

Abstract
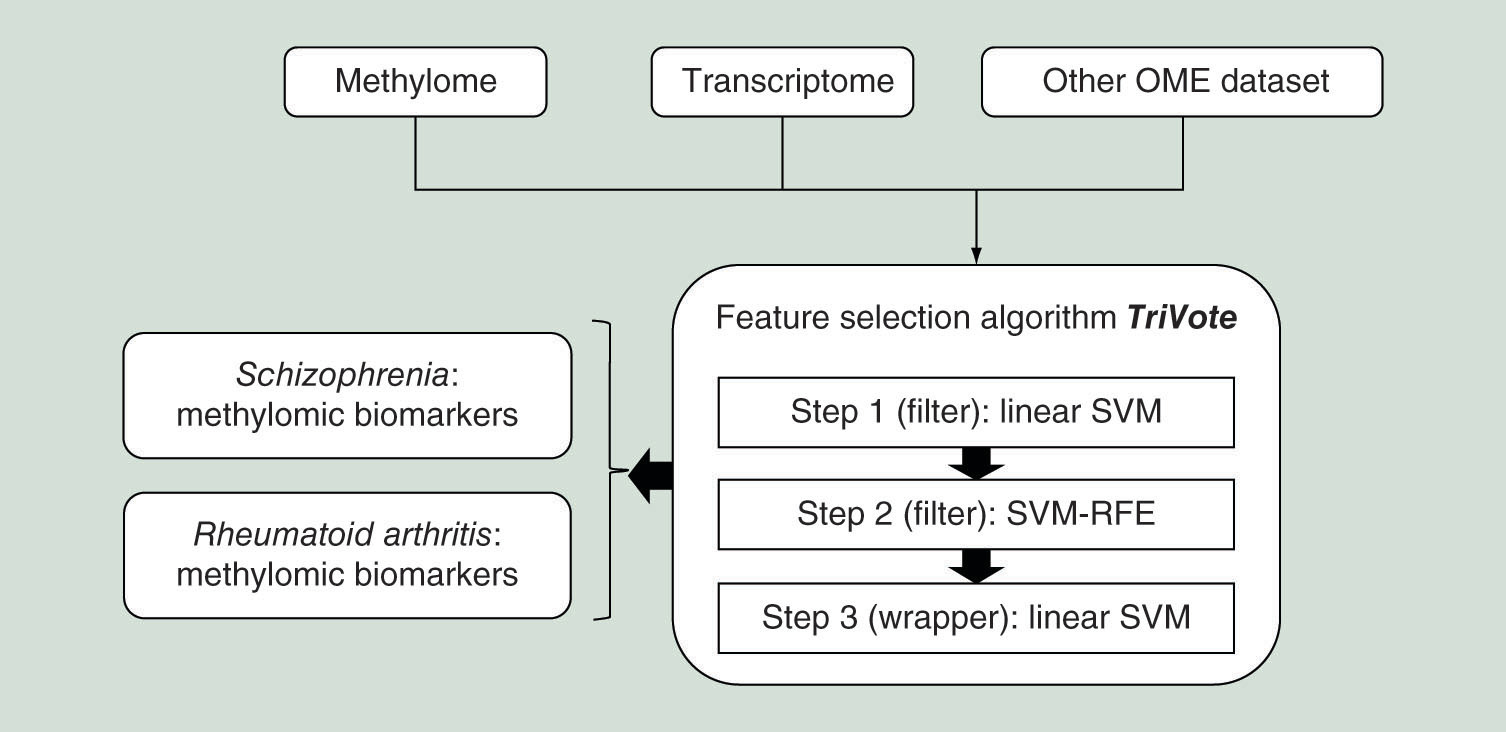
Aim: Transcriptomic and methylomic patterns represent two major OMIC data sources impacted by both inheritable genetic information and environmental factors, and have been widely used as disease diagnosis and prognosis biomarkers. Materials & methods: Modern transcriptomic and methylomic profiling technologies detect the status of tens of thousands or even millions of probing residues in the human genome, and introduce a major computational challenge for the existing feature selection algorithms. This study proposes a three-step feature selection algorithm, TriVote, to detect a subset of transcriptomic or methylomic residues with highly accurate binary classification performance. Results & conclusion: TriVote outperforms both filter and wrapper feature selection algorithms with both higher classification accuracy and smaller feature number on 17 transcriptomes and two methylomes. Biological functions of the methylome biomarkers detected by TriVote were discussed for their disease associations. An easy-to-use Python package is also released to facilitate the further applications.
Graphical abstract
Acknowledgements
The constructive comments from the anonymous reviewers were greatly appreciated.
Financial & competing interests disclosure
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13040400) and the startup grant from the Jilin University. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Notes
SVM: Support vector machine; SVM-RFE: Support vector machine recursive feature elimination.