

Abstract
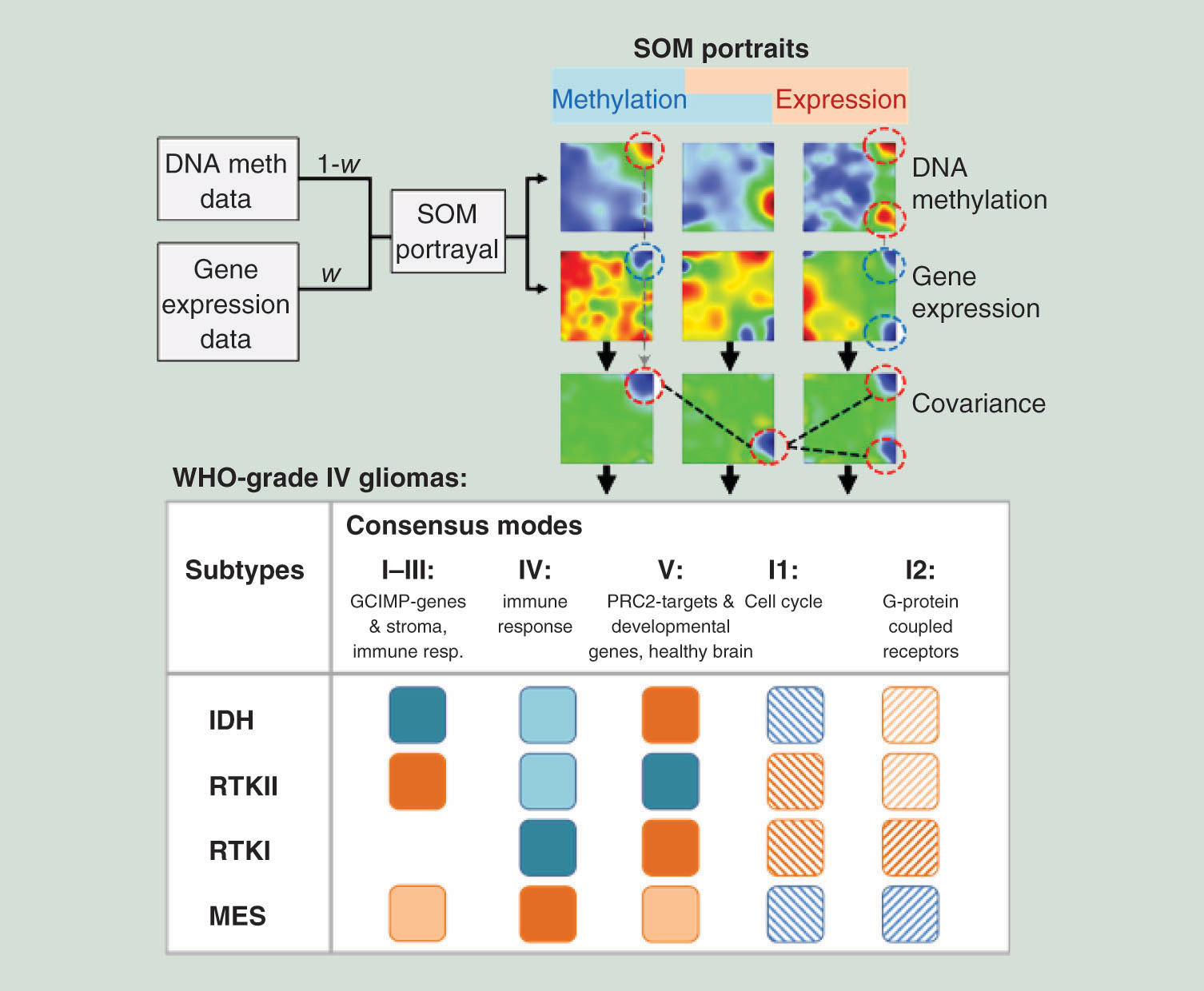
Aim: We present here a novel method that enables unraveling the interplay between gene expression and DNA methylation in complex diseases such as cancer. Materials & methods: The method is based on self-organizing maps and allows for analysis of data landscapes from ‘governed by methylation’ to ‘governed by expression’. Results: We identified regulatory modules of coexpressed and comethylated genes in high-grade gliomas: two modes are governed by genes hypermethylated and underexpressed in IDH-mutated cases, while two other modes reflect immune and stromal signatures in the classical and mesenchymal subtypes. A fifth mode with proneural characteristics comprises genes of repressed and poised chromatin states active in healthy brain. Two additional modes enrich genes either in active or repressed chromatin states. Conclusion: The method disentangles the interplay between gene expression and methylation. It has the potential to integrate also mutation and copy number data and to apply to large sample cohorts.
Graphical abstract
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/full/10.2217/epi-2017-0140
Financial & competing interests disclosure
This work is supported by the Federal Ministry of Education and Research (BMBF), project grant SysGlio and the DKTK joint funding project ‘Next generation molecular diagnostics of malignant gliomas’ and the German Glioma Network. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.