicated in many biological processes. However, due to the unique nature of lncRNAs and the consequential difficulties associated with their characterization, there is a growing disparity between the rate at which lncRNAs are being discovered and the assignment of biological function to these transcripts. Here we present a molecular biology toolbox equipped to help dissect aspects of lncRNA biology and reveal functionality. We outline an approach that begins with a broad survey of genome-wide, high-throughput datasets to identify potential lncRNA candidates and then narrow the focus on specific methods that are well suited to interrogate the transcripts of interest more closely. This involves the use of imaging-based strategies to validate these candidates and observe the behaviors of these transcripts at single molecule resolution in individual cells. We also describe the use of gene editing tools and interactome capture techniques to interrogate functionality and infer mechanism, respectively. With the emergence of lncRNAs as important molecules in healthy and diseased cellular function, it remains crucial to deepen our understanding of their biology.
First draft submitted: 4 May 2017; Accepted for publication: 4 July 2017; Published online: 6 September 2017
One of the greatest challenges in modern day genomics is ascribing function to genes and genetic elements. It has become apparent that most of the human genome is pervasively transcribed, but less than 2% of these outputs are putatively functional RNAs that encode for proteins [Citation1–4]. The functions of the remaining proportion of the transcriptome and the genomic regions from which they arise, mostly remain uncharacterized and make up the ‘dark matter’ of the genome. With the advent and development of RNA deep sequencing technologies, long noncoding RNAs (lncRNAs) have been revealed to emanate from these dark regions of the genome [Citation5]. For a long time, the biological utility of these transcripts remained unknown and highly controversial, with many believing that they were functionless due to their lack of any protein-coding potential. In short, they were thought to simply be the result of transcriptional noise. This skepticism was further compounded by evidence pointing to the poor specificity of RNA PolII binding and transcription initiation, their low abundance and poor conservation [Citation6–8]. However, over the last decade, several examples of well-studied lncRNAs have been identified that show they are indeed functional molecules and play important roles in many biological processes [Citation5].
LncRNAs are generally products of RNA PolII activity and are arbitrarily defined as being greater than 200 nucleotides in length [Citation9]. These transcripts can arise from genomic regions that intervene (lincRNAs) or are within protein-coding genes (i.e., introns, overlapping exons, antisense transcripts) as well as enhancer regions and promoter regions [Citation5]. Nascent lncRNA transcripts undergo post-transcriptional modifications resulting in products that are capped at the 5′-end and often spliced and polyadenylated [Citation9]. Furthermore, certain lncRNAs undergo special post-transcriptional processing to complete their maturation. For example, Malat1 requires RNase P-mediated cleavage of the 3′ terminus to generate a stable transcript that is functional [Citation10]. Alternatively, enhancer RNAs (eRNAs) require the nucleolytic activity of the integrator complex to terminate their transcription [Citation11]. After being processed and modified, mature lncRNAs assume specific secondary and higher-order structures. The resulting conformations are often highly conserved as they imbue the transcripts with lncRNA functionality by providing binding sequences or structural domains for the interaction with RNA, DNA and effector or adapter proteins [Citation12]. In this way, lncRNAs can serve as the crucial interface between these different types of biomolecules to form highly specific and functional combinations of RNA, DNA and protein complexes.
The current collection of known lncRNAs is curated into four classes that describe the main modes of lncRNA function (decoys, scaffolds, guides and enhancers). LncRNAs can operate as decoys that competitively bind to transcription factors or sequester miRNAs to attenuate their function [Citation13]. Lethe, a pseudogene lncRNA, functions in this way to negatively regulate NF-κB signaling by sequestering RelA, a subunit of NF-κB, and preventing DNA binding and gene activation [Citation14]. LncRNAs can also serve as scaffolds that mediate the assembly of multiprotein complexes, as well as guide and localize these complexes to specific genomic sites of function [Citation13]. Hottip is an example that carries out both these functions to regulate homeotic gene expression during development. Hottip is expressed from the HoxA gene cluster and recruits the WDR5/MLL complex to drive localized histone methylation and maintain an active chromatin state across its target genes [Citation15,Citation16]. eRNAs encompass another class of function that involves activating gene expression in a tissue specific manner in response to specific stimuli. Thus, through the formation of chromosomal loops via the recruitment of the mediator complex, the spatial proximity of genetic elements and their bound transcription factors is controlled to achieve highly precise spatiotemporal and tissue-specific regulation of gene expression [Citation17]. Despite the growing catalogue of lncRNAs, the majority of detected products of transcription remain functionally unannotated. This is because the ability to characterize lncRNAs and distinguish bona fide functional transcripts from transcriptional noise still remains very challenging. Importantly, lncRNA-specific effects may be attributed to the ‘enhancer-like’ activity of their promoters, the act of lncRNA transcription itself or the splicing of the transcript [Citation18]. Therefore, lncRNA classification requires specialized tools that go well beyond demonstrating correlation and instead, unambiguously prove causality.
Specialized tools are needed to study lncRNAs
Many of the difficulties associated with identifying functional lncRNAs, amid the noise of the transcriptome, has been the use of conventional methods established for the study of mRNAs, which are ineffective for lncRNAs. In order to overcome this, we must recognize and consider the unique features of lncRNAs so that they are more amenable for our study. For example, some lncRNAs provide allele-specific epigenetic modulation of gene expression in cis (, left). In order to do this, their effect must be spatially limited to their site of transcription. A feature of these lncRNAs is that they exist in very low copy numbers and can be quite unstable allowing for the transcript and its function to be restricted to the vicinity of its transcription. As a consequence, this makes their detection extremely difficult by population based methods, such as quantitative PCR or even sequencing. Furthermore, their residence in the nucleus makes them refractory to RNA interference (RNAi) for loss of function experiments. Therefore, methods used to ablate function of this type of lncRNA ideally need to be highly efficient in the nucleus and preferably effective at the site of transcription. The use of targeted nucleases such as CRISPR/Cas9 for such purposes is possible, however, their application has to be carefully considered. Mutations to the sequence are usually ineffective against noncoding transcripts, unless it critically alters the lncRNA structure or a binding site. More importantly, rearrangements in the genome and the chromosomal topology, as a side effect of genome engineering, could result in the emergence of confounding phenotypes. In order to study the biology of such lncRNAs, the development and use of tools that are sensitive to their detection and spatial positioning, as well as discrete enough to preserve the complex surrounding environment in which they reside, is critical.
(A) Publicly available databases provide transcriptomic, conformational and epigenetic information on the entire genome, aiding in the identification of lncRNAs and the generation of broad hypotheses for their function. (B) (left) LncRNAs are able to interface with DNA, RNA and proteins to exert their functions. Many lncRNAs are thought to modify the epigenetic state of chromatin and alter the transcription of genes in cis or trans (Right) lncRNAs can be visually detected by decorating the transcript with fluorescently labelled antisense oligonucleotides for smFISH. The inset shows an example of a lncRNA and a nearby protein coding gene detected by smFISH and fluorescent microscopy. These experiments complement high-throughput biochemical assays and provide useful details on the absolute abundance and the subcellular localization patterns of lncRNA transcripts with single cell resolution. (C) CRISPR/Cas9 tools are able to (i) ablate lncRNA expression by direct mutagenesis to the DNA sequence for loss of function studies (ii) function as a targeting module to recruit activators, inhibitors and chromatin modifiers for locus specific perturbation of lncRNA expression or (iii) target lncRNA transcripts to gene loci to investigate their spatial nature.
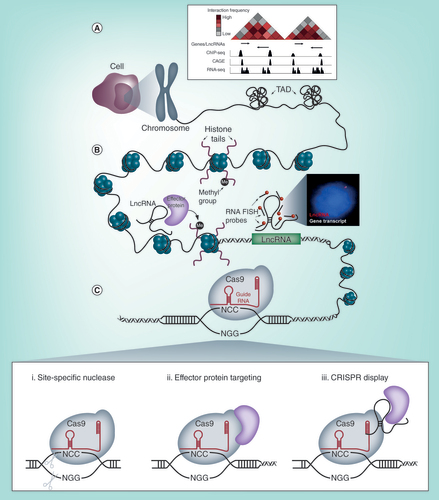
Perhaps the best studied example that illustrates the use of highly specialized tools to uncover lncRNA function is Xist and the discovery of its involvement in X-chromosome inactivation (XCI) in female mammals [Citation19]. This phenomenon has been widely studied and serves as an excellent model that demonstrates some of the current paradigms of lncRNA biology as well as the tools that were needed to establish them. Xist is transcribed from the X-inactivation center (Xic) on the X-chromosome – a locus that is also highly enriched with many other lncRNAs that serve as activators and inhibitors of XCI. Biochemical methods, such as RNA anti-sense purification (RAP) and RNA and chromatin immunoprecipitation (RIP and ChIP) have proven to be highly valuable in understanding the localization and the functional mechanism of Xist [Citation20–22]. The findings of these experiments have been further complemented by work done in transgenic knock-out mouse models where different parts of the Xic have been deleted [Citation23,Citation24]. From this, it is now known that Xist operates in cis and exploits the three-dimensional architecture of the genome to spread to distal sites that are spatially proximal to its transcription site [Citation21]. Through the recruitment of proteins, such as PRC2, the entire X-chromosome is epigenetically silenced [Citation20]. Through the use of these molecular tools, these studies have defined the functional mechanism of Xist, which has now been established as a common modality for a subclass of lncRNAs [Citation5,Citation25]. From the same Xic locus, Tsix emerges as a long anti-sense transcript that negatively regulates XCI. Examination of the higher order chromatin structure of the Xic locus by chromosome conformation capture carbon copy (5C) has revealed that Tsix and Xist, along with their respective positive regulators, inhabit neighboring topologically associated domains (TADs). This structure was further confirmed by observations made in single cells using super-resolution microscopy and fluorescence in situ hybridization (FISH) methods [Citation26]. Furthermore, by combining polymer modeling and imaging-based single cell analysis, the population averaged data from chromosome conformation capture techniques was deconvolved to reveal fluctuations in the structure of the Xic TADs, which are coupled to variations in the transcription of the genes from this locus [Citation27]. This highlighted aspects of the regulation of lncRNA expression and the important role of spatial organization in the nucleus for controlling gene transcription. Together, the extensive study of Xist has revealed many aspects of its function that are now considered paradigms of lncRNA biology and nuclear architecture. Furthermore, these studies have demonstrated the use of a myriad of molecular techniques to provide these insights and serve as lessons in the use of emerging tools to interrogate novel transcripts for functionality.
Here we describe some emerging methods and strategies that are well suited to the study of lncRNAs (). With the advances made in high-throughput sequencing technologies and efforts made by community based consortia, large datasets describing various elements of the genome are freely available. These databases can be examined to identify various lncRNAs across the entire genome. From these, we can narrow our focus on specific lncRNAs of interest, where we can use single cell and single molecule imaging approaches to observe some basic characteristics that cannot be revealed by bulk biochemical assays. Furthermore, we discuss the use of gene editing approaches using CRISPR/Cas9 tools to perturb lncRNA expression and subcellular positioning to uncover their function. Lastly, we discuss some methods used to capture and delineate the lncRNA interactome, from which the mechanism of function can be inferred. The development of our abilities to effectively identify and assign function to these elusive transcripts will not only greatly add to our understanding of the repertoire of regulatory mechanisms that exist in the cell, but potentially also identify novel therapeutic targets.
Table 1. Summary of the emerging methods used to study long noncoding RNA biology.
Searching the genome for lncRNAs
Publicly available databases, such as those provided by the ENCODE and FANTOM consortium, aim to catalogue all identified elements of the genome and transcriptome. Searching these resources can identify putative lncRNAs that can subsequently be tested in a hypothesis-driven manner (). Genome-wide next generation sequencing approaches, including RNA-seq [Citation28] and Cap Analysis Gene Expression (CAGE) [Citation29], allow for the unbiased detection and quantification of lncRNA transcripts. In addition to this, the development of 3C-based technologies [Citation30], such as Hi-C [Citation31] and ChIA-PET [Citation32] have allowed for the high resolution mapping of chromosomal interactions, allowing for spatially proximal genes to be identified. Furthermore, by overlaying ChIP-seq traces, the epigenetic status of the examined region is revealed, confirming the presence of lncRNAs, active or inactive genes and other regulatory elements through epigenetic signatures [Citation33]. These large datasets can be mined in numerous ways to identify candidate lncRNAs. Depending on the bioinformatic pipeline and the filter parameters, putative lncRNAs that are potentially involved in specific biological processes can be isolated. Together, these many layers of different information synergistically aid in the identification of potentially functional lncRNA transcripts that can be experimentally tested.
Imaging lncRNAs
One of the first steps in characterizing a novel lncRNA is to experimentally validate its expression in the cell type of interest, quantify its abundance and determine its subcellular location. As useful as the above mentioned high-throughput experiments may be in identifying candidate lncRNAs, they are fundamentally limited in describing these properties. With population averaged readouts, there is a diminished detection sensitivity (which can be further compounded by the low copy number of some lncRNAs) and the loss of the cell-to-cell variability of expression. These methods also do not provide critical spatial information on these transcripts, which often hints at their mode function. This has resulted in the majority of annotated lncRNAs missing fundamental data describing their single cell abundance and subcellular localization.
Owing to the development of RNA labeling techniques, microscopy can be used to complement high-throughput biochemical assays by revealing these characteristics in single cells. RNA single molecule FISH (smFISH) has been widely used to detect and quantify individual mRNA and lncRNA molecules (, right) [Citation34–36]. Furthermore, the subcellular localization of lncRNAs can be successfully resolved using this method, providing insights into their function [Citation35]. For example, lncRNA smFISH foci that appear in the cytoplasm suggests a role in translation or signal transduction, whereas those that solely occur in the nucleus are implicated in the regulation of gene transcription. Co-labeling of lncRNAs with coding mRNAs allow for the effect of the lncRNA on target gene transcription to be directly visualized and quantified. For lncRNAs that are spliced, intronic or exonic portions of the lncRNA can be exclusively labelled to distinguish sites of transcription as well as cis and trans modes of action. smFISH can also be applied concomitantly with immunofluorescence labeling (IF-FISH) [Citation37]. By co-labeling proteins involved in epigenetic modification or histone marks, lncRNAs can be associated with epigenetic regulation and different chromatin states.
However, the throughput of these labeling strategies are limited by the spectral bandwidth of conventional fluorescent microscopes, allowing for only a few different lncRNA species to be observed simultaneously. This low-throughput can potentially be overcome by adapting multiplexed error-robust fluorescence in situ hybridization (MERFISH) for lncRNAs. MERFISH is a high-throughput imaging strategy that makes use of smFISH readouts to generate single cell, spatially resolved transcriptomic profiles [Citation38]. MERFISH employs a complex barcoding scheme to encode RNAs, which are detected by multiple rounds of smFISH. This method has recently been improved to detect 130 unique RNAs in over 100,000 cells [Citation39]. The application of MERFISH to lncRNA transcriptome profiling would be extremely powerful in identifying lncRNA expression and localization patterns in different healthy and diseased cell types. The data generated would complement single cell RNA-seq readouts by adding the vital layer of spatial context.
Despite smFISH being very useful in studying lncRNA biology, its application to lncRNAs can be challenging. Some lncRNAs, particularly those that emanate from enhancers can be very small (less than 1000 nucleotides), leaving a shortage of ‘real estate’ for a sufficient number of smFISH probes to bind and produce a detectable focus (usually 30–48 fluorescently labelled probes are required). In such cases, the signal from a small number of probes (≥10 probes) can be observed after tyramine signal amplification (TSA) [Citation40]. The caveat to this, however, is that the quantitative nature of smFISH is sacrificed. Furthermore, the formation of strong ribonucleoprotein (RNP) complexes, extreme low abundance and frequent repeat sequences associated with lncRNAs can contribute to the difficulties in obtaining robust smFISH signals. LncRNAs commonly associated with protein complexes may be occluded from being successfully labeled. In our laboratory, we have found that we can gain accessibility to the lncRNA transcript by denaturing these RNP complexes by methanol fixation. The low abundance and frequent repeats of lncRNAs may result in off-target probe binding, creating poor signal-to-noise ratio. In order to overcome this and validate the correct binding of a probe set, a dual labelling system can be applied, whereby the even probes are labelled in a different color to the odd probes. Co-localization of these two colors at the same focus suggests that that the hybridization is on target [Citation36]. Even though there are numerous strategies to optimize smFISH for lncRNA detection, there are occasions where the lncRNA may not be amenable to detection by this method. In these extreme cases, it may be worth investigating the use of gene editing technologies to tag lncRNA transcripts with RNA mimics of GFP [Citation41,Citation42]. However, the consequences of adding such structured sequences to lncRNAs are unknown and may very well affect the function of the transcript.
Observations made by imaging techniques are an imperative first step in the experimental pipeline for the characterization of lncRNAs. These experiments provide key details into the biology of the lncRNA and help foster hypotheses that can be tested further.
Determining function using genome & epigenome editing tools
Perhaps the most direct method for elucidating the function of what a gene does, is to observe the impact upon its removal. This is as true in classical fly genetics studies as it is in unraveling the function of lncRNAs. As discussed, RNAi has been an important contributor in observing the effects of temporal knockdown of lncRNA transcripts. Though some disadvantages can be overcome, RNAi presents limitations, in that the effect is often temporal, requiring repetitive exposure, as well as being inefficient, due to the low abundance of lncRNA transcripts. In addition, RNAi is not as reliable in the nucleus where many lncRNAs function [Citation43]. The use of antisense oligonucleotides (ASOs) and their derivatives has proven to be much more effective than RNAi methods and are being widely adopted for targeting lncRNA transcripts [Citation44]. ASOs, such as gapmers, form very specific DNA-RNA hybrid duplexes with the target lncRNA transcript, resulting in their efficient degradation by nuclear resident RNase H. Similar to RNAi, however, their effects are transient.
The age of genome engineering, in particular, the discovery of CRISPR/Cas9 [Citation45], has provided a much needed tool with which to address these and several other problems associated with the elucidation of lncRNA function. Foremost, is the discrete and permanent nature of CRISPR-based lncRNA knockout (i). However, straightforward insertions/deletions caused by a single double strand break are unlikely to cause functional ablation of a noncoding gene – therefore one has to consider a more comprehensive approach with respect to lncRNA function. One strategy which ensures complete ablation would be to delete the entire genomic region associated with a lncRNA. For example, deletion of the 6kb lncRNA HPAT5 in pluripotent stem cells revealed a critical role for this lncRNA in pluripotency and primate preimplantation development [Citation46]. This approach has been highly effective and expanded to create highly informative genome-wide scales of lncRNA depletion screens [Citation47]. However, deletion of the genomic DNA can confound the issue of whether the phenotypic effect is due to loss of the noncoding transcript or potential regulatory sequences of the genomic region itself [Citation48,Citation49]. This was exemplified by the systematic CRISPR-based deletion and insertion of the lncRNA Haunt [Citation50]. In this study, the authors demonstrated the dependence of HoxA gene activation on the presence of the Haunt-encompassing DNA locus upon ES cell differentiation. Strikingly, however, interruption of Haunt transcription via insertion of a transcriptional stop signal, increased HoxA activation. Furthermore, insertion of the Haunt cDNA within genomic knock outs of the same region, failed to restore HoxA activation, ultimately demonstrating that the Haunt genomic region has an enhancing regulatory function, while the noncoding transcript itself appears to act as a repressor. As outlined above, introducing a transcriptional stop signal can be highly effective in ablating the lncRNA transcript without significant effect on the role of the DNA sequence itself, as shown by others [Citation51]. An alternative approach is to target the promoter region of the lncRNA. In deleting the region surrounding the promoter of lncRNA-CSR, Basu and colleagues demonstrated that the absence of the transcript itself, (despite an intact genomic region) disrupted long range chromatin contacts over 2.6 Mb away, leading to dysregulation of isotype switching in B cells [Citation52]. A further advantage of targeting the promoter is the small size that needs to be deleted, increasing the efficiency of editing. As such, paired guide-based CRISPR deletions of lncRNA promoters have been adapted in large scale studies for screening the effects of lncRNAs [Citation53].
These genome engineering approaches can be further enhanced with the use of the iCRISPR system [Citation54]. By integrating an inducible Cas9 construct into the AAVS1 safe harbor locus of induced pluripotent stem cells (iPSCs), lncRNAs can be targeted by the simple addition of RNA guides. The power of this system lies within the ability to subsequently differentiate these iPSCs to specific cell types that may be relevant to the lncRNA function. Furthermore, lncRNAs responsible for lineage specification can also be identified using this system through the systematic ablation of lncRNAs and observing the resultant cell fates.
Going beyond classical genome editing
In our laboratory, we have developed a strategy to obtain single cell, in situ functional readouts after lncRNA ablation by using a combination of genome editing and imaging techniques. By exploiting the homologous-directed repair (HDR) pathway, we have been able to replace a lncRNA sequence with the coding sequence of green fluorescent protein (GFP). The GFP repair template is designed such that the endogenous promoter of the lncRNA is preserved after repair and is responsible for driving the transcription of the newly inserted GFP cassette. We are able to detect these cells using smFISH probes targeted to the GFP RNA transcript. By co-labeling the mRNA transcripts of the genes regulated by the deleted lncRNA, we are able to quantify the effect of the lncRNA deletion on its target genes and their subcellular localization in single cells (publication under review).
Though the approaches outlined above harness the endonuclease ability of the CRISPR-Cas system, it is, in fact the malleability of the tool that provides researchers with further manipulations with which to elucidate lncRNA function. One such modification has been the development of the dCas9 (dead Cas9) variant, in which the nuclease capability of Cas9 has been disabled and replaced with the loading of a repressor, termed CRISPR interference (CRISPRi; [Citation55]) or enhancer, termed CRISPRa (CRISPR activator; [Citation56]) of transcription (ii). CRISPRi leads to the repression of the chromatin surrounding the target sequence – and thus transcription – in a highly sequence specific manner, negating the need to edit the genome directly. The most powerful example of this epigenome strategy was recently shown by creating a large CRISPRi library targeting lncRNAs [Citation57]. This delicate strategy revealed the impact of single nucleotide polymorphisms (SNPs) within lncRNAs, their contributions to expression and important associations with cancer phenotypes. The corollary of CRISPRi is the use of dCas9 variants with a transcription factor payload of enhancement rather than repression. As with CRISPRi, this has been expanded, termed CRISPR-on, to include modifications that would allow for multiple genomic regions to be epigenetically modified to enhance expression of transcripts, discretely [Citation58]. The flexibility associated with dCas9 variants is no way restricted to traditional ‘on/off’ expression switches. Indeed, understanding the roles of specific chromatin marks overlaying the genomic DNA encompassing lncRNA transcripts is of equal importance. In this regard, several variants of the CRISPR system which allow for site specific deposition of chromatin modifiers have been developed (extensively summarized in [Citation59]) and will no doubt reveal remarkable insight about lncRNA control and function.
Despite the myriad advantages associated with dCas9 variants of temporal or cell type-specific control of lncRNA expression, a recent study suggests only 38% of lncRNAs may be amenable to CRISPR-based dysregulation without affecting proximal gene expression [Citation60]. As such, this necessitates the need for careful analysis of the genetic neighborhood, as well as complementary knockdown studies.
Nonetheless, while these variations of CRISPR/Cas9 may address the unravelling of the temporal nature of lncRNA expression to elucidate function, there remains the question of spatial control. One of the key questions involved in lncRNA function is whether they act in cis or in trans. The development of CRISPR-DISPLAY involved re-engineering the CRISPR/Cas9 (iii) system to ‘carry’ a specific lncRNA transcript and deposit it at any region within the genome [Citation61]. Though the results of this study showed modest effects as a result of depositing known lncRNAs at arbitrary loci, we see this as the evolution of the then, highly innovative BoxB experiment [Citation15], to now being able to demonstrate lncRNA function endogenously, with the ability of spatial control.
While none of these encompass a ‘one shoe fits all’ CRISPR-based tool, each modification provides researchers with an ability to address numerous aspects of lncRNA function, and indeed, are rapidly evolving. Furthermore, it remains challenging to apply these tools to more physiologically relevant systems, such as primary cell cultures or animal models. Primary cells, compared with immortalized cell lines, are often refractory to conventional lipid-based transfection methods and are limited in their lifespan, making the generation of isogenic mutants very difficult, if not impossible. It is in these situations where methods and strategies to identify and make measurements in the few cells that have successfully undergone the targeted genomic alteration(s) are currently lacking and are in great need of development. The difficulty of delivering these tools to in vivo systems is even greater, and continues to limit many of our observations of lncRNA biology to the petri-dish.
Determining the mechanism of lncRNA function
The ablation of lncRNAs from the cell enables functionality to be ascribed to the transcript by observing the resultant phenotype. However, in order to gain insight into the mechanism of function, the interacting partners of the lncRNA need to be identified and closely examined. The mechanism of lncRNA function is often dependent on the network of interactions that the lncRNA establishes with DNA, RNA and proteins. The composition of this interactome is dictated by the availability of binding sites for these partner molecules, once the lncRNA has assumed its mature secondary and higher-order structures. The mapping of this interactome can help infer the mechanism of lncRNA function by identifying associated genomic locations of the lncRNA, sequestered mRNAs or miRNAs and protein effectors. With the development of cross-linking methods, the complex and delicate state of lncRNA complexes can be preserved in vivo and subsequently isolated. Thereafter by, for example, tiling antisense oligonucleotide probes along the lncRNA to capture the transcript, components of the complex can be isolated and determined by numerous approaches. Variations to this RNA-centric purification method exist and include chromatin isolation by RNA purification (ChIRP) [Citation62], capture hybridization analysis of RNA targets (Chart) [Citation63] and RAP [Citation21]. Using these methods in conjunction with high-throughput sequencing and mass spectrometry, interacting DNA, RNA and proteins can be detected. Alternatively, protein-centric capture methods such as photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP) and its more sophisticated variants like iCLIP, can be used to identify lncRNA transcripts associated with particular proteins of known function and interest [Citation64,Citation65].
Central to the establishment of the lncRNA interactome is the secondary structure of the mature lncRNA transcript. It is this conformation that determines the availability of nucleic acids for base-pairing with DNA or RNA, as well as the formation of structural motifs for protein and DNA binding. By resolving this, a deeper insight into the molecular properties that are required for the lncRNA to mediate these interactions and its functions, is revealed. For example, by combining the structures of SHAPE [Citation66], DMS [Citation67] and terbium structure probing [Citation68], it has been shown that Hotair forms intricate structural modules that are essential in binding to PRC2 to carry out its function [Citation69]. Coolair is another lncRNA with a complex structure that includes multi-helix junctions and two right-hand turn motifs, which appear to impact its function [Citation70]. By developing a picture of the partners that interact with lncRNAs, their mechanism of function can be inferred and further tested, through the removal or occlusion of the interacting domains, as revealed by the structure of the lncRNA.
Conclusion & future perspective
The discovery of functional lncRNAs has fundamentally changed our appreciation and perception of the transcriptome. With the correct tools, we can systematically mine this space and identify biologically functional molecules that serve as a new layer in the complex circuitry of life. We envision that over time, the methods and technologies will evolve to be better tailored to studying lncRNA biology. Eventually, a richly annotated catalogue of all the lncRNAs will be established, which we can integrate into and use to widen our current understanding of the cell. We can also revisit disease associated polymorphisms, of which many reside outside of coding regions. With this, these important transcripts could possibly be adopted as mainstream diagnostic markers for disease and serve as targets for the next generation of therapeutics.
A key challenge in lncRNA biology is proving biological function over transcriptional noise.
We discuss some tools that are well suited for studying lncRNA biology.
High-throughput biochemical methods and publicly available datasets are useful in identifying lncRNAs which can be further tested with hypotheses-driven experiments.
smFISH strategies can provide information about the absolute abundance of lncRNAs and the subcellular localization of lncRNAs in single cells.
CRISPR/Cas9 tools are useful in creating permanent or locus specific disruptions in lncRNA expression to interrogate function.
Methods to capture the lncRNA interactome can be used to infer the mechanism of function.
Financial & competing interests disclosure
The authors of this publication are supported by the Biomedical Translational and Research Initiative (BTRI) of the Council of Scientific and Industrial Research (CSIR) and The University of Cape Town (UCT) (project V6GBT60). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Lander ES , LintonLM , BirrenBet al. Initial sequencing and analysis of the human genome . Nature409 ( 6822 ), 860 – 921 ( 2001 ).
- Bertone P , StolcV , RoyceTEet al. Global identification of human transcribed sequences with genome tiling arrays . Science306 ( 5705 ), 2242 – 2246 ( 2004 ).
- Consortium EP , BirneyE , StamatoyannopoulosJAet al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project . Nature447 ( 7146 ), 799 – 816 ( 2007 ).
- Kapranov P , ChengJ , DikeSet al. RNA maps reveal new RNA classes and a possible function for pervasive transcription . Science316 ( 5830 ), 1484 – 1488 ( 2007 ).
- Kung JT , ColognoriD , LeeJT . Long noncoding RNAs: past, present, and future . Genetics193 ( 3 ), 651 – 669 ( 2013 ).
- Wang J , ZhangJ , ZhengHet al. Mouse transcriptome: neutral evolution of ‘non-coding’ complementary DNAs . Nature431 ( 7010 ), 1 p following 757; discussion following 757 ( 2004 ).
- Struhl K . Transcriptional noise and the fidelity of initiation by RNA polymerase II . Nat. Struct. Mol. Biol.14 ( 2 ), 103 – 105 ( 2007 ).
- Mercer TR , GerhardtDJ , DingerMEet al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome . Nat. Biotechnol.30 ( 1 ), 99 – 104 ( 2011 ).
- Quinn JJ , ChangHY . Unique features of long non-coding RNA biogenesis and function . Nat. Rev. Genet.17 ( 1 ), 47 – 62 ( 2016 ).
- Wilusz JE , FreierSM , SpectorDL . 3’ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA . Cell135 ( 5 ), 919 – 932 ( 2008 ).
- Lai F , GardiniA , ZhangA , ShiekhattarR . Integrator mediates the biogenesis of enhancer RNAs . Nature525 ( 7569 ), 399 – 403 ( 2015 ).
- Mercer TR , MattickJS . Structure and function of long noncoding RNAs in epigenetic regulation . Nat. Struct. Mol. Biol.20 ( 3 ), 300 – 307 ( 2013 ).
- Wang KC , ChangHY . Molecular mechanisms of long noncoding RNAs . Mol. Cell43 ( 6 ), 904 – 914 ( 2011 ).
- Rapicavoli NA , QuK , ZhangJJ , MikhailM , LabergeRM , ChangHY . A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics . Elife2 ( 2013 ).
- Wang KC , YangYW , LiuBet al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression . Nature472 ( 7341 ), 120 – 124 ( 2011 ).
- Yang YW , FlynnRA , ChenYet al. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency . Elife3 , e02046 ( 2014 ).
- Lai F , OromUA , CesaroniMet al. Activating RNAs associate with mediator to enhance chromatin architecture and transcription . Nature494 ( 7438 ), 497 – 501 ( 2013 ).
- Engreitz JM , HainesJE , PerezEMet al. Local regulation of gene expression by lncRNA promoters, transcription and splicing . Nature539 ( 7629 ), 452 – 455 ( 2016 ).
- Nguyen DK , DistecheCM . Dosage compensation of the active X chromosome in mammals . Nat. Genet.38 ( 1 ), 47 – 53 ( 2006 ).
- Zhao J , SunBK , ErwinJA , SongJJ , LeeJT . Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome . Science322 ( 5902 ), 750 – 756 ( 2008 ).
- Engreitz JM , Pandya-JonesA , McdonelPet al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome . Science341 ( 6147 ), 1237973 ( 2013 ).
- Mchugh CA , ChenCK , ChowAet al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3 . Nature521 ( 7551 ), 232 – 236 ( 2015 ).
- Penny GD , KayGF , SheardownSA , RastanS , BrockdorffN . Requirement for Xist in X chromosome inactivation . Nature379 ( 6561 ), 131 – 137 ( 1996 ).
- Wutz A , RasmussenTP , JaenischR . Chromosomal silencing and localization are mediated by different domains of Xist RNA . Nat. Genet.30 ( 2 ), 167 – 174 ( 2002 ).
- Engreitz JM , OllikainenN , GuttmanM . Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression . Nat. Rev. Mol. Cell Biol.17 ( 12 ), 756 – 770 ( 2016 ).
- Nora EP , LajoieBR , SchulzEGet al. Spatial partitioning of the regulatory landscape of the X-inactivation centre . Nature485 ( 7398 ), 381 – 385 ( 2012 ).
- Giorgetti L , GalupaR , NoraEPet al. Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription . Cell157 ( 4 ), 950 – 963 ( 2014 ).
- Wang Z , GersteinM , SnyderM . RNA-Seq: a revolutionary tool for transcriptomics . Nat. Rev. Genet.10 ( 1 ), 57 – 63 ( 2009 ).
- Kodzius R , KojimaM , NishiyoriHet al. CAGE: cap analysis of gene expression . Nat. Methods3 ( 3 ), 211 – 222 ( 2006 ).
- Dekker J , RippeK , DekkerM , KlecknerN . Capturing chromosome conformation . Science295 ( 5558 ), 1306 – 1311 ( 2002 ).
- Lieberman-Aiden E , Van BerkumNL , WilliamsLet al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome . Science326 ( 5950 ), 289 – 293 ( 2009 ).
- Fullwood MJ , LiuMH , PanYFet al. An oestrogen-receptor-alpha-bound human chromatin interactome . Nature462 ( 7269 ), 58 – 64 ( 2009 ).
- Guttman M , AmitI , GarberMet al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals . Nature458 ( 7235 ), 223 – 227 ( 2009 ).
- Raj A , Van Den BogaardP , RifkinSA , Van OudenaardenA , TyagiS . Imaging individual mRNA molecules using multiple singly labeled probes . Nat. Methods5 ( 10 ), 877 – 879 ( 2008 ).
- Cabili MN , DunaginMC , McclanahanPDet al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution . Genome Biol.16 , 20 ( 2015 ).
- Dunagin M , CabiliMN , RinnJ , RajA . Visualization of lncRNA by single-molecule fluorescence in situ hybridization . Methods Mol. Biol.1262 , 3 – 19 ( 2015 ).
- Hinten M , MaclaryE , GayenS , HarrisC , KalantryS . Visualizing long noncoding RNAs on chromatin . Methods Mol. Biol.1402 , 147 – 164 ( 2016 ).
- Chen KH , BoettigerAN , MoffittJR , WangS , ZhuangX . RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells . Science348 ( 6233 ), aaa6090 ( 2015 ).
- Moffitt JR , HaoJ , WangG , ChenKH , BabcockHP , ZhuangX . High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization . Proc. Natl Acad. Sci. USA113 ( 39 ), 11046 – 11051 ( 2016 ).
- Shibayama Y , FanucchiS , MhlangaMM . Visualization of enhancer-derived noncoding RNA . Methods Mol. Biol.1468 , 19 – 32 ( 2017 ).
- Paige JS , WuKY , JaffreySR . RNA mimics of green fluorescent protein . Science333 ( 6042 ), 642 – 646 ( 2011 ).
- Filonov GS , MoonJD , SvensenN , JaffreySR . Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution . J. Am. Chem. Soc.136 ( 46 ), 16299 – 16308 ( 2014 ).
- Zeng Y , CullenBR . RNA interference in human cells is restricted to the cytoplasm . RNA8 ( 7 ), 855 – 860 ( 2002 ).
- Lennox KA , BehlkeMA . Cellular localization of long noncoding RNAs affects silencing by RNAi more than by antisense oligonucleotides . Nucleic Acids Res.44 ( 2 ), 863 – 877 ( 2016 ).
- Jinek M , ChylinskiK , FonfaraI , HauerM , DoudnaJA , CharpentierE . A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity . Science337 ( 6096 ), 816 – 821 ( 2012 ).
- Durruthy-Durruthy J , SebastianoV , WossidloMet al. The primate-specific noncoding RNA HPAT5 regulates pluripotency during human preimplantation development and nuclear reprogramming . Nat. Genet.48 ( 1 ), 44 – 52 ( 2016 ).
- Zhu S , LiW , LiuJet al. Genome-scale deletion screening of human long noncoding RNAs using a paired-guide RNA CRISPR-Cas9 library . Nat. Biotechnol.34 ( 12 ), 1279 – 1286 ( 2016 ).
- Bassett AR , AkhtarA , BarlowDPet al. Considerations when investigating IncRNA function in vivo . Elife3 , e03058 ( 2014 ).
- Paralkar VR , TabordaCC , HuangPet al. Unlinking an lncRNA from its associated cis element . Mol. Cell62 ( 1 ), 104 – 110 ( 2016 ).
- Yin YF , YanPX , LuJLet al. Opposing roles for the lncRNA haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation . Cell Stem Cell16 ( 5 ), 504 – 516 ( 2015 ).
- Liu YY , HanX , YuanJTet al. Biallelic insertion of a transcriptional terminator via the CRISPR/Cas9 system efficiently silences expression of protein-coding and noncoding RNA genes . J. Biol. Chem.292 ( 14 ), 5624 – 5633 ( 2017 ).
- Pefanis E , WangJG , RothschildGet al. RNA exosome-regulated long noncoding RNA transcription controls super-enhancer activity . Cell161 ( 4 ), 774 – 789 ( 2015 ).
- Aparicio-Prat E , ArnanC , SalaI , BoschN , GuigoR , JohnsonR . DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs . BMC Genomics16 , 846 ( 2015 ).
- Zhu ZR , GonzalezF , HuangfuD . The iCRISPR platform for rapid genome editing in human pluripotent stem cells . Use of Crispr/Cas9, Zfns, and Talens in Generating Site-Specific Genome Alterations546 , 215 – 250 ( 2014 ).
- Qi LS , LarsonMH , GilbertLAet al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression . Cell152 ( 5 ), 1173 – 1183 ( 2013 ).
- Maeder ML , LinderSJ , CascioVM , FuYF , HoQH , JoungJK . CRISPR RNA-guided activation of endogenous human genes . Nat. Methods10 ( 10 ), 977 – + ( 2013 ).
- Liu SJ , HorlbeckMA , ChoSWet al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells . Science355 ( 6320 ), ( 2017 ).
- Cheng AW , WangHY , YangHet al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system . Cell Res.23 ( 10 ), 1163 – 1171 ( 2013 ).
- Stricker SH , KoferleA , BeckS . From profiles to function in epigenomics . Nat. Rev. Genet.18 ( 1 ), 51 – 66 ( 2017 ).
- Goyal A , MyachevaK , GrossM , KlingenbergM , Duran ArqueB , DiederichsS . Challenges of CRISPR/Cas9 applications for long non-coding RNA genes . Nucl. Acids Res.45 ( 3 ), e12 ( 2016 ).
- Shechner DM , HacisuleymanE , YoungerST , RinnJL . Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display . Nat. Methods12 ( 7 ), 664 – 670 ( 2015 ).
- Chu C , QuK , ZhongFL , ArtandiSE , ChangHY . Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions . Mol. Cell44 ( 4 ), 667 – 678 ( 2011 ).
- Simon MD , WangCI , KharchenkoPVet al. The genomic binding sites of a noncoding RNA . Proc. Natl Acad. Sci. USA108 ( 51 ), 20497 – 20502 ( 2011 ).
- Hafner M , LandthalerM , BurgerLet al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP . Cell141 ( 1 ), 129 – 141 ( 2010 ).
- Bose DA , DonahueG , ReinbergD , ShiekhattarR , BonasioR , BergerSL . RNA binding to CBP stimulates histone acetylation and transcription . Cell168 ( 1–2 ), 135.e122 – 149.e122 ( 2017 ).
- Wilkinson KA , MerinoEJ , WeeksKM . Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution . Nat. Protoc.1 ( 3 ), 1610 – 1616 ( 2006 ).
- Tijerina P , MohrS , RussellR . DMS footprinting of structured RNAs and RNA-protein complexes . Nat. Protoc.2 ( 10 ), 2608 – 2623 ( 2007 ).
- Hargittai MR , Musier-ForsythK . Use of terbium as a probe of tRNA tertiary structure and folding . RNA6 ( 11 ), 1672 – 1680 ( 2000 ).
- Somarowthu S , LegiewiczM , ChillonI , MarciaM , LiuF , PyleAM . HOTAIR forms an intricate and modular secondary structure . Mol. Cell58 ( 2 ), 353 – 361 ( 2015 ).
- Hawkes EJ , HennellySP , NovikovaIV , IrwinJA , DeanC , SanbonmatsuKY . COOLAIR antisense RNAs form evolutionarily conserved elaborate secondary structures . Cell Rep.16 ( 12 ), 3087 – 3096 ( 2016 ).