
Abstract
Aim: To examine the effects of genetic variation, parental age and BMI on parental allele-specific methylation of imprinted genes in fetal cord blood samples. Methodology: We have developed SNP genotyping and deep bisulphite sequencing assays for six imprinted genes to determine parental allele-specific methylation patterns in diploid somatic tissues. Results: Multivariate linear regression analyses revealed a negative correlation of paternal age with paternal MEG3 allele methylation in fetal cord blood. Methylation of the maternal PEG3 allele showed a positive correlation with maternal age. Paternal BMI was positively correlated with paternal MEST allele methylation. In addition to parental origin, allele-specific methylation of most imprinted genes was largely dependent on the underlying SNP haplotype. Conclusion: Our study supports the idea that parental factors can have an impact, although of small effect size, on the epigenome of the next generation, providing an additional layer of complexity to phenotypic diversity.
In highly developed countries, the number of couples who postpone their wish for parenthood for social, economic and/or cultural reasons has been constantly increasing over the last decades. The shift in parental age is associated with an increased use of assisted reproductive technologies and prenatal diagnostics as well as an increased risk for medical problems of the offspring. It is well known that maternal aging leads to a decreased pool of follicles (from approximately 200,000 at menarche to approximately 1000 at the onset of menopause) and an increased rate of aneuploid oocytes [Citation1–3]. Oocyte aneuploidy can cause fertility problems, spontaneous abortions and children with Down syndrome [Citation4]. Mouse knockout experiments suggest that this maternal age effect is due to an age-dependent loss of cohesins and/or DNA repair proteins in the meiotically arrested oocyte [Citation5,Citation6]. Although the abortion risk of a >35-year-old woman and a >40-year-old male is approximately two-times higher than that of a >35-year-old woman and a younger partner [Citation7], the contribution of paternal factors to reproductive problems of older couples has been largely neglected so far.
Although life-long spermatogenesis can provide a life-long period of male fertility, the developmental potential of sperm from aging men is reduced. Fertilization, blastocyst formation and implantation rates decrease with paternal age [Citation8]. Moreover, advanced paternal age is associated with an increased risk of spontaneous abortions, rare de novo dominant conditions and neurodevelopmental disorders such as autism and schizophrenia in the offspring [Citation9]. Recent genome-wide sequencing studies provided compelling evidence for higher de novo genetic mutation rates in the offspring of older males [Citation10]. The number of spermatogonial cell divisions prior to spermatogenesis increases from 35 at puberty to 840 at 50 years [Citation11]. During each cell division, not only the DNA sequence but also its epigenetic modifications must be copied to the daughter cells. Considering that the error rate during this copying process is at least one order of magnitude higher for epigenetic information than for genetic information [Citation12], the sperm epigenome can be expected to acquire 10- to 100-times more age-related epimutations than DNA sequence mutations. Mouse studies have associated age-related changes in sperm DNA methylation with alterations in brain gene expression and abnormal behavior in the offspring [Citation13,Citation14], providing a mechanism for transgenerational epigenetic effects. Subsequently, age-dependent sperm DNA methylation [Citation15] and transmission to the offspring [Citation16] were also observed in humans. Similar to father’s age, paternal obesity also has an impact on sperm DNA methylation [Citation17,Citation18] and offspring health [Citation19,Citation20]. Little is known about possible epigenetic effects of maternal aging. The oocytes and embryos of aged mice displayed genome-wide DNA methylation changes, which may be due to reduced expression of DNA methyltransferases [Citation21].
Deep bisulphite sequencing (DBS) is an amplicon-based next-generation sequencing technique which allows one to determine the DNA methylation levels of many thousands of individual DNA molecules (alleles), each from multiple genes and samples. Here, we have combined DBS with genotyping of informative single nucleotide polymorphisms (SNPs) to distinguish between paternal and maternal allele methylation in fetal cord blood (FCB) samples. Both paternal and maternal age, respectively, can have an impact on allele-specific methylation in the offspring. To study the effects of parental factors on the next generation, we have used imprinted genes as a model. Imprinted genes escape epigenetic reprogramming after fertilization and, therefore, any stochastic or environmentally induced epigenetic changes in the germ cells are directly transmitted to the offspring [Citation22,Citation23].
Methods
Study samples
The study on FCB samples was approved by the ethics committee at the medical faculty of Würzburg University (number 117/11 and 212/15). Written informed consent was obtained from couples undergoing treatment at the Fertility Center Wiesbaden. All analyzed FCB samples were from newborns conceived through in vitro fertilization (IVF) or intracytoplasmic sperm injection (ICSI) in a single fertility center and were collected by collaborating obstetric clinics throughout Germany. The vast majority of the couples undergoing IVF/ICSI treatment were of middle European descent. Only offspring without any medical problems at birth were included in the study. A total of 121 FCBs (including 11 twin pairs) were initially genotyped for each of the six analyzed amplicons in order to identify informative samples. Usually, only one twin from each pair was included. The clinical parameters of the studied samples are listed in Supplementary Table 1 (Supplementary table 1). Blood samples were pseudonymized and stored at -80°C until further use. Genomic DNA was isolated with the FlexiGene kit (Qiagen, Hilden, Germany). DNA quality and concentration were determined by a NanoDrop 2000c spectrophotometer (Thermo Scientific, MA, USA). Bisulphite conversion of 1 μg aliquots of genomic DNA was performed using EpiTect Fast 96 Bisulphite kit (Qiagen).
Genotyping
To distinguish between parental alleles in informative FCB samples, SNPs with high heterozygosity rate (with the highest minor allele frequency within the region of interest) were identified in the H19 intergenic differentially methylated region (IG DMR), the IGF2 DMR0, the MEG3 IG DMR, MEST (PEG1), NNAT (PEG5), and PEG3 (PW1) (Supplementary table 2). PCR and sequencing primers for bisulphite converted DNA were designed using PyroMark Assay Design 2.0 software (Qiagen). PCRs were performed in 25 μl reactions consisting of 2.5 μl 10× PCR buffer with MgCl2, 0.5 μl (10 mM) of PCR grade nucleotide mixture, 0.2 μl (5 U/μl) FastStart Taq DNA polymerase (Roche Diagnostics, Mannheim, Germany), 1.25 μl (10 pmol/ml) of forward and reverse primers (Metabion, Martinsried, Germany), 1 μl (∼25 ng) bisulphite converted genomic DNA, and 18.3 μl PCR grade water. PCR amplifications were carried out with an initial denaturation at 95°C for 5 min, 40 cycles of 95°C for 30 s, primer-specific annealing temperature (Supplementary table 2) for 30 s, and 72°C for 45 s, and a final extension step at 72°C for 10 min. Pyrosequencing was done on PyroMark Q96 MD system using PyroMark Gold Q96 CDT reagent kit and Pyro Q-CpG software (Qiagen).
Deep bisulphite sequencing
First-round gene-specific PCRs were performed in 50 μl reactions consisting of 5 μl 10X PCR buffer with MgCl2, 1 μl (10 mM) of PCR grade nucleotide mixture, 0.4 μl (5 U/μl) FastStart Taq DNA polymerase, 2.5 μl (10 pmol/ml) of forward and reverse primers (Supplementary table 3), 2 μl (∼50 ng) bisulphite converted genomic DNA, and 36.6 μl PCR grade water. Artificially methylated (0, 50 and 100%) DNA standards (Qiagen, #59695) were processed along with FCB samples. They served as controls for assessing the reliability of the methylation measurements for each DBS assay/amplicon. PCR products were purified with Agencourt AMPure XP Beads (Beckman Coulter, Krefeld, Germany), quantified using a Qubit Fluorometer and the Qubit dsDNA BR Assay kit (Invitrogen, Karlsruhe, Germany), and diluted to a concentration of 0.2 ng/μl. The six different amplicons for each sample were pooled together and endowed with a unique multiplex identifier (of a total of 48 MIDs). For adapter ligation with NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1), A-tailing was performed with Klenow fragment and subsequent ligation with T4 DNA ligase. The final amplification was for sample-specific barcoding. Touchdown PCR thermocycling conditions were adapted to achieve homogenous amplification of varying PCR product sizes. The final PCR pools were purified again with Agencourt AMPure XP Beads and quantified using the High Sensitivity DNA Reagent kit (Agilent Technologies, Böblingen, Germany). The pools for each sample were diluted to a concentration of 4 nM and 3 μl of this dilution from each of the 48 MIDs were pooled together into one final pool.
Next-generation sequencing was performed using the Reagent kit v2 (500 cycles) on the Illumina MiSeq (Illumina, CA, USA) platform following the manufacturer’s instructions. The sequencing reaction was performed with 250 base pair paired-end sequencing. Illumina Genome Analyzer was used to process the sequence reads and FASTQ files were further analyzed with the Amplikyzer2 software [Citation24], which provides a detailed nucleotide-level analysis including the calculation of CpG methylation rates. Briefly, all sequences were aligned to the genomic sequence of each amplicon using default settings; allele splitting was performed based on the SNPs described above. For the subsequent extraction of reads and CpG-wise methylation status, only reads with an overall bisulphite conversion rate of >95% were considered. Further downstream processing of Amplikyzer output files and subsequent analyses of methylation rates were performed using in-house R scripts.
Statistical analyses
Statistical analyses were performed with the statistical software package R (version 3.2.2; https://www.R-project.org). For each amplicon allele-specific methylation β values were obtained for both (paternal and maternal) alleles separately, by averaging the methylation status across the reads and CpG sites for each amplicon. CpG sites outside the imprinted regions as well as sites with a SNP in a large fraction of the samples were removed from further analyses. To estimate the effect of parental age and BMI on fetal methylation, multivariate linear regression models were fitted to the allele-specific β values separately for each amplicon. All models were adjusted for paternal and maternal age and BMI, the sex of the child, and sequence haplotype, as determined by the base at the variant position used to separate the alleles. P-values of < 0.05 were considered statistically significant.
Results
Parental age&BMI effects
SNP genotyping (Supplementary table 2) of 121 FCBs (for each locus) identified 46 heterozygous samples each for H19 IG DMR, IGF2 DMR0, MEST, and PEG3, 44 for the MEG3 IG DMR, and 39 for NNAT. Since the total number of MIDs for each amplicon was 48, a maximum of 46 heterozygous samples (the remaining two being controls) per amplicon could be considered for a DBS run. Linear regression models were used for an in-depth statistical analysis of the allele-specific methylation data. The regression coefficients of the final model were adjusted for parental age, parental BMI, fetal sex, and SNP effects.
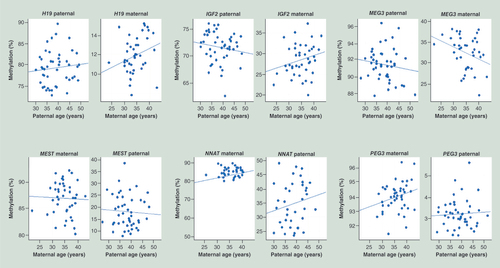
Based on the informative SNPs, parental allele-specific methylation of three paternally imprinted (H19 IG DMR, IGF2 DMR0 and MEG3 IG DMR) and three maternally imprinted (MEST, NNAT and PEG3) loci were determined by DBS and plotted against paternal and maternal age, respectively (). For most genes, the parental age-related methylation changes on the paternal and maternal alleles were in the same direction, consistent with additive effects. Methylation of the paternal and maternal H19, NNAT and PEG3 alleles increased with paternal and maternal age, respectively, whereas methylation of the paternal and maternal MEG3 and MEST alleles decreased with parental age (). We observed a trend towards negative correlation (regression estimate -0.001, p = 0.055) between paternal age and paternal FCB allele methylation for the MEG3 IG DMR and a positive correlation (regression estimate +0.001, p = 0.024) between maternal age and maternal allele methylation for PEG3 amplicon (). In addition, there was a positive correlation (regression estimate +0.004, p = 0.005) between paternal BMI and paternal MEST allele methylation (). MEST FCB methylation was not influenced by fetal sex, but strongly by the underlying genetic variation (SNP haplotype), which was accounted for in the regression models.
The upper panel shows three paternally imprinted/methylated (H19 IG DMR, IGF2 DMR0 and MEG3 IG DMR) regions and the lower panel shows three maternally imprinted/methylated (MEST, NNAT and PEG3) amplicons. Each dot represents an informative FCB sample. Regression lines indicate the direction of age-related changes.
FCB: Fetal cord blood; IG DMR: Intergenic differentially methylated region.
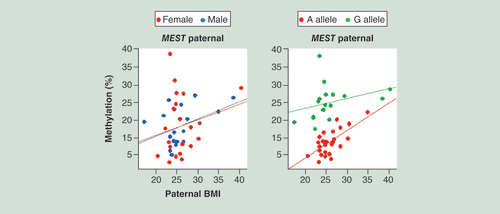
The left diagram shows identical BMI effects on paternal allele methylation in female and male FCBs. The right diagram displays the impact of the underlying genetic variation (A vs G SNP) on paternal MEST methylation.
FCB: Fetal cord blood.
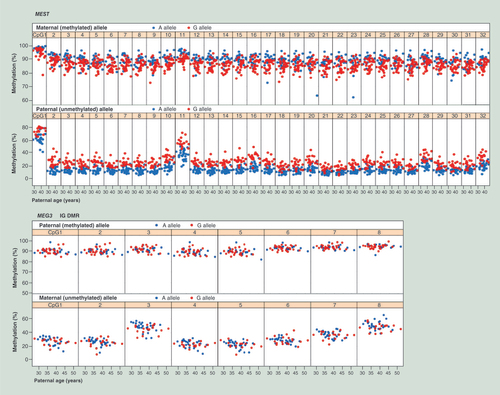
The single nucleotide polymorphism which is necessary to distinguish paternal and maternal alleles in informative samples was the strongest confounding factor in our analysis. For H19, IGF2, MEST, and NNAT, the methylation values of both parental alleles were significantly dependent on the SNP haplotype (). For example, all 32 CpGs in the MEST amplicon showed a higher methylation on maternally methylated A alleles than on maternal G alleles, whereas paternally unmethylated A alleles displayed a lower methylation than paternal G alleles (). The mean methylation difference between G and A alleles was 11.3% (p < 0.0001) for the paternal unmethylated, and -3.4% (p = 0.001) for maternal methylated allele in MEST. MEG3 was the only of six studied DMRs, which did not show a significant SNP effect ().
Table 1. SNP haplotype effects on the allele-specific methylation of imprinted genes.
For each gene, the upper panel shows single CpG methylation levels of the methylated allele and the lower panel of the unmethylated allele. A (Adenine) alleles are indicated by blue and G (Guanine) alleles by red dots. All 32 CpGs in the MEST target region display a clear SNP effect. Maternally methylated A alleles consistently show a higher methylation than maternally methylated G alleles, whereas paternally unmethylated A alleles show a lower methylation than paternally unmethylated G alleles. In contrast, none of the 8 targeted CpGs in the MEG3 IG DMR displays a clear SNP effect.
IG DMR: Intergenic differentially methylated region; SNP: Single nucleotide polymorphism.
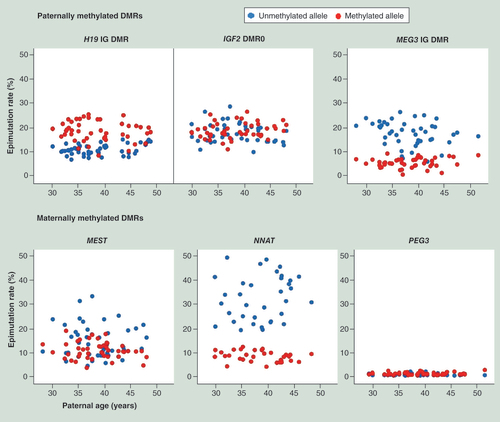
Allele-specific epimutation rates
Epimutations are defined as alleles showing >50% abnormally (de)methylated CpGs. For an unmethylated allele (e.g., paternal allele for MEST, NNAT, PEG3 and the maternal allele for H19 IG DMR, IGF2 DMR0, MEG3 IG DMR), CpGs displaying >50% methylation values were considered as epimutations. For a methylated allele (e.g., maternal allele for MEST, NNAT, PEG3 and the paternal allele for H19 IG DMR, IGF2 DMR0, MEG3 IG DMR), CpGs with <50% methylation levels were taken as epimutations. Epimutation rates were subsequently calculated by multiplying the number of epimutations divided by the number of reads with 100.
Previously, we have shown that the epimutation rates of the unmethylated allele of the paternally imprinted MEG3 IG DMR and the maternally imprinted MEST promoter are highly variable among individuals, and are significantly higher than those on the respective methylated allele [Citation25]. This was confirmed here for both MEG3 IG DMR (p < 0.0001) and MEST (p = 0.008) and was additionally detected in the maternally imprinted NNAT promoter (p < 0.0001) (). In contrast, for the paternally imprinted H19 IG DMR (p < 0.0001) and IGF2 DMR0 (p = 0.05), the methylated paternal allele showed a higher epimutation rate than the unmethylated maternal allele. The difference between parental alleles was much higher for the primary H19 IG DMR than for the secondary IGF2 DMR0. Unlike all the other studied imprinted genes, the PEG3 promoter displayed very low epimutation rates on both the parental alleles ().
Blue and red dots indicate the epimutation rates on the unmethylated and methylated alleles, respectively, for a given gene and sample.
Discussion
Parental effects on the next generation
The developmental origins of health and disease hypothesis suggest that adverse environmental exposures during the periconceptional and intrauterine period increase the life-long risk for complex (metabolic, cardiovascular and other civilization) diseases [Citation26,Citation27]. Moreover, adverse parental factors such as advanced age or obesity can even affect the nonexposed offspring across multiple generations [Citation28,Citation29]. Epigenetic mechanisms in other words, DNA methylation, histone modifications and/or noncoding RNAs, underlying this transgenerational inheritance through gametic epigenetic alterations remain to be elucidated. It is generally assumed that inherited epigenetic modifications can lead to persistent changes in gene regulation and pathways.
In this study, we have used DBS to identify parental effects in the next generation. DBS combined with SNP typing is a very powerful technique to determine parental-allele specific methylation patterns in somatic tissues of the offspring. It is true that a single sperm and a single oocyte cell both transmit binary methylation information at an individual CpG position. Therefore, at first glance, it seems plausible to assume 0, 50 or 100% methylation at an individual CpG site in the diploid zygote and the resulting offspring. However, it is generally the methylation density of several contiguous CpGs in a cis-regulatory region rather than methylation at individual CpG sites that turn a gene on or off [Citation30,Citation31]. The designed DBS assays for imprinted genes analyzed up to 32 CpGs per target region (Supplementary table 3) across many thousand individual paternal and/or maternal DNA molecules (number of reads per sample) in a single FCB sample. Thus, the methylation value of a given region in a given sample represents the overall mean of several thousand DNA molecules across several CpGs that can display any value between 0 and 100%. At a population level, there is a considerable variation of measured methylation values between individuals and between group differences of a few percentage points. Methylation of the paternal MEG3 IG DMR allele in FCB was inversely correlated with paternal age, whereas methylation of the maternal PEG3 allele was positively correlated with maternal age. For both genes, the imprinted (methylated) allele was subject to reprogramming by parental age. The observed effect sizes were small with parental age-related methylation changes in the order of 1–2 percentage points.
MEG3 is located next to the paternally expressed DLK1 on chromosome 14q32. Imprinting of the DLK1–MEG3 locus is controlled by a primary germline IG DMR and a secondary DMR. While the primary IG DMR lies 13 kb upstream in the intergenic region, the secondary DMR lies 1.5 kb upstream of the transcription start site of the MEG3 promoter [Citation32]. DLK1 encodes a transmembrane protein, which plays a role in cell differentiation processes and tumorigenesis [Citation33,Citation34]. The DLK1–MEG3 locus encodes microRNAs and small nucleolar RNAs which are downregulated by methylation in pancreatic islets of Type 2 diabetes mellitus patients [Citation35].
PEG3 encodes a Krüppel C2H2-type zinc finger protein. It is highly expressed in embryos, placentas and brains, controlling fetal growth and maternal behavior [Citation36]. It is endowed with three alternative promoters upstream of its main promoter and may be controlled by long-range regulatory mechanisms in addition to imprinting. Deletion of the repressed maternal allele of the main promoter leads to an upregulation of promoters on the opposite allele [Citation37].
Human MEST is endowed with two promoters generating transcripts with alternative first exons. The imprinted promoter DMR studied here is thought to control the paternal expression of isoform 1, whereas the isoform 2 under control of the second promoter, is biallelically expressed [Citation38]. In FCB studied here, methylation of the paternal promoter DMR positively correlated with paternal BMI. MEST is a primary candidate gene for developmental programming of metabolic phenotypes. Mest upregulation in mice [Citation39] and MEST hypomethylation in humans [Citation40] have been linked to an early overnutritional environment. The imprinted isoform 1 is upregulated in fat tissue of obese individuals in both species [Citation41,Citation42].
Due to our study design, all analyzed FCB samples were from newborns conceived through IVF/ICSI. There is concern that assisted reproductive technology (ART) procedures may affect the methylation status of imprinted genes, leading in rare cases to imprinting disorders [Citation43]. Several previous studies have reported aberrant methylation patterns in the imprinted LIT1 [Citation44], PLAG2 [Citation45] and SNRPN [Citation46] genes in ART offspring, whereas other studies did not find significant methylation differences between ART newborns and controls [Citation47,Citation48]. In a previous methylome study on FCBs from ICSI children and naturally conceived controls, we found a limited number of ART-associated methylation changes, however, none of them was of large effect size [Citation49]. Moreover, none of the imprinted genes studied here showed a differentially methylated CpG with genome-wide significance. Both the methylation changes observed in our previous genome-wide study [Citation49] and in the present candidate gene study were within the normal range of methylation variation and are not associated with imprinting defects. Although we cannot exclude the formal possibility that some of our findings are restricted to ART children, similar age, BMI and SNP effects are most likely also present in naturally conceived children.
SNP haplotypes influencing methylation signatures
Although allele-specific methylation of imprinted genes largely depends on parental origin, five out of six studied genes showed significant effects of cis-regulatory sequence polymorphisms. One impressive example was the effect of SNP rs3778859 in the MEST promoter (). The A variant of the SNP haplotype was associated with an increased (>3 percentage points) methylation of the imprinted maternal and a decreased (>10 percentage points) methylation of the nonimprinted paternal MEST allele. For MEST, the average methylation difference between parental AA alleles was approximately 15% higher than between GG alleles. This is by far larger than the effect of paternal BMI (this study) or maternal gestational diabetes mellitus [Citation40] on the offspring’s epigenome. All other studied imprinted genes except MEG3 exhibited a strong SNP effect. Thus, when studying the possible impact of parental factors on FCB methylation in the next generation, cis-regulatory SNPs must be considered as an important confounding factor.
Previous studies demonstrated that allele-specific methylation (ASM) (affecting mainly nonimprinted genes) is largely determined by cis-acting polymorphisms [Citation50]. One plausible explanation is sequence-specific DNA-binding proteins or other sequence-dependent regulatory mechanisms influencing DNA methylation. Genotype-dependent ASM was associated with allele-specific expression across the human genome [Citation51]. Consequently, the haplotype concept should be extended with epigenetic modifications, yielding epihaplotypes. Genome-wide analyses revealed that ASM is a widespread phenomenon across the genome, enlarging interindividual variation [Citation52–54]. Overall, our DBS data show that most imprinted genes are subject to ASM. Both methylation of the nonimprinted (unmethylated) and imprinted (methylated) parental allele is more variable than previously thought and influenced by the underlying SNP haplotype.
Allele-specific epimutations
Because DBS generates allele-specific methylation information on thousands of individual DNA molecules of a target gene per sample, it also allows one to determine allele-specific epimutation rates. Imprinted genes are endowed with one methylated (imprinted) and one unmethylated (nonimprinted) parental allele. We have noted previously that regardless of parental origin, the nonimprinted (unmethylated) parental allele usually shows a much higher epimutation rate than the imprinted (methylated) allele [Citation25]. Here, we observed for the first time that an imprinted region, IGF2 DMR0–H19 IG DMR, where the imprinted (methylated) allele displayed an increased epimutation rate when compared with the nonimprinted allele, leading to hypomethylation of both DMRs on the paternal allele. Previously, it has been shown that the H19 IG DMR influences methylation at the IGF2 DMR0 in cis [Citation55]. Hypomethylation of this DMR0 was associated with biallelic IGF2 expression and was observed in Wilms tumor [Citation56] and colorectal cancer [Citation57]. Loss of IGF2 imprinting in normal tissue, in other words, normal colonic mucosa is thought to predispose to cancer [Citation58]. Increased susceptibility of the imprinted H19–IGF2 allele to epimutations may be the mechanism underlying biallelic IGF2 expression in 10–20% of blood samples from normal healthy individuals [Citation59,Citation60]. Moreover, the difference between allele-specific epimutation rates was much higher for the primary H19 IG DMR than for the secondary IGF2 DMR0 in our data. This supports the idea that similar to the MEG3–DLK1 region [Citation25], the primary H19 IG DMR may become at least partially redundant once the imprinting of the secondary promoter DMR0 has been established.
Conclusion
Collectively, our results show that paternal and maternal factors such as age and BMI leave epigenetic signatures on the respective parental allele in somatic tissue of the next generation. Although the observed effect sizes were small (compared with sequence-dependent methylation variation), many more loci susceptible to programming by parental and/or stochastic factor may occur across the genome, contributing to individual phenotypic differences and life-long complex disease risk.
We have combined deep bisulphite sequencing with genotyping of informative single nucleotide polymorphisms to distinguish between paternal and maternal allele’s methylation in fetal cord blood (FCB) samples.
We used imprinted genes as a model to delineate the effects of parental factors on the next generation since these differentially methylated regions (DMRs) are known to escape the epigenetic reprogramming after fertilization and therefore, stochastic or environmentally induced epigenetic changes in the germ cells are directly transmitted to the offspring.
We observed a trend towards negative correlation of paternal age with paternal MEG3 allele’s methylation and a positive correlation of maternal age with maternal PEG3 allele’s methylation in FCBs.
We also detected a significant positive correlation between paternal BMI and paternal MEST allele’s methylation in FCBs.
In addition to parental origin, we noticed that allele-specific methylation of H19, IGF2, MEST, and NNAT imprinted DMRs was largely dependent on the underlying genetic variation (SNP haplotype).
When epimutations were defined as alleles with >50% abnormally (de)methylated CpGs, the MEST, NNAT and MEG3 epimutation rates were significantly higher on the nonimprinted unmethylated alleles than on the respective imprinted methylated alleles, leading to hypermethylation of the nonimprinted allele.
In contrast, for H19 and IGF2 DMRs, the imprinted methylated alleles displayed higher epimutation rates when compared with the nonimprinted unmethylated alleles, leading to hypomethylation of the imprinted allele.
Collectively, our results show that paternal and maternal factors leave epigenetic signatures on the corresponding parental allele in somatic tissue of the next generation, although of small effect size compared with sequence-dependent methylation variation.
Author’ contributions
T Haaf and N El Hajj designed the study, T Haaf and R Potabattula wrote the manuscript, R Potabattula performed all experiments and analyzed the data, M Dittrich and T Müller performed statistical analyses, J Böck and L Haertle established the methodology, M Schorsch and T Hahn contributed the samples. All authors read and approved the final manuscript.
Ethical conduct
The study was approved by the ethics committee at the medical faculty of Würzburg University (no. 117/11 and 212/15). Written informed consent was obtained from couples undergoing treatment at the Fertility Center Wiesbaden.
Supplemental application 1
Download PDF (117.8 KB)Acknowledgments
We thank all pregnant women and obstetric clinics participating in the study.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/full/10.2217/epi-2018-0059
Financial&competing interests disclosure
R Potabattula was supported by a fellowship of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg. This study was supported by the German Research Foundation (grant EL 742/1-1 to N El Hajj) and the European Union (ERAofART to T Haaf).The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Nelson SM , TelferEE , AndersonRA . The aging ovary and uterus: new biological insights . Hum. Reprod. Update19 ( 1 ), 67 – 83 ( 2013 ).
- Fragouli E , WellsD , DelhantyJD . Chromosome abnormalities in the human oocyte . Cytogenet. Genome Res.133 ( 2-4 ), 107 – 118 ( 2011 ).
- Eichenlaub-Ritter U . Oocyte aging and its cellular basis . Int. J. Dev. Biol.56 ( 10-12 ), 841 – 852 ( 2012 ).
- Maclennan M , CrichtonJH , PlayfootCJ , AdamsIR . Oocyte development, meiosis and aneuploidy . Semin. Cell Dev. Biol.45 , 68 – 76 ( 2015 ).
- Jessberger R . Age-related aneuploidy through cohesion exhaustion . EMBO Rep.13 ( 6 ), 539 – 546 ( 2012 ).
- Titus S , LiF , StobezkiRet al. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans . Sci. Transl. Med.5 ( 172 ), 172ra121 ( 2013 ).
- De La Rochebrochard E , ThonneauP . Paternal age and maternal age are risk factors for miscarriage; results of a multicentre European study . Hum. Reprod.17 ( 6 ), 1649 – 1656 ( 2002 ).
- Luna M , FinklerE , BarrittJet al. Paternal age and assisted reproductive technology outcome in ovum recipients . Fertil. Steril.92 ( 5 ), 1772 – 1775 ( 2009 ).
- Liu K , CaseA ; Reproductive Endocrinology and Infertility Committee . Advanced reproductive age and fertility . J. Obstet. Gynaecol. Can.33 ( 11 ), 1165 – 1175 ( 2011 ).
- Kong A , FriggeML , MassonGet al. Rate of de novo mutations and the importance of father’s age to disease risk . Nature488 ( 7412 ), 471 – 475 ( 2012 ).
- Crow JF . The origins, patterns and implications of human spontaneous mutation . Nat. Rev. Genet.1 ( 1 ), 40 – 47 ( 2000 ).
- Bennett-Baker PE , WilkowskiJ , BurkeDT . Age-associated activation of epigenetically repressed genes in the mouse . Genetics165 ( 4 ), 2055 – 2062 ( 2003 ).
- Milekic MH , XinY , O’DonnellAet al. Age-related sperm DNA methylation changes are transmitted to offspring and associated with abnormal behavior and dysregulated gene expression . Mol. Psychiatry20 ( 8 ), 995 – 1001 ( 2015 ).
- Smith RG , ReichenbergA , KemberRLet al. Advanced paternal age is associated with altered DNA methylation at brain-expressed imprinted loci in inbred mice: implications for neuropsychiatric disease . Mol. Psychiatry18 ( 6 ), 635 – 636 ( 2013 ).
- Jenkins TG , AstonKI , PfluegerC , CairnsBR , CarrellDT . Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility . PLoS Genet.10 ( 7 ), e1004458 ( 2014 ).
- Atsem S , ReichenbachJ , PotabattulaRet al. Paternal age effects on sperm FOXK1 and KCNA7 methylation and transmission into the next generation . Hum. Mol. Genet.25 ( 22 ), 4996 – 5005 ( 2016 ).
- Donkin I , VersteyheS , IngerslevLRet al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans . Cell Metab.23 ( 2 ), 369 – 378 ( 2016 ).
- Soubry A , GuoL , HuangZet al. Obesity-related DNA methylation at imprinted genes in human sperm: results from the TIEGER study . Clin. Epigenetics8 , 51 ( 2016 ).
- Chen YP , XiaoXM , LiJ , ReichetzederC , WangZN , HocherB . Paternal body mass index (BMI) is associated with offspring intrauterine growth in a gender dependent manner . PLoS ONE7 ( 5 ), e36329 ( 2012 ).
- Freeman E , FletcherR , CollinsCE , MorganPJ , BurrowsT , CallisterR . Preventing and treating childhood obesity: time to target fathers . Int. J. Obes.36 ( 1 ), 12 – 15 ( 2012 ).
- Yue MX , FuXW , ZhouGBet al. Abnormal DNA methylation in oocytes could be associated with a decrease in reproductive potential in old mice . J. Assist. Reprod. Genet.29 ( 7 ), 643 – 650 ( 2012 ).
- Reik W , DeanW , WalterJ . Epigenetic reprogramming in mammalian development . Science293 ( 5532 ), 1089 – 1093 ( 2001 ).
- Denomme MM , MannMR . Genomic imprints as a model for the analysis of epigenetic stability during assisted reproductive technologies . Reproduction144 ( 4 ), 393 – 409 ( 2012 ).
- Leitão E , BeygoJ , ZeschnigkM , Klein-HitpassL , BargullM , RahmannS , HorsthemkeB . Locus-Specific DNA methylation analysis by targeted deep bisulfite sequencing . Methods Mol. Biol.1767 , 351 – 366 ( 2017 ).
- Haertle L , MaierhoferA , BöckJet al. Hypermethylation of the non-imprinted maternal MEG3 and paternal MEST alleles is highly variable among normal individuals . PLoS ONE12 ( 8 ), e0184030 ( 2017 ).
- Gillman MW . Developmental origins of health and disease . N. Engl. J. Med.353 ( 17 ), 1848 – 1850 ( 2005 ).
- Barker DJ . The origins of the developmental origins theory . J. Intern. Med.261 ( 5 ), 412 – 417 ( 2007 ).
- Wei Y , SchattenH , SunQY . Environmental epigenetic inheritance through gametes and implications for human reproduction . Hum. Reprod. Update21 ( 2 ), 194 – 208 ( 2015 ).
- Blake GE , WatsonED . Unravelling the complex mechanisms of transgenerational epigenetic inheritance . Curr. Opin. Chem. Biol.33 , 101 – 107 ( 2016 ).
- Weber M , HellmannI , StadlerMBet al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome . Nat. Genet.39 ( 4 ), 457 – 466 ( 2007 ).
- Sontag LB , LorinczMC , Georg LuebeckE . Dynamics, stability and inheritance of somatic DNA methylation imprints . J. Theor. Biol.242 ( 4 ), 890 – 899 ( 2006 ).
- Murphy SK , WylieAA , CovelerKJet al. Epigenetic detection of human chromosome 14 uniparental disomy . Hum. Mutat.22 ( 1 ), 92 – 97 ( 2003 ).
- Laborda J . The role of the epidermal growth factor-like protein dlk in cell differentiation . Histol. Histopathol.15 ( 1 ), 119 – 129 ( 2000 ).
- Zhou Y , ZhongY , WangYet al. Activation of p53 by MEG3 non-coding RNA . J. Biol. Chem.282 ( 34 ), 24731 – 24742 ( 2007 ).
- Kameswaran V , BramswigNC , MckennaLBet al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets . Cell Metab.19 ( 1 ), 135 – 145 ( 2014 ).
- Li L , KeverneEB , AparicioSA , IshinoF , BartonSC , SuraniMA . Regulation of maternal behavior and offspring growth by paternally expressed Peg3 . Science284 ( 5412 ), 330 – 333 ( 1999 ).
- Perera BP , KimJ . Sex and tissue specificity of Peg3 promoters . PLoS ONE11 ( 10 ), e0164158 ( 2016 ).
- Riesewijk AM , HuL , SchulzUet al. Monoallelic expression of human PEG1/MEST is paralleled by parent-specific methylation in fetuses . Genomics42 ( 2 ), 236 – 244 ( 1997 ).
- Kozak LP , NewmanS , ChaoPM , MendozaT , KozaRA . The early nutritional environment of mice determines the capacity for adipose tissue expansion by modulating genes of caveolae structure . PLoS ONE5 ( 6 ), e11015 ( 2010 ).
- El Hajj N , PliushchG , SchneiderEet al. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus . Diabetes62 ( 4 ), 1320 – 1328 ( 2013 ).
- Kamei Y , SuganamiT , KohdaTet al. Peg1/Mest in obese adipose tissue is expressed from the paternal allele in an isoform-specific manner . FEBS Lett.581 ( 1 ), 91 – 96 ( 2007 ).
- Kosaki K , KosakiR , CraigenWJ , MatsuoN . Isoform-specific imprinting of the human PEG1/MEST gene . Am. J. Hum. Genet.66 ( 1 ), 309 – 312 ( 2000 ).
- Uyar A , SeliE . The impact of assisted reproductive technologies on genomic imprinting and imprinting disorders . Curr. Opin. Obstet. Gynecol.26 ( 3 ), 210 – 221 ( 2014 ).
- Zechner U , PliushchG , SchneiderEet al. Quantitative methylation analysis of developmentally important genes in human pregnancy losses after ART and spontaneous conception . Mol. Hum. Reprod.16 ( 9 ), 704 – 713 ( 2010 ).
- Vincent RN , GoodingLD , LouieK , Chan WongE , MaS . Altered DNA methylation and expression of PLAGL1 in cord blood from assisted reproductive technology pregnancies compared with natural conceptions . Fertil. Steril.106 ( 3 ), 739.e3 – 748.e3 ( 2016 ).
- Whitelaw N , BhattacharyaS , HoadG , HorganGW , HamiltonM , HaggartyP . Epigenetic status in the offspring of spontaneous and assisted conception . Hum. Reprod.29 ( 7 ), 1452 – 1458 ( 2014 ).
- Tierling S , SourenNY , GriesJet al. Assisted reproductive technologies do not enhance the variability of DNA methylation imprints in human . J. Med. Genet.47 ( 6 ), 371 – 376 ( 2010 ).
- Oliver VF , MilesHL , CutfieldWS , HofmanPL , LudgateJL , MorisonIM . Defects in imprinting and genome-wide DNA methylation are not common in the in vitro fertilization population . Fertil. Steril.97 ( 1 ), 147.e7 – 153.e7 ( 2012 ).
- El Hajj N , HaertleL , DittrichMet al. DNA methylation signatures in cord blood of ICSI children . Hum. Reprod.32 ( 8 ), 1761 – 1769 ( 2017 ).
- Schilling E , El ChartouniC , RehliM . Allele-specific DNA methylation in mouse strains is mainly determined by cis-acting sequences . Genome Res19 ( 11 ), 2028 – 2035 ( 2009 ).
- Kerkel K , SpadolaA , YuanEet al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation . Nat. Genet.40 ( 7 ), 904 – 908 ( 2008 ).
- Schalkwyk LC , MeaburnEL , SmithRet al. Allelic skewing of DNA methylation is widespread across the genome . Am. J. Hum. Genet.86 ( 2 ), 196 – 212 ( 2010 ).
- Meaburn EL , SchalkwykLC , MillJ . Allele-specific methylation in the human genome: implications for genetic studies of complex disease . Epigenetics5 ( 7 ), 578 – 582 ( 2010 ).
- Cheung WA , ShaoX , MorinAet al. Functional variation in allelic methylomes underscores a strong genetic contribution and reveals novel epigenetic alterations in the human epigenome . Genome Biol.18 ( 1 ), 50 ( 2017 ).
- Murrell A , ItoY , VerdeGet al. Distinct methylation changes at the IGF2-H19 locus in congenital growth disorders and cancer . PLoS ONE3 ( 3 ), e1849 ( 2008 ).
- Sullivan MJ , TaniguchiT , JheeA , KerrN , ReeveAE . Relaxation of IGF2 imprinting in Wilms tumours associated with specific changes in IGF2 methylation . Oncogene18 ( 52 ), 7527 – 7534 ( 1999 ).
- Cui H , OnyangoP , BrandenburgS , WuY , HsiehCL , FeinbergAP . Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2 . Cancer Res.62 ( 22 ), 6442 – 6446 ( 2002 ).
- Cui H , Cruz-CorreaM , GiardielloFMet al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk . Science299 ( 5613 ), 1753 – 1755 ( 2003 ).
- Sakatani T , WeiM , KatohMet al. Epigenetic heterogeneity at imprinted loci in normal populations . Biochem. Biophys. Res. Commun.283 ( 5 ), 1124 – 1130 ( 2001 ).
- Rancourt RC , HarrisHR , BaraultL , MichelsKB . The prevalence of loss of imprinting of H19 and IGF2 at birth . FASEB J.27 ( 8 ), 3335 – 3343 ( 2013 ).