
Abstract
Exposure times and dosage required for dietary components to modify DNA methylation patterns are largely unknown. Aim: This exploratory research represents the first genome-wide analysis of DNA methylation changes during a randomized-controlled-trial (RCT) for dietary supplementation with broad spectrum vitamins, minerals and amino acids in humans. Methods: Genome-wide changes in methylation from paired, peripheral blood samples were assessed using the Infinium Methylation EPIC 850 K array. Results: Methylation increased at 84% of the most significant differentially methylated CpGs; however, none showed significance after adjustment for genome-wide testing. Conclusion: Micronutrient supplementation is unlikely to have a substantial biological effect on DNA methylation over 10 weeks; however, the trend toward hypermethylation that we observed is likely to become more marked with longer exposure periods.
Keywords::
Epigenetic modification of DNA provides a mechanistic basis through which environmental stimuli can modulate human gene activity. Changes in the pattern of epigenetic marks on the DNA, such as the methylation of cytosine, have been widely implicated in the development of many key diseases, including cancer, neuropsychiatric disorders, cardiovascular disease and obesity [Citation1–5]. Because epigenetic modifications are dynamic, there is substantial interest and speculation surrounding the potential development of targeted epigenetic manipulations for disease treatment. Understanding the conditions that contribute to DNA methylation changes and the implications of such changes is fundamental to the aetiology and successful treatment of disease, and is one of the major contemporary challenge in human genetics [Citation6–8].
DNA methylation patterns are initially established during gametogenesis, fertilization and in utero development and enable genes to be expressed in a tissue or cell specific manner, which is important in cell growth, differentiation, immunity and aging [Citation9–16]. However, a subset of marks are dynamic throughout life and can be modified by interactions with environmental stimuli such as cigarette smoke, pollutants, oxidative stress and alcohol consumption [Citation17–23]. Most inferences of dietary effects on the human methylome are drawn from animal studies or past naturalistic events, such as the Dutch Hunger Winter and similar events [Citation24]. Although it is accepted that environmental factors including nutrition may promote epigenomic modifications, very little is known about the exposure time and dosage required for changes to occur. Furthermore, few studies of methylation change have been performed using gene-specific resolution, which can provide insight into potential biological implications.
It has been hypothesized that during the slow growth period (ages 8–12 years) gene expression is particularly responsive to environmental stimuli such as diet, and this is likely to reflect epigenetic changes [Citation25–27]. We investigated if micronutrient treatment (consisting mainly of vitamins, minerals and amino acids) correlated with genomic DNA methylation changes measured in peripheral blood, which could potentially occur through one carbon metabolism pathways [Citation28]. This study was performed in a sample of children between the ages of 7 and 12 years, diagnosed with attention-deficit/hyperactivity disorder (ADHD). These individuals were a subset of children who had participated in a 10-week double-blind randomized controlled trial (RCT) comparing a broad spectrum micronutrient treatment with a placebo to investigate therapeutic effects on ADHD symptoms [Citation29]. In this study, micronutrient administration was associated with improved overall function, reduced impairment and improved inattention, emotional regulation and aggression relative to placebo [Citation29].
This systematic analysis is unique as the treatment was administered using a RCT design, which enables the direct comparison of pre- and postmethylation levels. Our analysis also provides a significant advance over traditional techniques because it assesses DNA methylation levels at approximately 800,000 CpG sites across the human genome, with nucleotide resolution. This approach enables assessment of individual increases and decreases in DNA methylation levels, at specific gene loci.
Methods
Randomized control trial layout
The original study design followed a standard double-blind RCT, where children diagnosed with ADHD were randomized to either placebo or treatment groups at a 1:1 ratio. Two blood samples were taken from each participant, one sample at baseline and one sample post-treatment. Due to technical difficulties paired blood samples were available for only 36 participants from this larger study. Although the sample size was reduced, the randomization was not influenced. Methylation analysis was performed on each blood sample, equaling a total of 72 samples. All participants were under supervised administration of capsules, where the treatment group received a formulation which consists of a blend of vitamins, minerals, amino acids and antioxidants (see Supplementary Table 1 for ingredients of both treatment and placebo capsules). Participants began by taking one capsule, three-times each day, increasing the dose by three capsules every two days up to a target dose of 12 capsules per day: four taken at three different intervals. An in-depth description of the full study design can be found in Rucklidge et al. [Citation29].
Data preprocessing & quality control
DNA was extracted from whole blood by automated magnetic particle processing, using the KingFisher Nucleic Acid purification system (Thermo Fisher Scientific, MA, USA) as per the manufacturer’s protocol. Sodium bisulfite treatment of genomic DNA and genome wide DNA methylation profiles were assayed with the Illumina Infinium MethylationEPIC 850 K kit, at the Australian Genome Research Facility (NSW, Australia). Analysis was performed on all samples in a single batch.
Data analysis was performed using the Minfi and Limma Bioconductor software packages within the R statistical program (www.R-project.org). Custom scripts were used to compute raw methylation values; however, the workflow was based upon published scripts [Citation30]. Sample sex was checked by performing estimates using X/Y chromosome methylation within Minfi. Correct sample matching of DNA from pre- and post-treatment analysis was ensured by extracting genotypes from the 65 illumina SNP probes using Minfi, and performing hierarchical cluster analysis. Hierarchical clustering was performed within R using the ‘Minkowski’ distance measure, which defines a distance between two points in a distance matrix. All samples passed QC and were normalized together using Subset-quantile Within Array Normalization (SWAN) [Citation31] and variance-stabilizing transformation [Citation32,Citation33]. Probes containing detection p-values >0.05 for 1% or more samples, were excluded from further analysis. Because the study sample contained both males and females, probes mapping to sex chromosomes were removed as were probes with polymorphic hybridizing potential and homology to common SNPs [Citation34]. The final data frame contained 792,347 probes available for analysis.
Estimation of cell fractions
Changes in the distribution of white blood cell fractions between subpopulations were estimated within Minfi, using the algorithms developed by Houseman et al. [Citation35]. This analysis characterizes cell composition based upon the DNA methylation signatures of each of the principal immune components, and was performed using data from whole blood, developed for the Illumina 450 K array [Citation36]. There were no statistical differences between estimated cell fractions at either the initial or final time points for the treatment and control groups. This indicated that the methylation signals were not influenced by variation in cell fractions between each collection time point and therefore, this variability was not incorporated into the final statistical model (Supplementary Figure 1).
Identification of differentially methylated CpGs
The methylation status of each probe was calculated using normalized probe signals represented as methylation values (M-values) and β-values. M-values were generated within Minfi as the logit transformation in log2 scale of signal intensity values for methylated divided by unmethylated intensity values. β-values (average DNA methylation level for each probe) were used for data visualization and range from 0 (unmethylated) to 1 (methylated). β-values were generated by dividing the methylated probe signal with the sum of the methylated and unmethylated probe signals. Unless stated otherwise, statistical analyses were performed using the change in methylation status of each probe. This was calculated by subtracting the baseline measurement (methylation initial) from the post-treatment measurement (methylation final) (mf-mi), for each of the 36 individuals. Differentially methylated positions which correlated with treatment were identified using a linear regression model within the Limma package, with adjustment for multiple testing. This statistical analysis was performed using the change in methylation (mf-mi) values + treatment (active vs control) + gender + age + smoking during pregnancy. Pathway analysis was performed by comparison with the Kyoto Encyclopedia of Genes and Genomes (KEGG) [Citation37] and gene ontology databases (GO) [Citation38] using the missmethyl R package [Citation39], with correction for probe bias.
Global DNA methylation change
Global DNA methylation change was calculated using mean β-values (post-pre) from all probes for each individual in the active and control groups. The absolute mean difference between control and treatment groups was then calculated. Probe positions which demonstrated a mean difference in β-values greater than 10% were assessed in the full dataset (72 samples) using the analysis of covariance (ANCOVA) model: methylation post–methylation pre + treatment. The significance threshold was adjusted for probe wide multiple testing using BY correction (n = 792,347).
Differentially methylated regions
Differentially methylated regions (DMRs) were interrogated within the Minfi package using the statistical package DMRcate [Citation40]. A methylation differential cut off of 10 was used, and unless stated otherwise significance was determined using a false discovery rate (FDR) of 0.05 in conjunction with a p-value cut off of 0.05.
Blood measurements
At baseline and study completion, hematological variables, biochemical variables, thyroid function, prolactin, fasting glucose, homocysteine, iron, zinc, vitamin D, vitamin B12, copper, blood pressure, height and weight were recorded [Citation29].
Genotyping MTHFR
PCR amplification was carried out in a total reaction volume of 25 μl containing 1 × PCR reaction buffer with 1.5 mM MgCl2 (Roche Diagnostics, Basel, Switzerland), 0.5 μM of each primer (IDT, IA, USA)(), 0.2 μM each deoxynucleoside triphosphate, 1 M betaine, 0.5 U Fisher Taq-ti polymerase (Fisher Biotec, Wembley, Australia) and approximately 20 ng of genomic DNA. Standard thermal cycling conditions consisted of an initial denaturation step of 95°C for 2 min, followed by 35 cycles of 95°C for 30 s, 63°C annealing for 15 s and 72°C for 45 s with a final extension of 72°C for 5 min.
Table 1. Clinical description of cohort.
Table 2. Oligonucleotide sequences.
Sanger DNA sequencing was carried out on PCR products purified using AcroPrep (PALL Corporation, NY, USA) 96-well filter plates (omega 30 K) then resuspended in water. Purified PCR amplicons (∼10 ng) were sequenced with the appropriate primer () using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, CA, USA), following the manufacturers protocol. Sequencing reaction products were run on an AB3130xl fragment analysis system equipped with a 50 cm capillary using POP7 polymer.
Results
Characteristics of the participants
Participants ranged between ages of seven and 12 years, and consisted of six females and 30 males (). Ethnicity included 29 New Zealand Europeans and seven New Zealand Maori participants. The groups were well matched and there were no group differences at baseline in severity of ADHD behaviors, socioeconomic status, BMI, dietary patterns and presence of other comorbid disorders. Consistent with the results reported in the larger sample [Citation29], based on the Clinical Global Impression (CGI-I), there were more responders in the treatment group relative to the placebo group (50% vs 17% respectively; p < 0.05). There were no significant differences between the subsample and the larger sample for the measured variables presented in (p > 0.05). Adherence to pill taking was measured by parent reports and returned pill counts. Parents’ reports indicated dherence for the active group was 94.04% (±3.58), and 93.53% (±7.14) for the placebo group. Pill counts indicated that adherence in the active group was 85.15% (±12.54) and 92.89% (±8.06) for the placebo group. Both measures demonstrated excellent adherence with no significant difference between the groups (p > 0.05).
Global DNA methylation change
There was no significant change in global methylation between the treatment and placebo group. The treatment group demonstrated a mean increase by 0.0022 (SD = 0.0081), and the placebo group demonstrated a decrease by 0.0004 (SD = 0.0069). Eleven probes demonstrated a mean difference in β-values greater than 10%, between the two conditions; however, these did not reach individual significance (Supplementary Table 2) (adj p = 0.9).
Individual CpG analysis
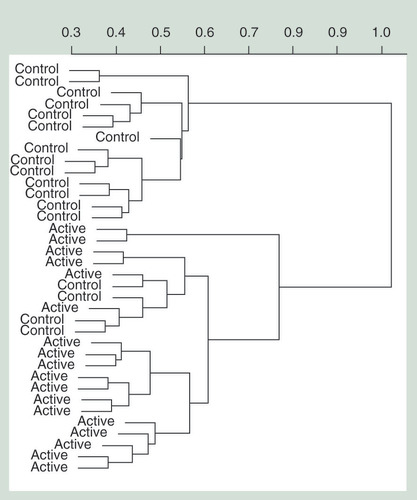
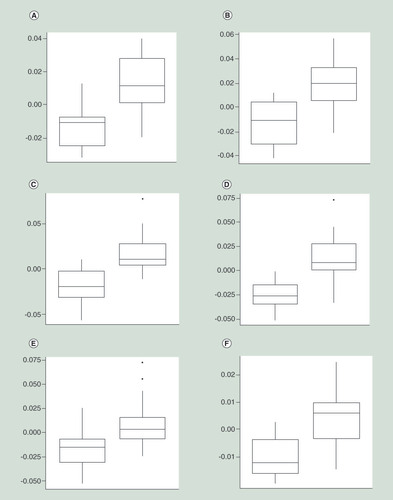
The association between micronutrient treatment and DNA methylation changes was determined for each individual probe site. From the top 100 most significant differentially methylated CpGs (Supplementary Table 3), 84% demonstrated an increase in methylation from baseline to post-treatment (). Hierarchical clustering of these probes had partial success at differentiating the control and treatment groups (). However, no individual CpG site reached significance, after adjustment for multiple testing (adj p > 0.5) () (for full list of top 100 CpGs refer to Supplementary Table 3, and the Manhattan plot in Supplementary Figure 2). The top most significant CpG sites demonstrated less than 5% change in methylation ().
Table 3. Differentially methylated probes identified by Minfi.
Hierarchical clustering performed using the top 100 most significant probes (Supplementary Table 3). The change in relative methylation values (β) is represented on the y-axis and individuals from each treatment are represented on the x-axis.
Change in methylation values is represented on the y-axis, in each instance the boxplot on the left represents the control group and the boxplot on the right represents the treatment group. (A) Chr1:204985932; (B) Chr3:183913285; (C) Chr8:25829073; (D) Chr11: 16430764; (E) Chr17: 39120012; (F) Chr10:8092775.
The genes corresponding to the top 100 CpG sites were assessed for significant biological relevance by pathway analysis within the GO and KEGG databases. Comparison against the KEGG database identified a significant (FDR = 0.04; p = 0.0001) association of three genes with the olfactory transduction pathway (path:hsa04740). However, this should be interpreted with caution as the olfactory transduction pathway is estimated to involve 388 genes.
Differentially methylated regions
We next investigated if multiple probes mapping to the same genomic loci demonstrated consistent methylation changes. Individually significant CpGs were not detected using the default settings (FDR = 0.05), which were consistent with earlier observations (), and consequently DMRs were not detected using these settings. The most significant differentially methylated regions demonstrated a small (∼5%) mean β-fold change value and did not meet significance according to the Stouffer transformation ().
Table 4. Differentially methylated regions.
Blood phenotype measures & MTHFR genotyping
Multiple measures were recorded from each peripheral blood sample, including folate, homocysteine and B12 (for the full list see [Citation29]). Homocysteine levels reduced with micronutrient administration (Supplementary Figure 3), which is a common observation [Citation41,Citation42]. A subset of individuals demonstrated elevated plasma folate and B12 levels after micronutrient supplementation in the treatment group (Supplementary Figures 4 & 5). We investigated if this effect was due to a genetic polymorphism in the MTHFR gene. MTHFR is a rate limiting enzyme of one carbon metabolism, which catalyses the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, a co-substrate for homocysteine remethylation to methionine. We genotyped two variants of the MTHFR gene (C677T and A1298C), as they are commonly observed in the population and have direct relevance to MTHFR enzyme activity [Citation43,Citation44]. The study did not contain individuals who were double homozygous for the minor SNPs, and allele frequencies were consistent with the European population in the Ensembl database () [Citation45]. When analyzed together, MTHFR genotypes did not appear to have a significant effect on baseline homocysteine or folate levels, in the treatment or placebo groups (p > 0.2). However, when each genotype was analyzed individually, the C677T genotype was weakly associated with folate change (post- vs pretreatment) of the treatment group during the trial (p = 0.07).
Table 5. Comparison of the MTHFR genotype frequencies observed in our attention-deficit/hyperactivity disorder study sample, with the wider population of similar descent.
Identifying differentially methylated regions in relation to MTHFR gene variants
It has been suggested that MTHFR gene variants may impact on DNA methylation profiles by modulating folate metabolism; however, there is limited experimental data detailing the effects. Therefore, we assessed baseline methylation levels using a linear model that accounts for the interaction between the MTHFR C677T and A1298C variants. Using the default settings (FDR = 0.05), five probes within a 155 bp region upstream of the PM20D1 gene on chromosome 1 were identified that displayed significant differential methylation (). Two individuals with the T677T genotype, and one individual with the C1298C genotype were excluded to avoid bias. Therefore, the analysis compared the methylation levels between the common homozygote and the heterozygous genotypes. However, when the analysis was performed on all the samples, the results indicated that this effect may be further increased by having two copies of the 1298C rare allele (Supplementary Table 4 & Supplementary Figure 6). Because our genotyping by sequencing analysis cannot provide phase information for the alleles, we also performed separate statistical analysis for each variant. No DMRs were significant after Stouffer transformation when each genotype was individually assessed.
Table 6. Significant differentially methylated regions.
Top most significant individual CpG sites
We then investigated the association between MTHFR genotypes and differential methylation for each individual probe (; for the top 100 probes see Supplementary Table 5). C1298C or T677T homozygote individuals were excluded from the analysis. Analysis demonstrated two genes (ZNF827 and FAM160A2) displayed significant differential methylation after adjusting for multiple testing (p = 0.018). Separate analyses of each C677T and A1298C MTHFR variants failed to identify significant probes (adj. p > 0.35) and the top 100 probes did not bind to comparable gene regions in each separate analysis (data not shown).
Table 7. 10 most significant probes with respect to A1298C* and C677T genotypes.
The gene names from the top 100 most significant probes presented in were analyzed for biological relevance against the KEGG database and six significant pathways were identified (FDR < 0.01) (). Four of these pathways are related to metabolism, one to platinum drug resistance and one to cellular energy (AMPK signaling pathway).
Table 8. KEGG gene pathway analysis of top 100 probes demonstrating differential methylation.
Discussion
The one carbon metabolism cycle provides an appealing pathway through which environmental factors, such as diet, could directly influence DNA methylation patterns. S-adenosylmethionine (SAM) is an important co-substrate that is regenerated through one carbon metabolism, and facilitates the conversion of cytosine to 5-methylcytosine by providing DNA methyltransferase (DNMT) enzymes with a labile methyl group [Citation46,Citation47]. Cellular fluctuations in the levels of SAM have been shown to affect methyltransferase activity with potential epigenetic consequences [Citation48–50]. Many dietary micronutrients, including folate, betaine, choline, riboflavin, vitamin B6 and vitamin B12 act as co-factors and regulators of one carbon metabolism, and several studies have demonstrated that global DNA methylation levels correlate with blood folate concentrations [Citation51–53]. Additionally, gene mutations that decrease enzyme activity such as the methylenetetrahydrofolate reductase (MTHFR) gene may also alter genome wide methylation profiles, although experimental evidence for this is limited and contradictory [Citation44,Citation53,Citation54]. Disrupting the bioavailability of SAM may lead to an altered methylation profile, which provides a mechanism through which environmental factors can modulate DNA methylation and may influence gene activity [Citation55–57].
Our analysis measured genomic methylation changes from peripheral blood cells during a 10-week double-blind RCT trial comparing micronutrient treatment and placebo. We believe this exploratory research represents the first genome-wide analysis of DNA methylation changes associated with controlled nutrient manipulation in humans. For each individual, the change in methylation was assessed using the Illumina Methylation EPIC850 K array by subtracting the pretreatment values from the post-treatment values. The correlation between changes in methylation and treatment were then assessed using a linear model that also accounted for the effects of smoking during pregnancy, gender and age. We then extended this analysis to investigate the effect of two common variants observed within the MTHFR gene on pretreatment DNA methylation profiles.
Methylomic changes during the 10 week micronutrient trial
From the top 100 probes, 84% demonstrated an increase in methylation, which is substantially higher than expected if the effects were random. This observation was suggestive of a general trend to increased global methylation in the treatment group, compared with the placebo group. However, this observation was not consistent for genome wide methylation levels (p > 0.05). Given the trend toward increased global methylation, hierarchical clustering of the top 100 most significant probes was carried out, which demonstrated moderate success at differentiating the treatment and control groups based on DNA methylation change. However, the individual CpG sites exhibited less than 5% change in the magnitude of methylation and did not reach significance after correcting for genome wide multiple testing. Only 11 probes displayed a mean difference in methylation greater than 10% between the treatment and control groups. These probes were not significantly associated with treatment; however, this demonstrates that random fluctuations in genomic methylation were very minor for this sample, over the 10-week period.
The two most significant CpG sites occurred within the promoter of the NFASC gene and within noncoding region of chromosome 3, downstream of the ABCF3 gene. NFASC is a cell adhesion molecule that is largely expressed in the brain where it is involved in neurite outgrowth [Citation58], and likely plays a role in axon potential through an interaction with voltage-gated sodium channels [Citation59]. From the top 20 most significant differentially methylated genes, CDH13 and PLK4 have previously been reported to respond to folate levels [Citation60,Citation61]. CDH13 expression was found to associate with folate concentration in cancer cells [Citation60,Citation61], and has also been identified as a candidate risk gene for ADHD in several GWAS studies [Citation62–64]. Cadherins are calcium-dependent proteins, which play a role in cell–cell adhesion. CDH13 is expressed in the brain where it regulates growth of neural cells [Citation65] and has also been proposed as a strong candidate gene for participating in brain mechanisms relevant for developing and quitting addictions [Citation66].
It is possible that small changes at multiple genes converging on the same or similar biological pathways can have a combined biological effect. From the top 100 most significant probes, we observed a significant association with gene families involved in olfactory transduction pathways. The olfactory sense is biologically complex and plays a role in perception of food and flavor [Citation67]. The relevance of a role for methylation and micronutrient supplementation is not known; however, the pathway is highly reliant on ions, such as Ca2+.
Analysis of methylomic differences with respect to MTHFR genotype
The MTHFR gene codes for a rate-limiting enzyme of one carbon metabolism and MTHFR genetic variants have been demonstrated to cause reduced enzymatic function [Citation43,Citation68], which can correspond with an accumulation of folates in the blood [Citation69]. In our sample, we observed that the 677T variant was weakly associated with elevated folate (p = 0.07), which suggests this variant may be partially explaining the observed accumulation of folate. However, MTHFR is one of several enzymes involved in folate metabolism, and it is likely that variation in other genes, such as MTR or MTRR, may also contribute to elevated folate levels in this sample. There has been substantial speculation involving the contribution of MTHFR gene mutations in neuropsychiatric disease, such as autism, schizophrenia and ADHD. This hypothesis is not strongly supported by the scientific literature [Citation70], and in our sample of ADHD children, we did not observe unusual genotype frequencies.
Reduced functionality of the MTHFR gene has also been proposed to influence global DNA methylation [Citation44,Citation51,Citation71]. We therefore investigated if MTHFR genotype correlated with abnormal DNA methylation in our sample group. One region upstream of the gene PM20D1 displayed differential methylation between common homozygotes and heterozygotes. From the top 100 differentially methylated probes, the genes ZNF827 and FAM160A2 demonstrated significant differential methylation (adj p = 0.017). ZNF827 has been associated with autism [Citation72–74] and FAM160A2 with schizophrenia [Citation75]; however, there is no clear evidence for an interaction between these genes and MTHFR. MTHFR variants have previously been demonstrated to influence methylation at the SLC6A4 gene [Citation76], and our top 100 probes included four genes of the solute carrier superfamily, SLC9C2, SLC41A1, SLC43A3 and SLC2A8.
KEGG pathway analysis demonstrated that there was a significant enrichment in genes corresponding to metabolic pathways, platinum drug resistance and also the AMPK signaling pathway. In addition to the central role that MTHFR has in carbon and folate metabolism, it has also been implicated in survival outcome after platinum/5-Fu-based neoadjuvant chemotherapy [Citation77,Citation78], and AMPK signaling [Citation79]. Two MTHFR specific probes were identified within the top 100 probes, and the binding site of these probes is approximately 2 kb away from our genotyped region, indicating this is not a SNP specific artifact. The biological relevance of the identified pathways provides support for the correlation between MTHFR genotype and differential methylation at these gene regions.
Strengths & limitations
The results suggest that the interaction of both MTHFR genotypes is a determining factor of DNA methylation patterns. This is likely to indicate that compound heterozygosity is driving the observed effects and future studies need to incorporate appropriate haplotyping methods for these variants. Our investigations of micronutrient supplementation had several strengths, including the paired sample design which enabled an evaluation of within-individual variation during the trial period. Although the large number of CpG sites of the methylation array substantially decreased statistical power after correction for multiple testing, this enabled a genome-wide assessment. This was partially offset by the highly conserved methylation levels observed within our study, which is likely due to the young age of participants and the small age range across the study (five years from youngest to oldest). The conserved methylation profiles of the study sample enabled detection of small methylation changes, and even with increased statistical power it seems unlikely we would have observed larger effect sizes for the micronutrient study.
There are several limitations to this exploratory analysis which may have masked potential findings. The observed methylation changes are reflective of genomic DNA isolated from peripheral blood, and the results of our study may not represent the tissues which are most affected by dietary supplementation. There has been limited research on the methylomic impacts of diet, and the correlation between blood methylation profiles and metabolically relevant tissues has not received investigation. However, there is increasing evidence for meaningful correlations between DNA methylation changes in peripheral blood cells and central tissues, such as the brain [Citation80,Citation81]. The short 10-week treatment time was a substantial limitation, especially as the majority of large epigenetic changes are associated with long-term exposure to environmental stressors, such as smoking [Citation19,Citation82–84]. However, our findings have a comparable effect size to similar studies performed in humans [Citation85,Citation86]. Furthermore, individuals from our study did not have nutritionally restricted diets, and therefore the magnitude of methylomic changes is expected to be less than observed in extreme cases such as those described by Waterland et al. [Citation87].
Conclusion
The results were suggestive of a general trend toward increased methylation in the treatment group; however, no gene specific associations were identified after adjusting for multiple testing. This suggests that the potential effects of micronutrient supplementation on DNA methylation are likely to correspond with small genome wide changes, rather than individual gene specific effects, and therefore cytosine methylation is not likely to play an immediate role in the observed beneficial impacts of dietary supplementation of ADHD [Citation60,Citation88]. The magnitude of the observed changes may increase proportionally with exposure time, which should be considered for future studies. Future studies in this area should also consider increasing the sample size and incorporating the effects of genotype on epigenetic analysis, as it is likely that both diet and MTHFR genotype interact to determine epigenetic outcomes.
Although it is accepted that environmental factors including nutrition may promote epigenomic modifications, very little is known about the exposure time and dosage required for changes to occur.
We performed a pilot study to investigate if micronutrient treatment (consisting mainly of vitamins, minerals and amino acids) correlated with genomic DNA methylation changes measured in peripheral blood.
Our approach assessed pre- and post-DNA methylation levels, at approximately 800,000 specific loci.
The results were suggestive of a general trend toward increased methylation in the treatment group; however, no specific methylation changes remained significant after adjusting for multiple testing.
MTHFR genotype appears to determine epigenetic outcomes, and should be incorporated in future studies.
Author contributions
AJ Stevens carried out experimental work, data analysis and manuscript preparation. JJ Rucklidge conceived the study, recruited and managed patients, coordinated funding, and contributed toward manuscript editing. KA Darling coordinated sample collection. MJF Eggleston aided in patient management and manuscript editing. JF Pearson provided statistical advice and verification. MA Kennedy contributed toward study design, coordinated funding and manuscript preparation.
Ethical disclosure
Ethical approval for this study was approved by the Southern Health and Disability Ethics Committee (New Zealand). Ethics ref: 13/STH/45/AM05. The trial was prospectively registered ACTRN12613000896774.
Supplemental Table 1
Download MS Word (305.8 KB)Acknowledgements
The authors are grateful to G Wiggins (University of Otago, Christchurch, New Zealand) for constructive advice with the bioinformatic analysis, and the authors are also grateful to the participants of this trial.
Supplementary data
The original scripts used for data analysis are available on request. To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2217/epi-2018-0029
Financial & competing interests disclosure
This work was supported by Vic Davis Memorial Trust, the GAMA Foundation, the Foundation for Excellence in Mental Health Research, the Canterbury Medical Research Foundation, The Carney Centre for Pharmacogenomics, the Lotteries Health Research Commission, and the Health Research Council. The authors are also grateful to Gravida for providing PhD funding support to K Darling. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Pidsley R , MillJ . Epigenetic studies of psychosis: current findings, methodological approaches, and implications for postmortem research . Biol. Psychiatry69 ( 2 ), 146 – 156 ( 2011 ).
- Nilsson E , JanssonPA , PerfilyevAet al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes . Diabetes63 ( 9 ), 2962 – 2976 ( 2014 ).
- Kebir O , ChaumetteB , RivollierFet al. Methylomic changes during conversion to psychosis . Mol. Psychiatry22 ( 4 ), 512 ( 2017 ).
- Wockner LF , NobleEP , LawfordBRet al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients . Transl. Psychiatry4 ( 1 ), e339 ( 2014 ).
- Van Dijk SJ , MolloyPL , VarinliHet al. Epigenetics and human obesity . Int. J. Obes. (Lond.)39 ( 1 ), 85 – 97 ( 2015 ).
- Goldberg AD , AllisCD , BernsteinE . Epigenetics: a landscape takes shape . Cell128 ( 4 ), 635 – 638 ( 2007 ).
- Hou L , ZhangX , WangD , BaccarelliA . Environmental chemical exposures and human epigenetics . Int. J. Epidemiol.dyr154 ( 2011 ).
- Bird A . Perceptions of epigenetics . Nature447 ( 7143 ), 396 – 398 ( 2007 ).
- Bloushtain-Qimron N , YaoJ , SnyderELet al. Cell type-specific DNA methylation patterns in the human breast . Proc. Natl Acad. Sci. USA105 ( 37 ), 14076 – 14081 ( 2008 ).
- Wu H , SunYE . Epigenetic regulation of stem cell differentiation . Pediatr. Res.59 , 21R – 25R ( 2006 ).
- Sakamoto H , SuzukiM , AbeTet al. Cell type-specific methylation profiles occurring disproportionately in CpG-less regions that delineate developmental similarity . Genes Cells12 ( 10 ), 1123 – 1132 ( 2007 ).
- Rakyan VK , DownTA , ThorneNPet al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs) . Genome Res.18 ( 9 ), 1518 – 1529 ( 2008 ).
- Mcmillen IC , RobinsonJS . Developmental origins of the metabolic syndrome: prediction, plasticity, and programming . Physiol. Rev.85 ( 2 ), 571 – 633 ( 2005 ).
- Gillman MW . Prenatal famine and developmental origins of Type 2 diabetes . Lancet Diabetes Endocrinol.3 ( 10 ), 751 ( 2015 ).
- Olsen J . David Barker (1938–2013)–a giant in reproductive epidemiology . Acta Obstet. Gynecol. Scand.93 ( 11 ), 1077 – 1080 ( 2014 ).
- Karouzakis E , RengelY , JüngelAet al. DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts . Genes Immun.12 ( 8 ), 643 ( 2011 ).
- Anderson OS , SantKE , DolinoyDC . Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation . J. Nutr. Biochem.23 ( 8 ), 853 – 859 ( 2012 ).
- Rönn T , VolkovP , DavegårdhCet al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue . PLoS Genet.9 ( 6 ), e1003572 ( 2013 ).
- Wan ES , QiuW , BaccarelliAet al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome . Hum. Mol. Genet.21 ( 13 ), 3073 – 3082 ( 2012 ).
- Jaenisch R , BirdA . Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals . Nat. Genet.33 ( 3S ), 245 – 254 ( 2003 ).
- Breitling LP , YangR , KornB , BurwinkelB , BrennerH . Tobacco-smoking-related differential DNA methylation: 27 K discovery and replication . Am. J. Respir. Crit. Care Med.88 ( 4 ), 450 – 457 ( 2011 ).
- Breton CV , ByunH-M , WentenM , PanF , YangA , GillilandFD . Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation . Am. J. Res. Crit. Care Med.180 ( 5 ), 462 – 467 ( 2009 ).
- Waterland RA , JirtleRL . Transposable elements: targets for early nutritional effects on epigenetic gene regulation . Mol. Cell. Biol.23 ( 15 ), 5293 – 5300 ( 2003 ).
- Stevens AJ , RucklidgeJJ , KennedyMA . Epigenetics, nutrition and mental health. Is there a relationship?Nutr. Neurosci. doi:10.1080/1028415X.2017.13315241 – 12 ( 2017 ) ( Epub ahead of print ).
- Kaati G , BygrenLO , EdvinssonS . Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period . Eur. J. Hum. Genet.10 ( 11 ), 682 – 688 ( 2002 ).
- Pembrey ME , BygrenLO , KaatiGet al. Sex-specific, male-line transgenerational responses in humans . Eur. J. Hum. Genet.14 ( 2 ), 159 – 166 ( 2006 ).
- Bygren LO , KaatiG , EdvinssonS . Longevity determined by paternal ancestors’ nutrition during their slow growth period . Acta Biotheor.49 ( 1 ), 53 – 59 ( 2001 ).
- Locasale JW . Serine, glycine and one-carbon units: cancer metabolism in full circle . Nat. Rev. Cancer13 ( 8 ), 572 – 583 ( 2013 ).
- Rucklidge JJ , EgglestonMJ , JohnstoneJM , DarlingK , FramptonCM . Vitamin-mineral treatment improves aggression and emotional regulation in children with ADHD: a fully blinded, randomized, placebo-controlled trial . J. Child Psychol. Psychiatry59 ( 3 ), 232 – 246 ( 2017 ).
- Maksimovic J , PhipsonB , OshlackA . A cross-package Bioconductor workflow for analysing methylation array data . F1000Res.5 , 1281 ( 2016 ).
- Maksimovic J , GordonL , OshlackA . SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips . Genome Biol.13 ( 6 ), R44 ( 2012 ).
- Shi W , OshlackA , SmythGK . Optimizing the noise versus bias trade-off for Illumina whole genome expression BeadChips . Nucleic Acids Res.38 ( 22 ), e204 – e204 ( 2010 ).
- Lin SM , DuP , HuberW , KibbeWA . Model-based variance-stabilizing transformation for Illumina microarray data . Nucleic Acids Res.36 ( 2 ), e11 – e11 ( 2008 ).
- Pidsley R , ZotenkoE , PetersTJet al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling . Genome Biology17 ( 1 ), 208 ( 2016 ).
- Houseman EA , AccomandoWP , KoestlerDCet al. DNA methylation arrays as surrogate measures of cell mixture distribution . BMC bioinformatics13 ( 1 ), 86 ( 2012 ).
- Reinius LE , AcevedoN , JoerinkMet al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility . PLoS ONE7 ( 7 ), e41361 ( 2012 ).
- Kanehisa M , GotoS . KEGG: kyoto encyclopedia of genes and genomes . Nucleic Acids Res.28 ( 1 ), 27 – 30 ( 2000 ).
- Carlson M , FalconS , PagesHet al. A set of annotation maps describing the entire Gene Ontology . R package version 3.0.0 ( 2007 ).
- Phipson B , MaksimovicJ , OshlackA . missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform . Bioinformatics32 ( 2 ), 286 – 288 ( 2016 ).
- Peters TJ , BuckleyMJ , StathamALet al. De novo identification of differentially methylated regions in the human genome . Epigenetics Chromatin8 ( 1 ), 6 ( 2015 ).
- Collaboration HLT . Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials . BMJ316 ( 7135 ), 894 – 898 ( 1998 ).
- Shi M , CaprauD , RomittiP , ChristensenK , MurrayJC . Genotype frequencies and linkage disequilibrium in the CEPH human diversity panel for variants in folate pathway genes MTHFR, MTHFD, MTRR, RFC1, and GCP2 . Birth Defects Res. A Clin. Mol. Teratol.67 ( 8 ), 545 – 549 ( 2003 ).
- Weisberg I , TranP , ChristensenB , SibaniS , RozenR . A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity . Mol. Genet. Metabol.64 ( 3 ), 169 – 172 ( 1998 ).
- Friso S , ChoiS-W , GirelliDet al. A common mutation in the 5, 10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status . Proc. Natl Acad. Sci. USA99 ( 8 ), 5606 – 5611 ( 2002 ).
- Yates A , AkanniW , AmodeMRet al. Ensembl 2016 . Nucleic Acids Res.44 ( D1 ), D710 – D716 ( 2016 ).
- Bird A . DNA methylation patterns and epigenetic memory . Genes Dev.16 ( 1 ), 6 – 21 ( 2002 ).
- Jones PA , LiangG . Rethinking how DNA methylation patterns are maintained . Nat. Rev. Genet.10 ( 11 ), 805 – 811 ( 2009 ).
- Sassone-Corsi P . When metabolism and epigenetics converge . Science339 ( 6116 ), 148 – 150 ( 2013 ).
- Teperino R , SchoonjansK , AuwerxJ . Histone methyl transferases and demethylases; can they link metabolism and transcription?Cell Metabolism12 ( 4 ), 321 – 327 ( 2010 ).
- Katada S , ImhofA , Sassone-CorsiP . Connecting threads: epigenetics and metabolism . Cell148 ( 1 ), 24 – 28 ( 2012 ).
- Fenech M , AitkenC , RinaldiJ . Folate, vitamin B12, homocysteine status and DNA damage in young Australian adults . Carcinogenesis19 ( 7 ), 1163 – 1171 ( 1998 ).
- Jacob RA , GretzDM , TaylorPCet al. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women . J. Nutrition128 ( 7 ), 1204 – 1212 ( 1998 ).
- Shelnutt KP , KauwellGP , GregoryJFet al. Methylenetetrahydrofolate reductase 677C→ T polymorphism affects DNA methylation in response to controlled folate intake in young women . J. Nutr. Biochem.15 ( 9 ), 554 – 560 ( 2004 ).
- Kok RM , SmithDE , BartoRet al. Global DNA methylation measured by liquid chromatography-tandem mass spectrometry: analytical technique, reference values and determinants in healthy subjects . Clin. Chem. Lab. Med.45 ( 7 ), 903 – 911 ( 2007 ).
- Dolinoy DC . The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome . Nutr. Rev.66 ( suppl 1 ), S7 – S11 ( 2008 ).
- Jirtle RL , SkinnerMK . Environmental epigenomics and disease susceptibility . Nat. Rev. Genet.8 ( 4 ), 253 – 262 ( 2007 ).
- Feil R , FragaMF . Epigenetics and the environment: emerging patterns and implications . Nat. Rev. Genet.13 ( 2 ), 97 – 109 ( 2012 ).
- Koticha D , BabiarzJ , Kane-GoldsmithN , JacobJ , RajuK , GrumetM . Cell adhesion and neurite outgrowth are promoted by neurofascin NF155 and inhibited by NF186 . Mol. Cell. Neurosci.30 ( 1 ), 137 – 148 ( 2005 ).
- Kriebel M , WuchterJ , TrinksS , VolkmerH . Neurofascin: a switch between neuronal plasticity and stability . Int. J. Biochem. Cell Biol.44 ( 5 ), 694 – 697 ( 2012 ).
- Jin M , KawakamiK , FukuiYet al. Different histological types of non-small cell lung cancer have distinct folate and DNA methylation levels . Cancer Science100 ( 12 ), 2325 – 2330 ( 2009 ).
- Gagnon A , SirardM-A , RichardFO , LaforestJ-P . Effects of folic acid and vitamin B12 supplementation on granulosa cells of the dominant follicle of dairy cows . Biology of Reproduction87 ( Suppl. 1 ), 504 ( 2012 ).
- Mavroconstanti T , JohanssonS , WingeI , KnappskogPM , HaavikJ . Functional properties of rare missense variants of human CDH13 found in adult attention deficit/hyperactivity disorder (ADHD) patients . PLoS ONE8 ( 8 ), e71445 ( 2013 ).
- Lesch K-P , TimmesfeldN , RennerTJet al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies . J. Neural. Transm.115 ( 11 ), 1573 – 1585 ( 2008 ).
- Stergiakouli E , ThaparA . Fitting the pieces together: current research on the genetic basis of attention-deficit/hyperactivity disorder (ADHD) . Neuropsychiatr. Dis. Treat.6 , 551 ( 2010 ).
- Takeuchi T , MisakiA , LiangSBet al. Expression of T-Cadherin (CDH13, H-Cadherin) in human brain and its characteristics as a negative growth regulator of epidermal growth factor in neuroblastoma cells . J. Neurochem.74 ( 4 ), 1489 – 1497 ( 2000 ).
- Uhl GR , DrgonT , JohnsonCet al. Molecular genetics of addiction and related heritable phenotypes . Ann. N. Y. Acad. Sci.1141 ( 1 ), 318 – 381 ( 2008 ).
- Boesveldt S . Olfaction and eating behavior . In : Springer Handbook of Odor . Springer International Publishing , Cham, Switzerland , 109 – 110 ( 2017 ).
- Frosst P , BlomH , MilosRet al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase . Nat. Genet.10 ( 1 ), 111 – 113 ( 1995 ).
- Bagley PJ , SelhubJ . A common mutation in the methylenetetrahydrofolate reductase gene is associated with an accumulation of formylated tetrahydrofolates in red blood cells . Proc. Natl Acad. Sci. USA95 ( 22 ), 13217 – 13220 ( 1998 ).
- Ergul E , SazciA , KaraI . Methylenetetrahydrofolate reductase gene polymorphisms in Turkish children with attention-deficit/hyperactivity disorder . Genet. Test. Mol. Biomark.16 ( 1 ), 67 – 69 ( 2012 ).
- Castro R , RiveraI , RavascoPet al. 5, 10-methylenetetrahydrofolate reductase (MTHFR) 677C→ T and 1298A→ C mutations are associated with DNA hypomethylation . J. Med. Genet.41 ( 6 ), 454 – 458 ( 2004 ).
- Anney R , KleiL , PintoDet al. Individual common variants exert weak effects on the risk for autism spectrum disorders . Hum. Mol. Genet.21 ( 21 ), 4781 – 4792 ( 2012 ).
- Butler MG , RafiSK , ManzardoAM . High-resolution chromosome ideogram representation of currently recognized genes for autism spectrum disorders . Int. J. Mol. Sci.16 ( 3 ), 6464 – 6495 ( 2015 ).
- Turner TN , CoeBP , DickelDEet al. Genomic patterns of de novo mutation in simplex autism . Cell171 ( 3 ), 710 – 722 . e712 ( 2017 ).
- Purcell SM , MoranJL , FromerMet al. A polygenic burden of rare disruptive mutations in schizophrenia . Nature506 ( 7487 ), 185 – 190 ( 2014 ).
- Devlin AM , BrainU , AustinJ , OberlanderTF . Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth . PLoS ONE5 ( 8 ), e12201 ( 2010 ).
- Wang Z , ChenJ-Q , LiuJ-L , QinX-G , HuangY . Polymorphisms in ERCC1, GSTs, TS and MTHFR predict clinical outcomes of gastric cancer patients treated with platinum/5-Fu-based chemotherapy: a systematic review . BMC Gastroenterol.12 ( 1 ), 137 ( 2012 ).
- Li Z , XingX , ShanFet al. ABCC2-24C> T polymorphism is associated with the response to platinum/5-Fu-based neoadjuvant chemotherapy and better clinical outcomes in advanced gastric cancer patients . Oncotarget7 ( 34 ), 55449 ( 2016 ).
- Cięszczyk P , ZarębskaA , JastrzębskiZet al. Does the MTHFR A1298C Polymorphism Modulate the Cardiorespiratory Response to Training? J. Hum. Kinet. 54 ( 1 ), 43 – 53 ( 2016 ).
- Walton E , HassJ , LiuJet al. Correspondence of DNA methylation between blood and brain tissue and its application to schizophrenia research . Schizophr. Bull.sbv074 ( 2015 ).
- Edgar RD , JonesMJ , MeaneyMJ , TureckiG , KoborMS . BECon: A tool for interpreting DNA methylation findings from blood in the context of brain . bioRxivorg111609 ( 2017 ).
- Shenker NS , PolidoroS , Van VeldhovenKet al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking . Hum. Mol. Genet.22 ( 5 ), 843 – 851 ( 2012 ).
- Guida F , SandangerTM , CastagnéRet al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation . Hum. Mol. Genet.24 ( 8 ), 2349 – 2359 ( 2015 ).
- Joubert BR , HåbergSE , NilsenRMet al. 450 K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy . Environ. Health Perspect.120 ( 10 ), 1425 ( 2012 ).
- Steegers-Theunissen RP , Obermann-BorstSA , KremerDet al. Periconceptional maternal folic acid use of 400 μg per day is related to increased methylation of the IGF2 gene in the very young child . PLoS ONE4 ( 11 ), e7845 ( 2009 ).
- Dominguez-Salas P , MooreSE , BakerMSet al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles . Nat. Commun.5 , 3746 ( 2014 ).
- Waterland RA , KellermayerR , LaritskyEet al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles . PLoS Genet.6 ( 12 ), e1001252 ( 2010 ).
- Rucklidge J , TaylorM , WhiteheadK . Effect of micronutrients on behavior and mood in adults with ADHD: Evidence from an 8-week open label trial with natural extension . J. Atten. Disord.15 ( 1 ), 79 – 91 ( 2011 ).