
Abstract
Global DNA hypomethylation promoting genomic instability leads to cancer and deterioration of human health with age. Aim: To invent a biotechnology that can reprogram this process. Methods: We used Alu siRNA to direct Alu interspersed repetitive sequences methylation in human cells. We evaluated the correlation between DNA damage and Alu methylation levels. Results: We observed an inverse correlation between Alu element methylation and endogenous DNA damage in white blood cells. Cells transfected with Alu siRNA exhibited high Alu methylation levels, increased proliferation, reduced endogenous DNA damage and improved resistance to DNA damaging agents. Conclusion: Alu methylation stabilizes the genome by preventing accumulation of DNA damage. Alu siRNA could be useful for evaluating reprograming of the global hypomethylation phenotype in cancer and aging cells.
DNA methylation at interspersed repetitive sequences (IRSs) plays an important role in maintaining genome stability. Cells with IRS hypomethylation exhibit increased mutation rates [Citation1,Citation2]. Here, we tested whether DNA damage, an alteration in the chemical structure of DNA and a precursor to mutation [Citation3], plays a role in mediating how global hypomethylation promotes genomic instability. Our recent study found that global hypomethylation is associated with plasma 8-hydroxy-2′-deoxyguanosine (8-OHdG) in biliary atresia patients [Citation4]. Moreover, urinary 8-OHdG, DNA strand breaks and global DNA hypomethylation are associated with low serum folate [Citation5] and oxidative stress [Citation6]. Therefore, we hypothesized that the human genome may use DNA methylation in IRSs to prevent DNA damage.
This study developed a technology to add DNA methylation at Alu elements. The human genome contains greater than one million Alu elements [Citation7]. Several reports demonstrated de novo methylation by siRNA in plants [Citation8–10]. In human cells, small RNA was used to promote DNA methylation, shRNA for long interspersed element-1 (LINE-1) and hepatitis B virus, and siRNA for HIV-1 promoter region [Citation11–13]. There are many types of IRS such as Alu elements, LINE-1, and human endogenous retrovirus [Citation14–17]. To increase DNA methylation, we tested siRNA to unique sequences and several IRS sequences. Our preliminary trial demonstrated that only Alu siRNA could increase methylation of the target sequences. Here, we evaluated whether Alu siRNA is a useful tool to explore the role of global hypomethylation in genomic instability [Citation18].
Alu hypomethylation may play a role in causing genomic instability in cancer and aging cells. Genomic instability, the main enabling characteristic of cancer and aging processes [Citation18,Citation19], may mainly be promoted by IRS hypomethylation. IRS hypomethylation is commonly observed both in aging [Citation14,Citation20] and cancer cells [Citation21]. IRS hypomethylation is noted in individuals who are exposed to carcinogens, such as benzene [Citation22] and smoke [Citation23]. In cancer cells, both IRS hypomethylation and mutations increase the multistep process of cancer progression [Citation24]. Interference with DNA methyltransferase function in cancer cells increases mutation accumulation [Citation25]. Finally, DNA hypomethylation and mutation are linked on the same chromosome [Citation26,Citation27]. In contrast to LINE-1s, Alu elements are IRSs that reduce methylation levels with age [Citation14]. In younger individuals, Alu methylation is directly correlated with growth rate [Citation28]. Alu hypomethylation is associated with the severity of conditions involved in the deterioration of human health with age, such as osteoporosis [Citation29] and diabetes mellitus [Citation30].
Here, we evaluated genomic instability by determining endogenous DNA damage lesions, including chemically changed bases, such as 8-OHdG and a base missing from the backbone of DNA, which is referred to as an abasic site (AP site) [Citation31]. Then, we tested how cells respond to DNA damage agents, including methyl methanesulphonate (MMS) and H2O2 [Citation32,Citation33].
Materials & methods
Cells & cultures
For double blind experiments using 12 samples of cases and controls, the tumor HeLa cell line, human embryonic kidney HEK293, a human immortalized proximal tubule epithelial cell line HK2, a nontumor cell line and the periodontal ligament (PDL) fibroblast were cultured in Dulbecco’s modified Eagle medium (DMEM) high glucose (Sigma-Aldrich, MO, USA) supplemented with 10% fetal bovine serum (Gibco by Thermo Scientific, MA USA) (working culture medium) and maintained at 37°C in a humidified atmosphere with 5% CO2.
Subjects
Our study population was peripheral blood mononuclear cell DNA selected from our earlier report [Citation14]. Age and sex were described in Supplementary Table 1.
siRNA transfection
Alu siRNA sequences are 5′-CUUUGGGAGGCCGAGGCGGGCGGAUCA-3′ (sense) and 5′-AUCCGCCCGCCUCGGCCUCCCAAAGUG-3′ (antisense). Scramble siRNA was from Bioneer. The day before transfection, cells were seeded at 5 × 104 cells per well in a 24-well plate in 0.5 ml of DMEM with 10% fetal bovine serum. For each well of transfection, 20 μM of siRNA was diluted into 25 μl Opti-MEM medium and mixed gently. For each transfection, dilute 2 μl of Lipofectamine reagent into 25 μl Opti-MEM and mix. Combined diluted DNA and diluted Lipofectamine reagent, mix gently, and incubate at room temperature for 20 min. While complexes were forming, replaced medium on the cells with 0.45 ml of DMEM growth medium. For each transfection, added complexes onto the cells, and a produced final concentration of Alu siRNA was 150 nM. Incubated cells with the complex at 37°C in a CO2 incubator for 2 days. After the incubation time, cells were collected by trypsinization.
DNA preparation & quantification of DNA methylation level by Combined Bisulfite Restriction Analysis
Cells were harvested by trypsinization and performed DNA extraction with 10% sodium dodecyl sulfate (Sigma Aldrich), lysis buffer II (0.75 M NaCl, 0.024M EDTA at pH 8) and 20 mg/ml proteinase K (USB, OH, USA), and incubated at 50°C over three nights until cell digestion. Lysed cells were extracted with phenol/chloroform and DNA was precipitated with absolute ethanol. Next, DNA sample was subjected to quantify methylation level using Combined Bisulfite Restriction Analysis (COBRA). Briefly, genomic DNA (750 ng) were treated with sodium bisulfite using EZ DNA methylation-Gold™ kit (Zymo Research, CA USA) based on the manufacturer’s instructions. To investigate Alu methylation, the bisulfite-treated DNA was subjected to 35 cycles of PCR with Alu-Forward (5′-GGYGYGGTGGTTTAYGTTTGT AA-3′) and Alu-Reverse (5′-CTAACTTTTTATATTTTTAATAAAAAC RAAATTTCACCA-3′) primers at an annealing temperature of 57°C to generate 133 bp amplicon. The PCR amplicons were digested with two units of TaqI (Thermo Scientific, MA USA) at 65°C overnight. The digested products were analyzed in an 8% nondenaturing polyacrylamide gel and stained with SYBR green (Lonza, Basel, Switzerland). The band intensity of COBRA Alu methylation was observed and measured by Strom840 and ImageQuanNT Software (Amersham Biosciences, Little Chalfont, UK).
In addition to methylation level, COBRA can distinguish methylation pattern (Supplementary Figure 1) [Citation14,Citation29,Citation34]. Alu methylation status of 2 CpG dinucleotide of Alu loci was separated into four groups including: Alu loci with two unmethylated CpGs (uCuC); Alu loci with two methylated CpGs (mCmC); Alu loci with 5′-methylated and 3′-unmethylated CpGs (mCuC); and Alu loci with 5′-unmethylated and 3′-methylated CpGs (uCmC). Four bands that differed in their Alu methylation statuses including 133 bp (uCuC), 90 (mCuC), 75 (uCmC), 58 and 32 (mCmC), were quantified (Supplementary Figure 1). The intensity of each band was grouped by its paired length as followed: 133 bp/133 (A), 58 bp/58 (B), 75 bp/73 (C), 90 bp/90 (D) and 43 bp/41 (E). The Alu methylation level (overall or total) was calculated as the percent of the methylated bands (mC) intensity divided by the sum of the mC and unmethylated bands (uC) intensities, ([E + B]/[E +B + 2A + C +D]) × 100. Similarly, to evaluate LINE-1 methylation, the bisulfite-treated DNA was used to amplify with LINE-1-Forward (5′-CCGTAAGGGGTTAGGGAGTTTTT-3′) and LINE-1- Reverse (5′-RTAAAACCCTCCRAACCAAATATA AA-3′) primers at an annealing temperature of 50°C to generate 92 bp PCR amplicon. PCR products were digested with two units of TaqI and TasI (Thermo Scientific) at 65°C overnight. The digested products were analyzed as described above. Four bands that differed in their LINE-1 methylation statuses comprised of 92 bp (mCuC), 60 bp (uCuC), 50 (mCmC) and 18 (uCmC) bp, were quantified (Supplementary Figure 2). The intensity of each band was divided by its paired length as follows: 92 bp/92 (A), 60/56 (B), 50/50 (C), 42/40 (D), 32/28 (E), 18/14 or ([D + E] - [B + C])/2 (F). The LINE-1 methylation level (overall or total) was calculated as the percent of the methylated bands (mC) intensity divided by the sum of the mC and unmethylated bands (uC) intensities, ([A + 2C + F]/[2A + 2B + 2C + 2F]) × 100 [Citation14,Citation29].
Alu methylation evaluation by Ion Torrent sequencing & sequence analysis
DNA from Alu siRNA transfected cells was extracted, bisulfite treated and amplified as described above. The PCR products were sequenced by Ion Torrent sequencer (Ion Torrent™ Personal Genome Machine®, Life Technologies, USA). For each sample, we employed Bismark [Citation35] to perform methylation calls at each CpG sites. Based on results from Bismark, we can calculate% identity to siRNA sequence for each read. Reads in each sample were divided into two groups: 100% identity and low identity to siRNA (% identity < 80%). Reads in the 100% identity group mean siRNA can bind well to a particular site that a read came from. Therefore, reads in the low identity group should have lower methylation level than the 100% identity group. In each read group of each sample, % methylation level at each CpG site were calculated then Student’s t-test on % methylation level were performed between samples with and without added siRNA at each CpG site.
Proliferation assay
Cell proliferation assay was performed. Briefly, 2000 cells (20,000 cell/ml) were seeded in 96-well plate containers in 100 μl media. Cell proliferation was assessed using the MTT reagent (MTT assay; Sigma Aldrich) for 4 days starting 24 h after cell seeding. Moreover, total cell number was counted after seeding Alu siRNA transfected cells over 4 days (cell count assay) [Citation32].
Determination of population doubling of cells
Population doublings (PDs) were determined using the following equation: D = ([B - A (2n)/(A (2n + 1) - A (2n) ]). Where A is number of cells plated, B is number of cells counted after growth period, C is old PDs, D is new PDs and n is the largest number that satisfies the equation A(2n) ≥ B [Citation36].
Cell cytotoxicity assay
Cells were seeded at 4000 cells in 100 μl (40,000 cells/ml) to a 96-well plate. Twenty-four hours later, cells were treated with medium containing an increasing concentration of MMS (Sigma-Aldrich; 0–2 mM) and hydrogen peroxide (H2O2; Sigma Aldrich) (0–150 μM) for one and 24 h, respectively, in a CO2 incubator. Then, the media containing MMS was replaced with normal working media. Cell growth was measured with MTT assay 48 h after treatment [Citation32,Citation33].
Determination of 8-OHdG
8-OHdG levels in both peripheral blood of healthy individuals and cell lines were analyzed using OxiSelect™ Oxidative DNA Damage ELISA Kit (8-OHG Quantitation; Cell BioLabs, CA, USA) according to the manufacturer’s protocol.
Determination of AP site
AP site levels in both peripheral blood of healthy people and cell lines were analyzed using OxiSelect™ Oxidative DNA Damage ELISA Kit (AP site Quantitation; Cell BioLabs) according to the manufacturer’s protocol.
Statistical analysis
For comparisons between two independent groups, the independent sample t-test was used for normally distributed data, and the Mann–Whitney test was used for skewed data. Pearson’s correlation test was used to assess the correlation between continuous variables. SPSS version 17.0 (SSPS Inc., IL, USA) and GraphPad Prism 5 (GraphPad Software Inc., CA, USA) were used for all calculations. A p-value of 0.05 was considered statistically significant.
Results
Correlation between Alu methylation & endogenous DNA damage
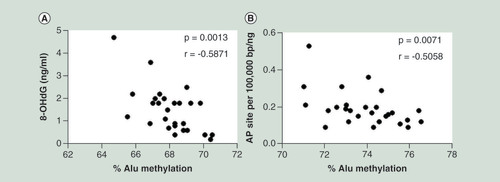
We hypothesized that the human genome may use DNA methylation in IRSs to prevent DNA damage. First, we evaluated whether Alu methylation in peripheral blood mononuclear cell of healthy people (Supplementary Table 1) determines endogenous DNA damage lesions, including 8-OHdG and AP site. We found a strong negative linear correlation between Alu methylation in circulating blood cells and 8-OHdG (r = -0.5871; p = 0.0013) () and AP sites (r = -0.5058; p = 0.0071) ().
(A) The 8-hydroxy-2′-deoxyguanosine and (B) abasic sites were linearly correlated with Alu methylation levels in peripheral blood cells of healthy people. The displayed p-values are from the Pearson’s correlation test.
8-OHdG: 8-Hydroxy-2′-deoxyguanosine; AP site: Abasic site; r: Pearson’s correlation coefficient.
Increase in Alu methylation level was observed in cells with Alu siRNA transection
We transfected Alu siRNA to HeLa, a cervical cancer cell line, and HEK293 and HK2 cell, human kidney cell lines, and observed an increase in the level of Alu methylation ( & Supplementary Figures 2 & 3). Alu siRNA increased Alu methylation significantly (p = 0.005) (). However, scramble siRNA slightly increased Alu methylation (p = 0.019) (). To evaluate roles of Alu methylation, we used lipofectamine transfection as a control in our next experiments. Alu methylation was sustainable because transfected HEK293 also resulted in an increase in Alu methylation level after continuing cell culture for 7 days (). There was a correlation between baseline Alu methylation status and the efficacy of Alu siRNA (Supplementary Figure 4A). The de novo methylation was sequence specific. Alu siRNA did not increase LINE-1 methylation, on the contrary, a trend of LINE-1 hypomethylation inversely associated with Alu methylation was observed (p = 0.04) (, Supplementary Figure 2D & H, & Supplementary Figure 4B). Next, we transfected Alu siRNA to PDL fibroblast (periodontal ligament), the level of Alu methylation significantly increased in siRNA transfected cells ().
(A) The percentage of Alu methylation level in HEK293 cells transfected with only Lipofectamine reagent (lipofect), negative control siRNA from Bioneer (Scramble) and Alu siRNA (100 nM). (B) The percentage of Alu methylation level in HeLa (gray) and HEK293 (white) cells transfected with Alu siRNA. (C) The percentage of LINE-1 methylation level in HeLa (gray) and HEK293 (white) cells transfected with Alu siRNA. (D) The percentage of Alu methylation level in periodontal ligament fibroblast (passage 10) transfected with Alu siRNA. (E–G) The percentage of Alu methylation level in Alu siRNA transfected HEK293 and control (lipofect) of five CpG sites categorized by % identity to siRNA sequence (%ID) including 100% ID (E), >90% ID (F) and <80% ID (G).
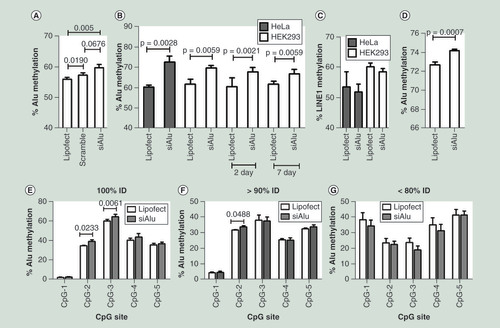
We hypothesized that Alu methylation occurred due to the binding of Alu siRNA to Alu elements. Therefore, the more homology between Alu siRNA sequence and Alu elements, the higher the methylation level in Alu elements. Bisulfite Alu PCR amplicons were evaluated by ion torrent sequencing. We compared DNA methylation levels between Alu siRNA transfected HEK293 and control. For each sample, Bismark [Citation35] was used to perform methylation calls at each CpG sites. We grouped bisulfite Alu PCR amplicons due to percent identity (%ID) value between Alu siRNA and amplicons sequence and evaluated methylation levels of five CpG sites (–G). Significant increase in CpG methylation was observed in 100% ID (). Trend of increase in methylation was observed in CpG-2 in >90% ID (), No significant methylation change was observed when ID less than 80%.
Alu methylation change altered proliferation
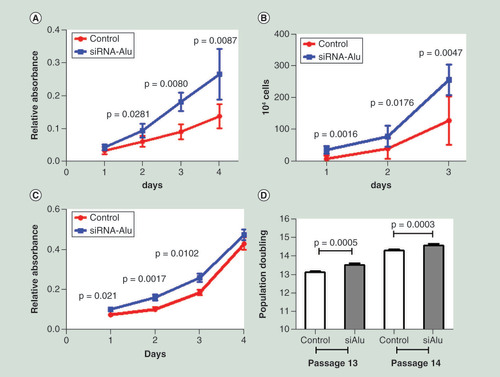
We therefore investigated the phenotypic change when increasing Alu methylation. First by an assessment of cell proliferation, using the MTT assay, we observed a significantly increased rate of proliferation in Alu siRNA transfected HEK293 cells (). We also confirmed this result by counting absolute cell numbers after seeding Alu siRNA transfected HEK293 cells over 3 days (), which again showed a similar increased rate of cell proliferation to Alu siRNA transfection. Of note, there was a correlation between Alu methylation level and rate of cell proliferation (Supplementary Figure 4C–F). To further support this result, we performed MTT assay in PDL fibroblast cells with Alu siRNA transfection (Alu methylation increase was detected), and the rate of proliferation in hypermethylated fibroblast cells was significantly faster than control cells (). Next, to increase the confidence of the proliferation results, we counted cell numbers of PDL cells after transfection at passage 13 and 14 and calculated the PDs value which is more accurate. Likewise, we found that cells with Alu siRNA generated a significantly higher PD value than control cells in both passage 13 and 14 (). Taken together, we proposed that Alu methylation altered cellular phenotypes. Hypermethylated Alu cells proliferated faster than control cells.
After transfection, cells were incubated for 48 h. Then, the transfected cells were seeded into 96-well plates for proliferation evaluation. (A & B) Cell proliferation in HEK293 was assessed by MTT over 4 days after Alu siRNA transfection (A) and absolute cell number was counted after seeding Alu siRNA transfected cells over 3 days (B). (C & D) Cell proliferation in periodontal ligament cells was assessed by MTT over 4 days after Alu siRNA transfection (C) and population doublings of Alu siRNA transfected cells were calculated at passage 13 and 14.
Increasing Alu methylation & genomic instability
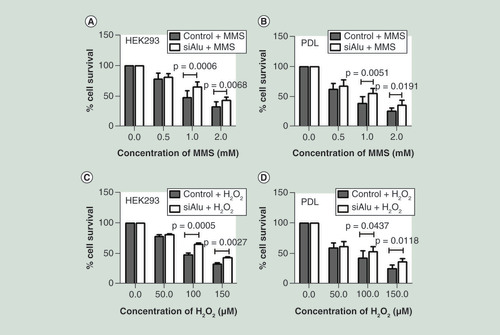
We hypothesized that Alu hypermethylation increased cellular resistance to DNA damage. To prove this possibility, control and Alu siRNA transfected cells with significantly increasing Alu methylation levels were exposed to an increased concentration of DNA damage agents including MMS and H2O2. Exposure of HEK293 and PDL cells to MMS showed that%cell survival of hypermethylated Alu cells was significantly higher than control cells at the concentration of 1.0 and 2.0 mM ( & B). Likewise, exposure of these cells to H2O2 demonstrated a significantly higher%cell survival in hypermethylated Alu cells than control cells ( & D). Our results can conclude that Alu hypermethylated cells increased resistance to DNA damaging agents.
After a significant increase in Alu methylation level confirmation, control and Alu siRNA transfected cells were exposed to the increasing concentration of methyl methanesulphonate for 1 h and H2O2 for 24 h. Then growth was assessed by MTT after 48 h. Error bars indicates ± SD. (A & B) Methyl methanesulphonate sensitivity of HEK293 (A) and periodontal ligament (B). (C & D) H2O2 sensitivity of HEK293 (C) and periodontal ligament (D).
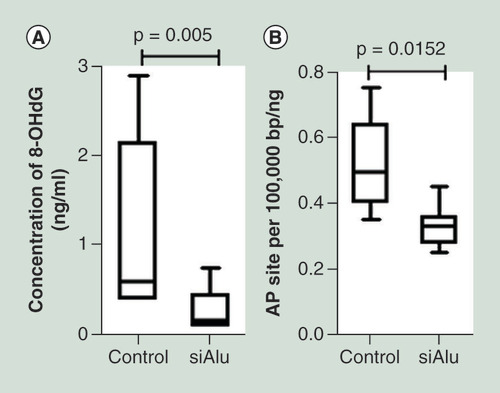
Finally, we monitored the level of endogenous DNA damage, 8-OHdG and AP site. The level of 8-OHdG and AP site were measured in control and Alu siRNA transfected cells. Interestingly, we found a significantly lower 8-OHdG and AP site in Alu hypermethylated cells than control cells ( & B). Of note, there was an inverse correlation between Alu methylation level and both endogenous DNA damage (Supplementary Figure 4G & H).
The level of 8-hydroxy-2′-deoxyguanosine (ng/ml) (A) and abasic site (per 100,000 bp/ng DNA) (B) was significantly increased in cells with Alu siRNA transfection.
8-OHdG: 8-Hydroxy-2′-deoxyguanosine; AP site: Abasic site.
Discussion
Here, we invented a molecular technique that reprograms specific DNA targets so the genome becomes more stable. Alu siRNA can increase Alu methylation levels and plays a role in DNA damage prevention by lowering endogenous DNA lesions and increasing resistance to DNA damaging agents. This technique may be important for cancer and aging prevention in the future.
Alu siRNA increased methylation specifically in Alu elements exhibiting sequence identity to the siRNA sequence without significantly methylating LINE-1, another IRS. This finding suggests that siRNA-induced methylation in humans using a mechanism similar to RNA-directed DNA methylation (RdDM) in plants [Citation37]. Of note, scramble siRNA could promote a variable degree of Alu methylation possible because the scramble siRNA may be able to bind some of Alu elements and consequently methylated CpGs. Another observation is that siRNA did not efficiently methylate human unique sequences and other human IRS. We hypothesize that the number of RdDM proteins in human cells is much lower than the RNA-induced silencing complex. Thus, most siRNAs were incorporated into RNA-induced silencing complex instead of RdDM machinery. Nevertheless, numerous Alu loci are located in the human genome, so Alu elements are easier targeted by RdDM than other IRSs or unique sequences. Another interesting observation was there were trend of LINE-1 hypomethylation in Alu siRNA transfected cells. We guess that Alu siRNA adding DNA methylation to Alu elements may reduce DNA methylation substrate for DNA methylation at LINE-1s.
Global hypomethylation, which promotes genomic instability, is a common molecular event in cancer and aging cells [Citation14,Citation21,Citation38,Citation39]. Here, we reported evidence that a hypomethylated genome is prone to DNA damage accumulation, and this characteristic helps explain why a hypomethylated genome is unstable. First, we observed an inverse correlation between Alu element methylation and endogenous DNA damage. We then observed that endogenous DNA damage was reduced after increased Alu methylation by Alu siRNA. Moreover, Alu hypermethylated cells exhibited resistance to DNA damaging agents. Therefore, Alu methylation prevents genomic DNA damage. DNA damage serves as a precursor of mutation. Thus, reductions in DNA damage accumulation explain why an increased genome-wide methylation level is associated with a reduced mutation rate in the genome. Other consequences of Alu siRNA, such as RNA degradation or translation delay of RNA containing Alu sequences, may be noted. Nevertheless, the inverse correlation between endogenous DNA damages and Alu methylation in Alu siRNA experiments suggested that Alu methylation is the most likely explanation of how Alu siRNA can improve DNA damage resistance.
Mechanisms of how Alu methylation increases DNA damage resistance require further study. We postulated several mechanisms. First, IRS methylation is associated with several epigenetic changes, such as heterochromatin formation [Citation40]. Heterochromatin may act as a shield to prevent DNA exposure to DNA damaging agents and radiation and also reduce DNA activity such as transcription and DNA replication [Citation41]. Second, we observed accumulation of replication-independent endogenous DNA double strand breaks (RIND-EDSBs) in hypermethylate genome [Citation42]. Our recent study indicated that RIND-EDSBs may possess an epigenetic role in reducing DNA strain similar to a small gap between two railway lines [Citation43–45]. Third, DNA repair activity is different depending on chromatin structure [Citation46,Citation47]. This phenomenon may result in variable accumulation of DNA damage between different epigenetic modification regions. Finally, increased Alu methylation may somehow reduce oxidative stress and consequently reduce DNA damage.
Each mechanism will result in different genomic distributions in DNA lesions compared with Alu methylation. If heterochromatin formation or DNA repair-associated chromatin structure can reduce DNA lesions, the reduction should be within the spreading of heterochromatin from Alu methylation. The role of RIND-EDSBs in reducing DNA tension is redundant to topoisomerase [Citation44]. Thus, strain reduction of Alu methylation containing RIND-EDSBs should affect regions more in distance from Alu methylation. Finally, if genome-wide Alu methylation reduces oxidative stress, the distribution of DNA damage should not be associated with Alu loci.
De novo Alu methylated cells in vitro explain Alu methylation-related phenotypes in humans. Here, Alu hypermethylated cells proliferated faster compared with cells that were not Alu siRNA transfected. This result is similar to a previous study demonstrating the positive correlation between the history of catch up growth and increased Alu methylation [Citation28]. In addition, the correlation between reduced Alu methylation levels and increased quantity of DNA lesions explained why individuals with Alu hypomethylation exhibit an increased risk of disabilities related to aging, such as osteoporosis and diabetes-related hypertension [Citation29,Citation30]. Cells with a normal p53 function can recognize DNA damage and consequently halt cell proliferation [Citation48–51]. The lower amount of endogenous DNA damage in Alu hypermethylated cells may explain why both Alu siRNA-transfected cells and individuals with Alu hypermethylation grew faster () [Citation28]. By contrast, accumulation of endogenous DNA damage may send cellular senescence signals [Citation52–54]. Most cancer cells resist cell death [Citation19]. The main mechanism is attributed to a lack of p53 recognition of the DNA damage network [Citation55]. This finding may also explain why Alu hypomethylation in cancer cells does not prevent cell proliferation. Given that Alu methylation can reduce endogenous DNA damage, it is interesting to further explore whether Alu siRNA transfection is able to reprogram the cellular senescence process.
Given that Alu siRNA may target several RNAs in addition to adding Alu methylation, applications using Alu siRNA transfection for therapeutic purposes must be performed with caution. Nevertheless, we have yet to observe harmful effects of this Alu siRNA in HEK293 and human fibroblasts.
Conclusion
In conclusion, Alu hypermethylated cells by in vivo epigenomic variation or by Alu siRNA transfection confers resistance to DNA damage, explaining the instability of the global hypomethylated genome. Reduction of endogenous DNA damage by Alu methylation improves cell proliferation and growth. Therefore, further studies on whether Alu siRNA has therapeutic potential for cellular senescence will be of great interest.
Alu interspersed repetitive element methylation plays a role in DNA damage prevention.
DNA damage accumulation in hypomethylate genome explains how global hypomethylation promoting genomic instability.
Alu siRNA transfection increased the methylation levels of Alu elements, increased proliferation, reduced both endogenous DNA damage and improved resistance to DNA damaging agents. Therefore, future research to assess the therapeutic potential of Alu siRNA on senescence or cancer cells should be considered.
Ethical conduct of research
The study protocol was reviewed and approved by the Ethics Committee of the Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand (IRB No. 461/59). Written informed consents were obtained from all participants.
Supplemental application 1
Download PDF (1.1 MB)Financial & competing interests disclosure
This work was supported by the Thailand Research Fund (DPG5980005 and TRG5880214); the Research Chair Grant 2011 from the National Science and Technology Development Agency (NSTDA), Thailand; the Anantara Siam Bangkok Hotel, Four Season Hotel Care for Cancer Fun Run in coordination with the Thai Red Cross Society; Grants for Development of New Faculty Staff, Ratchadaphiseksomphot Endowment Fund, Chulalongkorn University (GDNS 59-009-23-005) to M Pongpanich; and the Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (PHD/0131/2554 to M Patchsung [student] and Professor A Mutirangura [advisor]). Funding for open access charge: Chulalongkorn University. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/full/10.2217/epi-2017-0096
Additional information
Funding
References
- Gaudet F , HodgsonJG , EdenAet al. Induction of tumors in mice by genomic hypomethylation . Science300 ( 5618 ), 489 – 492 ( 2003 ).
- Chen RZ , PetterssonU , BeardC , Jackson-GrusbyL , JaenischR . DNA hypomethylation leads to elevated mutation rates . Nature395 ( 6697 ), 89 ( 1998 ).
- Tubbs A , NussenzweigA . Endogenous DNA damage as a source of genomic instability in cancer . Cell168 ( 4 ), 644 – 656 ( 2017 ).
- Udomsinprasert W , KitkumthornN , MutiranguraA , ChongsrisawatV , PoovorawanY , HonsawekS . Global methylation, oxidative stress, and relative telomere length in biliary atresia patients . Sci. Rep.6 , 26969 ( 2016 ).
- Wang T-C , SongY-S , WangHet al. Oxidative DNA damage and global DNA hypomethylation are related to folate deficiency in chromate manufacturing workers . J. Hazard Mater.213 , 440 – 446 ( 2012 ).
- Patchsung M , BoonlaC , AmnattrakulP , DissayabutraT , MutiranguraA , TosukhowongP . Long interspersed nuclear element-1 hypomethylation and oxidative stress: correlation and bladder cancer diagnostic potential . PLoS ONE7 ( 5 ), e37009 ( 2012 ).
- Deininger P . Alu elements: know the SINEs . Genome Biol.12 ( 12 ), 236 ( 2011 ).
- Chan SW-L , ZilbermanD , XieZ , JohansenLK , CarringtonJC , JacobsenSE . RNA silencing genes control de novo DNA methylation . Science303 ( 5662 ), 1336 – 1336 ( 2004 ).
- Hamilton A , VoinnetO , ChappellL , BaulcombeD . Two classes of short interfering RNA in RNA silencing . EMBO J.21 ( 17 ), 4671 – 4679 ( 2002 ).
- Mette M , AufsatzW , Van Der WindenJ , MatzkeM , MatzkeA . Transcriptional silencing and promoter methylation triggered by double-stranded RNA . EMBO J.19 ( 19 ), 5194 – 5201 ( 2000 ).
- Chen L , DahlstromJE , LeeSH , RangasamyD . Naturally occurring endo-siRNA silences LINE-1 retrotransposons in human cells through DNA methylation . Epigenetics7 ( 7 ), 758 – 771 ( 2012 ).
- Park HK , MinBY , KimNYet al. Short hairpin RNA induces methylation of hepatitis B virus covalently closed circular DNA in human hepatoma cells . Biochem. Biophys. Res. Commun.436 ( 2 ), 152 – 155 ( 2013 ).
- Castanotto D , TommasiS , LiMet al. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells . Mol. Ther.12 ( 1 ), 179 – 183 ( 2005 ).
- Jintaridth P , MutiranguraA . Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences . Physiol. Genomics41 ( 2 ), 194 – 200 ( 2010 ).
- Lavie L , KitovaM , MaldenerE , MeeseE , MayerJ . CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K (HML-2) . J. Virol.79 ( 2 ), 876 – 883 ( 2005 ).
- Aporntewan C , PhokaewC , PiriyapongsaJet al. Hypomethylation of intragenic LINE-1 represses transcription in cancer cells through AGO2 . PLoS ONE6 ( 3 ), e17934 ( 2011 ).
- Tongyoo P , AvihingsanonY , Prom-OnS , MutiranguraA , MhuantongW , HirankarnN . EnHERV: Enrichment analysis of specific human endogenous retrovirus patterns and their neighboring genes . PLoS ONE12 ( 5 ), e0177119 ( 2017 ).
- López-Otín C , BlascoMA , PartridgeL , SerranoM , KroemerG . The hallmarks of aging . Cell153 ( 6 ), 1194 – 1217 ( 2013 ).
- Hanahan D , WeinbergRA . The hallmarks of cancer . Cell100 ( 1 ), 57 – 70 ( 2000 ).
- Bollati V , SchwartzJ , WrightRet al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects . Mech. Ageing Dev.130 ( 4 ), 234 – 239 ( 2009 ).
- Chalitchagorn K , ShuangshotiS , HourpaiNet al. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis . Oncogene23 ( 54 ), 8841 ( 2004 ).
- Bollati V , BaccarelliA , HouLet al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene . Cancer Res.67 ( 3 ), 876 – 880 ( 2007 ).
- Puttipanyalears C , SubbalekhaK , MutiranguraA , KitkumthornN . Alu hypomethylation in smoke-exposed epithelia and oral squamous carcinoma . Asian Pac. J. Cancer Prev.14 ( 9 ), 5495 – 5501 ( 2013 ).
- Yegnasubramanian S , HaffnerMC , ZhangYet al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity . Cancer Res.68 ( 21 ), 8954 – 8967 ( 2008 ).
- Karpf AR , MatsuiS-I . Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells . Cancer Res.65 ( 19 ), 8635 – 8639 ( 2005 ).
- Maraschio P , ZuffardiO , Dalla FiorT , TiepoloL . Immunodeficiency, centromeric heterochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: the ICF syndrome . J. Med. Genetics25 ( 3 ), 173 – 180 ( 1988 ).
- Ji W , HernandezR , ZhangX-Yet al. DNA demethylation and pericentromeric rearrangements of chromosome 1 . Mutat. Res.379 ( 1 ), 33 – 41 ( 1997 ).
- Rerkasem K , RattanatanyongP , RerkasemAet al. Higher alu methylation levels in catch-up growth in twenty-year-old offsprings . PLoS ONE10 ( 3 ), e0120032 ( 2015 ).
- Jintaridth P , TungtrongchitrR , PreutthipanS , MutiranguraA . Hypomethylation of Alu elements in post-menopausal women with osteoporosis . PLoS ONE8 ( 8 ), e70386 ( 2013 ).
- Thongsroy J , PatchsungM , MutiranguraA . The association between Alu hypomethylation and severity of type 2 diabetes mellitus . Clin. Epigenetics9 , 93 ( 2017 ).
- De Bont R , Van LarebekeN . Endogenous DNA damage in humans: a review of quantitative data . Mutagenesis19 ( 3 ), 169 – 185 ( 2004 ).
- Dango S , MosammaparastN , SowaMEet al. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation . Mol. Cell44 ( 3 ), 373 – 384 ( 2011 ).
- Larrosa PF , GreccoMR , GómezDMet al. RAC3 more than a nuclear receptor coactivator: a key inhibitor of senescence that is downregulated in aging . Cell Death Dis.6 ( 10 ), e1902 ( 2015 ).
- Wangsri S , SubbalekhaK , KitkumthornN , MutiranguraA . Patterns and possible roles of LINE-1 methylation changes in smoke-exposed epithelia . PLoS ONE7 ( 9 ), e45292 ( 2012 ).
- Krueger F , AndrewsSR . Bismark: a flexible aligner and methylation caller for bisulfite-Seq applications . Bioinformatics27 ( 11 ), 1571 – 1572 ( 2011 ).
- Phipps SM , BerletchJB , AndrewsLG , TollefsbolTO . Aging cell culture: methods and observations . Biological Aging371 , 9 – 19 ( 2007 ).
- Wendte JM , PikaardCS . The RNAs of RNA-directed DNA methylation . Biochim. Biophys. Acta1860 ( 1 ), 140 – 148 ( 2017 ).
- Hoffmann MJ , SchulzWA . Causes and consequences of DNA hypomethylation in human cancer . Biochem. Cell Biol.83 ( 3 ), 296 – 321 ( 2005 ).
- Ross JP , RandKN , MolloyPL . Hypomethylation of repeated DNA sequences in cancer . Epigenomics2 ( 2 ), 245 – 269 ( 2010 ).
- Baylin SB , EstellerM , RountreeMR , BachmanKE , SchuebelK , HermanJG . Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer . Hum. Mol. Genetics10 ( 7 ), 687 – 692 ( 2001 ).
- Grewal SI , JiaS . Heterochromatin revisited . Nat. Rev. Genetics8 ( 1 ), 35 ( 2007 ).
- Pornthanakasem W , KongruttanachokN , PhuangphairojCet al. LINE-1 methylation status of endogenous DNA double-strand breaks . Nucleic Acids Res.36 ( 11 ), 3667 – 3675 ( 2008 ).
- Kongruttanachok N , PhuangphairojC , ThongnakAet al. Replication independent DNA double-strand break retention may prevent genomic instability . Mol. Cancer9 ( 1 ), 70 ( 2010 ).
- Thongsroy J , MatangkasombutO , ThongnakA , RattanatanyongP , JirawatnotaiS , MutiranguraA . Replication-independent endogenous DNA double-strand breaks in Saccharomyces cerevisiae model . PLoS ONE8 ( 8 ), e72706 ( 2013 ).
- Pongpanich M , PatchsungM , ThongsroyJ , MutiranguraA . Characteristics of replication-independent endogenous double-strand breaks in Saccharomyces cerevisiae . BMC Genomics15 ( 1 ), 750 ( 2014 ).
- Xu Y , XuC , PriceBD . Mechanistic links between ATM and histone methylation codes during DNA repair . Prog. Mol. Biol. Transl. Sci.110 , 263 – 288 ( 2012 ).
- Jakob B , SplinterJ , ConradSet al. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin . Nucleic Acids Res.39 ( 15 ), 6489 – 6499 ( 2011 ).
- Musich PR , ZouY . DNA-damage accumulation and replicative arrest in Hutchinson–Gilford progeria syndrome . Biochem. Soc. Trans.39 ( 6 ), 1764 – 1769 ( 2011 ).
- Liu Y , RusinolA , SinenskyM , WangY , ZouY . DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A . J. Cell Sci.119 ( 22 ), 4644 – 4649 ( 2006 ).
- Fritsche M , HaesslerC , BrandnerG . Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents . Oncogene8 ( 2 ), 307 – 318 ( 1993 ).
- Bunz F , DutriauxA , LengauerCet al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage . Science282 ( 5393 ), 1497 – 1501 ( 1998 ).
- Best BP . Nuclear DNA damage as a direct cause of aging . Rejuvenation Res.12 ( 3 ), 199 – 208 ( 2009 ).
- Freitas AA , De MagalhaesJP . A review and appraisal of the DNA damage theory of ageing . Mut. Res.728 ( 1 ), 12 – 22 ( 2011 ).
- Burhans WC , WeinbergerM . DNA replication stress, genome instability and aging . Nucleic Acids Res.35 ( 22 ), 7545 – 7556 ( 2007 ).
- Muller PA , VousdenKH . Mutant p53 in cancer: new functions and therapeutic opportunities . Cancer Cell25 ( 3 ), 304 – 317 ( 2014 ).