
Abstract
Cancer is a disease of global epigenetic dysregulation. Mutations in epigenetic regulators are common events in multiple cancer types and epigenetic therapies are emerging as a treatment option in several malignancies.
A major challenge for the clinical management of cancer is the heterogeneous nature of this disease. Cancers are composed of numerous cell types and evolve over time. This heterogeneity confounds decisions regarding treatment and promotes disease relapse.
The emergence of single-cell epigenomic technologies has introduced the exciting possibility of linking genetic and transcriptional heterogeneity in the context of cancer biology. The next challenge is to leverage these tools for improved patient outcomes. Here we consider how single-cell epigenomic technologies may address the current challenges faced by cancer clinicians.
The epigenome
The mammalian epigenome comprises numerous molecular mechanisms that regulate the accessibility of genomic information. Genes involved in the function of a particular cell type are maintained in an accessible state, while unrequired genes are epigenetically silenced. Thus, epigenetic mechanisms restrict the potential of each cell type and are essential for mammalian development [Citation1]. Epigenetic mechanisms include: regulation of 3D chromosome conformation, changes to nucleosome positioning and chromatin accessibility, chemical modifications to the histone proteins around which DNA is wrapped and chemical modifications to the DNA molecule itself. These modifications can have repressive or activating effects on transcription and the function of each modification can depend on the cellular and genomic context [Citation2]. The most intensely studied epigenetic modification is DNA methylation (5′-methylcytosine, 5mC), which occurs at CpG dinucleotides. In the context of promoters, DNA methylation generally acts as a transcriptional repressor. In contrast, DNA methylation of gene bodies has been linked to transcriptional elongation and regulation of alternative splicing [Citation3]. Similarly, histone modifications can be associated with either transcriptional repression or activation. H3K27ac is a marker of active enhancers, while H3K27me3 in promoters is associated with silent genes [Citation4].
The complex relationships between epigenetic mechanisms are balanced by a suite of regulatory enzymes [Citation5]. Epigenetic ‘writers’ add epigenetic marks to chromatin, while ‘erasers’ can remove these marks. The transcriptional consequences of epigenetic modifications are mediated by epigenetic ‘readers’, which bind to epigenetic marks and recruit transcriptional complexes (). DNMT enzymes act as epigenetic ‘writers’ by incorporating 5mC into the genome. DNMT1 acts during replication to copy 5mC marks from the template DNA strand to the de novo synthesized strand [Citation6]. This ensures the stable inheritance of DNA methylation during cell division and maintains cell identity. During differentiation, DNA methylation is dynamically regulated by addition of 5mC at new positions and removal of existing 5mC marks. The de novo DNMT3 enzymes do not require a hemimethylated substrate [Citation7] and can ‘write’ 5mC into new genomic positions, TET enzymes can initiate the erasure of DNA methylation by oxidizing 5mC to 5′-hydroxymethylcytosine (5hmC) [Citation8]. MBD proteins are examples of epigenetic ‘readers’ as they bind to 5mC and recruit the nucleosome remodeling and deacetylase (NuRD) complex, which can silence transcription [Citation9,Citation10]. In the case of histone acetylation, EP300 acts as ‘writer’ and HDAC enzymes are ‘erasers’. BRD4 is a ‘reader’ of histone acetylation and activates transcription via interaction with P-TEFb [Citation11].
Table 1. Epigenetic regulation in cancer.
In summary, the epigenome is a multi-layered system essential for mammalian genome organization and healthy cells rely on complex regulatory mechanisms for appropriate maintenance and interpretation of epigenetic information. In malignant cells, epigenetic regulation is frequently disrupted at many levels and cancer is consequently considered an epigenomic disease [Citation12].
Cancer as an epigenomic disease
The cancer epigenome is dysregulated on a global scale [Citation2]. In the case of DNA methylation, genome-wide loss is accompanied by focal increases at specific CpG-rich promoters. These changes can result from mutations in epigenetic regulators (discussed below), rapid proliferation and environmental factors. For example, wide-ranging epigenetic changes have been linked to replication timing, with late-replicating regions having reduced DNA methylation in rapidly proliferating cancer cells [Citation13]. Similarly, a hypoxic tumor microenvironment can lead to decreased 5hmC in cancer cells, since oxygen is required for TET-mediated DNA demethylation [Citation14]. A common event in many cancer types is gain of DNA methylation at promoters of tumor-suppressor genes. This results in transcriptional silencing and uncontrolled proliferation. For example, promoter hypermethylation of the cell cycle regulator, CDKN2A, occurs in colorectal cancer [Citation15], oral/oropharyngeal cancers [Citation16,Citation17], hepatocellular carcinoma [Citation18] and non-small-cell lung carcinoma [Citation19]. Similarly, promoter hypermethylation is observed at: APC in colorectal cancer [Citation15] and urothelial bladder cancer [Citation20]; RASSF1A in oral/oropharyngeal cancer [Citation17] and endometrial cancer [Citation21]; and TP53 [Citation22] and BRCA1 [Citation23] in breast cancers.
These changes to DNA methylation are matched by changes to histone modifications and nucleosome remodeling [Citation2]. For example, in colorectal cancer, H3K9 methylation positively correlates with promoter DNA methylation in CDKN2A, MLH1 and MGMT, whereas H3K9 acetylation inversely correlates with promoter DNA methylation of these genes [Citation24]. Histone acetylation at promoters, enhancers and gene bodies can lead to transcription of oncogenes (e.g., AR in prostate cancer [Citation25] and overexpression of HDACs can lead to silencing of tumor-suppressor genes (e.g., TP53) and cell cycle inhibitors (e.g., CDKN1A) [Citation26].
Enzymes involved in epigenetic regulation (e.g., DNMTs, HDACs) are commonly mutated in numerous cancer types () [Citation27]. For example, a third of renal clear cell carcinomas carry mutations in PBRM1, which is a component of the switch/sucrose nonfermentable nucleosome (SWI/SNF) remodeling complex. Mutations in epigenetic pathways can also occur very early in oncogenesis. Indeed, DNMT3A mutations have been observed in preleukemic hematopoietic stem cells in patients with acute myeloid leukemia (AML) [Citation28,Citation29] and may identify healthy individuals at increased risk of AML development [Citation30,Citation31]. Full leukemia results when a secondary driver mutation is acquired in the clonally expanded DNMT3Amut stem cell population [Citation28]. Mutations in epigenetic regulators are accompanied by global dysregulation of the cancer epigenome. For example, DNMT3Amut AML cases display loss of DNA methylation in CpG-poor genomic loci [Citation32] and CpG island shores [Citation33]. Moreover, TET2mut AML cases show hypermethylation at enhancer regions [Citation34].
Driven by the clinical challenges associated with cancer evolution, an increasing number of studies are also considering the dynamic regulation of epigenetics in cancer. A coarse-grained view of epigenetic changes during clonal evolution has been achieved through multiregion sampling and parallel analysis of copy number alterations and DNA methylation in prostate cancer [Citation35]. Other studies in hematological malignancies have monitored DNA methylation heterogeneity during cancer progression and identified links to patient outcome [Citation36,Citation37]. Excitingly, single-cell analyses are also linking genetic clonal evolution to epigenetic and transcriptional dysregulation during cancer progression [Citation38,Citation39].
Clinical relevance of cancer epigenetics
The cancer epigenome can be targeted for disease screening, monitoring and treatment. The relative stability of DNA methylation has led to the development of diagnostic biomarkers that detect cancer DNA in blood samples. The first US FDA approved blood-based biomarker for colorectal cancer screening was based on cancer-specific DNA hypermethylation at SEPTIN9 [Citation40]. Epigenetic profiling is also being used to develop generalized tests for the detection of numerous cancer types. For example, widespread epigenomic remodeling of circulating cell free DNA can be detected in the blood of patients with various solid cancer types [Citation41,Citation42]. In addition, DNA methylation signatures have also been studied in circulating tumor cells (CTCs) purified from the blood of breast and prostate cancer patients [Citation43,Citation44].
DNA methylation signatures differ between cancer sub-types and can predict prognosis. For example, a panel of differentially methylated loci can identify a sub-group of triple negative breast cancers with poor prognosis [Citation45]. In cancers with an unknown primary site, lineage-specific DNA methylation signatures can also guide appropriate diagnosis [Citation46]. Similarly, several tests are available for assessment of MGMT promoter methylation in glioblastoma, with methylated cancers having improved response to alkylating agents [Citation47]. Of course, the clinical value of such tests is limited by the availability of targeted therapies for every sub-group identified.
Epigenetic mechanisms are also being therapeutically targeted in several cancer types, with the aim of resetting aberrant transcriptional programs or increasing tumor immunogenicity [Citation12]. More than 30 agents are approved or undergoing clinical trials, including inhibitors of epigenetic ‘writers’, ‘readers’ and ‘erasers’ () [Citation12,Citation48]. The first epigenetic therapy to be used was the DNMT inhibitor (or hypomethylating agent), azacytidine. This cytidine analogue prevents the incorporation of 5mC during cell division and is used to treat certain patients with AML or myelodysplastic syndrome. Initial studies demonstrated durable responses, with azacytidine trebling the 2-year overall survival rate achieved by conventional care in a cohort of elderly AML patients [Citation49]. A number of other DNMT inhibitors have since been approved for use in AML and myelodysplastic syndrome [Citation12], but variability in patient response remains a clinical challenge [Citation50]. HDAC inhibitors demonstrate the utility of targeting epigenetic ‘erasers’ in cancer therapy. These agents were first used in T-cell lymphomas and have since been approved for use in multiple myeloma [Citation12]. Ongoing clinical trials are now applying HDAC inhibitors in other hematological and solid malignancies. Similarly, epigenetic ‘readers’ can be targeted using bromodomain and extra-terminal family inhibitors. These agents block the binding of BRD4 (and other bromodomain and extra-terminal family members) to acetylated histones and are being tested in several cancer types [Citation51]. Ongoing work is expanding the range of epigenetic therapies available and testing combination therapy approaches to improve the efficacy of existing therapies. For example, hypomethylating agents are being tested in combination with other epigenetic therapies (e.g., HDAC inhibitors), immunotherapies and targeted therapies [Citation50,Citation52,Citation53].
Single-cell epigenomics
The last 6 years have seen the emergence of single-cell sequencing technologies for analysis of epigenetic mechanisms including: DNA modifications, histone modifications, DNA accessibility and chromosome conformation [Citation54–56] (Supplementary Table 1). Whereas traditional analyses averaged measurements over thousands of cells, single-cell epigenomic methods can characterize rare cell populations of clinical relevance [Citation57] and detail the mechanisms of genetic regulation in heterogeneous cell populations [Citation58,Citation59].
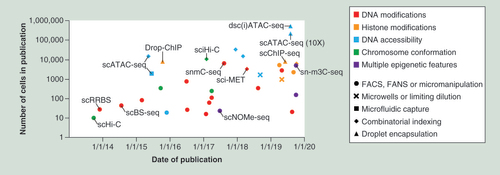
More than 30 single-cell epigenomic methods have been reported (Supplementary Table 1) and ongoing work continues to improve the capabilities and through-put of these methods. Early methods used FACS or micromanipulation to physically isolate single cells in separate reaction chambers and were able to process tens to hundreds of cells in parallel (). By encapsulating single cells in droplets that contain unique barcodes, more recent studies have processed thousands of single cells in parallel. A combinatorial indexing approach has also been used to identify cells through unique combinations of molecular tags without physically isolating individual cells. By combining combinatorial indexing with droplet encapsulation, a recent study demonstrated that tens of thousands of single-cell epigenomes can be measured in parallel [Citation60]. Another exciting advance has been the development of ‘multimodal’ methods that can measure several layers of molecular information in the same cell (Supplementary Table 1) [Citation61]. By coupling epigenetic information with parallel genomic and/or transcriptomic data, these methods facilitate the biological interpretation of epigenetic heterogeneity. For example, an early study performed parallel analysis of DNA methylation and gene expression in heterogeneous embryonic stem cells. This revealed specific enhancer sites where DNA methylation was associated with reduced expression of adjacent genes important for stem cell pluripotency [Citation58]. Another study has coupled analysis of DNA methylation and copy number alterations in colorectal cancer samples, to compare epigenomes from different genetic sub-clones [Citation39].
The publication dates and cell numbers are graphed for single-cell epigenomic methods, with the names of key methods shown. The epigenetic features assessed are indicated by colour and the means of cell isolation are indicated by symbol. This field has seen substantial growth in the diversity of epigenetic modifications that can be studied in single cells and significant improvements to through-put driven by combinatorial indexing and droplet encapsulation. Supplementary Table 1 provides further details.
FACS: Fluorescence activated cell sorting; FANS: Fluorescence activated nuclei sorting.
Despite the rapid advancements in single-cell epigenomics, this field remains immature. Epigenetic information is not encoded by simple nucleotide sequence, so these technologies rely on chemical conversions, enzymatic treatments or antibody-mediated enrichment to translate epigenetic information into sequencing libraries (Supplementary Table 1). Consequently, epigenomic analyses are more challenging and less established than similar transcriptomic and genomic methods. However, the first commercial assay for single-cell epigenomic analysis was recently released [Citation62] and the next 5 years will no doubt see rapid developments in this sector. This will involve adaptation of existing methods for implementation on user-friendly microfluidic devices, development of robust controls for quality assessment and streamlined data analysis pipelines. Critically, parallel indexing of DNA and RNA in droplet microfluidics devices will also be required for widespread uptake of multi-omic single-cell analyses.
The ultimate challenge for any new research technology is to find appropriate applications in biology. Single-cell epigenetic analyses have so far been focused on developmental biology where the epigenome is dynamically regulated [Citation63,Citation64]. However, the clinical implications of cellular heterogeneity in cancer make this an obvious subject for single-cell analyses.
Clinical relevance of single-cell epigenetics
Cellular heterogeneity
Cancers are known to comprise numerous cell types including malignant and normal cells from the tissue-of-origin, as well as infiltrating fibroblasts, immune cells, endothelial cells and nerves [Citation65]. Each of these cell lineages may also be represented by numerous cell states. For example, malignant cells can include cancer stem cells as well as more differentiated cell states and T cells can be either activated or exhausted. It is well understood that the cellular heterogeneity of individual cancers can influence important clinical considerations such as response to therapy and disease relapse. Indeed, pathologists routinely score solid tumors for the proportion of cells expressing clinically relevant molecules using immunohistochemistry (e.g., HER2 in breast cancer) [Citation66].
Thus, the majority of single-cell sequencing studies in cancer biology have been designed to profile cellular heterogeneity [Citation67]. For example, single-cell genomics has revealed the clonal evolution of human triple-negative breast cancers [Citation68] and single-cell transcriptomics has uncovered evidence of cancer initiating cells in human glioma [Citation69]. Another recent study has combined scRNA-seq with targeted single-cell genotyping in AML, revealing malignant cell types that were not apparent in clinical flow cytometric analyses [Citation70].
How can single-cell epigenomic analyses add to these transcriptomic studies of cancer cell heterogeneity? Pioneering work has used analysis of chromatin accessibility to look at the heterogeneity of cancer regulomes in leukemic cell lines and patient samples. In addition to describing different cell states, this allows the inference of transcription factors that drive those cell states. The first report of single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) analysed K562 and HL60 leukemia cell lines [Citation71]. While HL-60 cells are relatively homogeneous, K562 cells display epigenomic variability [Citation71,Citation72], with heterogeneous accessibility at GATA-binding motifs. Another study purified sub-populations based on CD24 expression, to investigate the functional relevance of variable GATA motif accessibility [Citation73]. CD24hi K562 cells were found to have lower accessibility at GATA-binding motifs and a less differentiated phenotype than CD24lo cells, demonstrating that key transcription factors can distinguish functionally distinct cell states. An elegant study by Corces and colleagues has applied similar analyses in patient samples [Citation74]. Chromatin accessibility was first assessed in several cell populations isolated from primary AML patient samples and compared with normal hematopoietic cell types [Citation74]. The chromatin accessibility profiles of AML cell populations were found to be intermediate between healthy cell states. For example, the leukemia stem cell populations from some patients did not match progenitor cells from healthy blood, but had some similarity to both lymphoid-primed multipotent progenitor cells and monocytes [Citation74]. This implied that AML cells either adopt a unique cell state intermediate between normal cell types or AML cell populations are a heterogeneous mix of cell states. To test these two possibilities, scATAC-seq was then applied to several patients. In one patient, single AML cells were found to have chromatin accessibility profiles distinct from any normal cell type, while in another patient, some AML cells resembled normal monocytes and others were in a cancer-specific cell state [Citation74]. Thus, the heterogeneity of AML regulomes can differ between patients. This study also compared normal hematopoietic stem cells from healthy donors to the premalignant hematopoietic stem cells from AML patients, in order to identify the very earliest changes contributing to AML development. HOX, RUNX and GATA motifs were found to have less accessible chromatin in AML stem cells, suggesting that leukemia is derived from a dysregulated stem cell pool [Citation74].
These studies demonstrate that analysis of epigenomic heterogeneity can complement scRNA-seq experiments by providing information on the regulatory networks that govern cell state. There is also evidence that epigenetic information can provide a more robust classification of cell states than transcriptomics [Citation74]. Future work will also consider the utility of DNA methylation analysis in defining cell heterogeneity, particularly in cancers where this epigenetic mark is influenced by mutations (e.g., DNMT3A, TET2) or targeted by epigenetic therapy.
Metastasis
Cancer dissemination is a major clinical challenge, as the majority of cancer deaths occur following metastasis. In the case of breast cancer, the 5-year survival rates are as high as 99% for disease confined to the breast, yet following metastasis to distant organs, this statistic drops dramatically to 27% [Citation75]. Metastasis is a complex biological process that occurs when cancer cells acquire invasive traits, escape the primary tumor site, travel through the circulation and colonize distant sites [Citation76,Citation77]. Epigenetic mechanisms are known to contribute to metastasis and may even drive this process [Citation78]. In order to escape the primary tumor site, epithelial cells must first undergo an epithelial to mesenchymal transition (EMT). Epigenetic mechanisms promote cell plasticity during this process, with reversible changes first allowing altered gene expression before stabilization of the mesenchymal state. For example, promoters of key epithelial genes first gain repressive histone modifications (e.g., H3K27me3), before DNA methylation is acquired in the mesenchymal state [Citation79]. Cell plasticity is also enabled by ‘bivalent’ chromatin, in which repressive and activating histone modifications occur at the same position. For example, ‘bivalent’ chromatin at the promoter of an EMT master regulator, ZEB1, allows breast cancer cells to switch between cell states. In the cancer stem cell state, increased metastatic capacity is associated with activating modifications ( H3K4me3) at the ZEB1 promoter [Citation80].
The clinical importance of metastasis has made this the subject of single-cell sequencing since early scRNA-seq analyses [Citation81]. Transcriptomic studies have defined transitional states during EMT [Citation82], identified transcripts specifically expressed in CTCs [Citation81,Citation83], described heterogeneity among CTCs with functional implications [Citation83,Citation84] and characterized metastasis initiating cells colonizing distant metastatic sites [Citation85].
Single-cell epigenomic analyses are also being applied to understand the metastatic process. In one study, multi-omic profiling of DNA methylation, somatic copy number alterations and gene expression was performed in the context of colorectal carcinoma [Citation39]. Single cells derived from the primary tumor and patient-matched liver metastasis were profiled and segregated into sub-lineages based on somatic copy number alteration analyses in five patients. While metastasis was associated with clonal selection, DNA methylation profiles remained largely stable within sub-lineages during metastasis. While some focal changes in DNA methylation were observed in gene promoters, these genes were not associated with metastatic processes or enriched for any other gene ontology. Another study applied single-cell DNA methylation analysis to single CTCs and CTC clusters purified from the blood of breast cancer patients [Citation86]. This study demonstrated hypomethylation at stem cell genes in CTC clusters, which was linked to increased expression of pluripotency networks. This suggests that the increased metastatic potential of CTC clusters is due to acquisition of a stem cell state. A subsequent screen for compounds that decreased the formation of CTC clusters, identified a candidate drug with encouraging results in xenograft models.
The importance of epigenetic mechanisms during metastasis and the clinical relevance of CTCs, will no doubt lead to further single-cell epigenomic analysis of these rare cells. Future studies will characterize DNA methylation patterns in CTCs to identify unique signatures in cells able to initiate distant metastasis. These signatures may form the basis of clinical biomarkers used to monitor patients at risk of progressive disease.
Therapeutic response
The recent growth of targeted therapeutic strategies has led to major improvements for patients in some cancer types. For example, the introduction of immunotherapy for treatment of advanced metastatic melanoma has seen 4-year overall survival rates more than double in recent years [Citation87]. However, patient response to therapy is variable, meaning that these expensive therapies have no benefit in some patients and other patients eventually develop resistance despite initially exciting results [Citation88]. Thus, strategies to predict as well as monitor patient response and prevent the development of resistance are clinical priorities.
A patient’s response to therapy can be dependent on the cellular heterogeneity within a tumor and/or its microenvironment. For example, single-cell transcriptomics has been used to identify a gene expression signature that can predict response to immune checkpoint inhibitors in melanoma patients [Citation89]. This study also identified a therapeutic strategy to target cells expressing the immune resistance program, with some promising preclinical results. Similarly, early scATAC-seq experiments demonstrated that epigenomic variability is modulated by pharmacological treatment (with CDK 4/6 inhibitor or TNF-α stimulation) [Citation71] and demonstrated that cell states with distinct epigenomic profiles can respond differently to targeted therapy (tyrosine kinase inhibitor) [Citation73]. More recently, single-cell analysis of DNA accessibility in the context of basal cell carcinoma has been used to understand the variability in patient response to PD-1 blockade [Citation62]. This study profiled the epigenomes of tumor-infiltrating T-cells to identify therapy-induced regulatory programs associated with T-cell exhaustion.
The clinical utility of epigenetic therapies (e.g., hypomethylating agents) is also limited by variable efficacy in patients and difficulty in predicting response [Citation90]. Therefore, a growing number of clinical trials are combining epigenetic therapies with hormonal therapies, immunotherapies and cytotoxic chemotherapies [Citation12,Citation50,Citation53]. Many of these trials are seeking to resensitize nonresponsive patients to standard treatments, but do not consider how epigenetic therapies influence different cell types. Future studies will apply single-cell epigenomic analyses to understand the heterogeneous response of cells (including cancer cells, immune cells and other cells of the tumor microenvironment) to epigenetic therapies. These studies will identify new strategies to improve drug efficacy or predict response. An initial single-cell study has already demonstrated increased DNA methylation variance following azacytidine treatment of the K562 leukemia cell line and reported that genomic loci resistant to loss of DNA methylation were enriched for repressive chromatin marks [Citation72]. This suggests that combination therapy with chromatin modifying drugs may increase the efficacy of azacytidine.
Future perspective
Toward clinical applications of single-cell epigenetics
How can the biological insights gleaned from single-cell epigenomic analyses (described above) be translated into meaningful clinical applications and improved outcomes for patients? This process will face many challenges (outlined below) familiar to biomedical research but will also encounter unique obstacles related to the nature of single-cell analyses. These challenges are equally relevant to single-cell transcriptomic studies which, despite their relative maturity, are only beginning to guide clinical decisions in pioneering case studies [Citation91].
Clinical implementation of single-cell sequencing technologies
For single-cell sequencing to be performed by pathology services, robust commercial platforms must be developed, including standardized procedures for cell preparation, data analysis and interpretation of results. It is encouraging to note that these challenges have already been overcome by the clinical use of flow cytometry and massive parallel sequencing. Much like single-cell sequencing technologies, flow cytometry involves careful preparation of cell suspensions and reports on the proportion of cells in a sample with a particular phenotype. For example, a clinical test based on flow cytometry can detect minimal residual disease in leukemia, with a sensitivity of <0.01% [Citation92,Citation93] and detection of minimal residual disease can be used to inform decisions about allogenic bone marrow transplantation [Citation94]. Massive parallel sequencing is also being implemented in clinical settings for molecular characterization of malignancy [Citation95,Citation96], monitoring of disease course [Citation97,Citation98] and to identify causative mutations in rare genetic diseases [Citation99]. Implementation of this technology has faced issues related to turn-around time, data analysis and interpretation of results. Similarly, clinical use of single-cell sequencing would require standardized and simplified reporting of complex data sets to distill clinically relevant information. Cost is another obstacle to clinical implementation of most new technologies. In the case of massive parallel sequencing, the cost has fallen by several orders of magnitude since the completion of the human genome project. The cost of single-cell transcriptomic analyses have also fallen substantially since the advent of droplet based cell indexing strategies [Citation100] and single-cell epigenomic technologies are following suit.
In summary, many obstacles to clinical implementation of single-cell sequencing are likely to be surmountable as illustrated by the precedent of similar technologies. Nonetheless, the cost of single-cell sequencing will always be higher than traditional molecular pathology methods, so simplified tests based on single-cell analyses, but compatible with existing clinical infrastructure, should be considered.
Adaptation of single-cell analyses for simplified tests using existing infrastructure
Single-cell sequencing analyses can disentangle complex cell mixtures to characterize specific cell types of clinical relevance. However, the molecular detail afforded may not be required for clinical applications. For example, while single-cell DNA sequencing can sequence the whole genome of individual cells, it is clinically more practical to study target genes using variant allele frequency analysis of sequencing data obtained from a mixed population of cells. Similarly, once single-cell transcriptomic studies have identified rare cell types with important clinical implications, tests based on flow cytometry could check for the presence of these cells in patient samples using cell surface markers identified in scRNA-seq data.
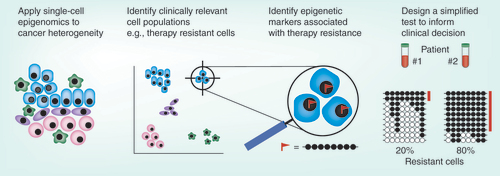
How can simplified tests be used to estimate the proportion of cells with a certain epigenetic status? In the case of DNA methylation, bisulphite sequencing is able to report on both methylated and unmethylated cytosines. In addition, the methylation status of a particular allele can be determined because neighbouring cytosines are linked in the same sequencing read. Since the days of clonal bisulphite sequencing, these properties have enabled estimation of the epigenetic heterogeneity in mixed cell populations. The advent of massive parallel sequencing has greatly increased the through-put of this approach, with amplicon bisulphite sequencing allowing efficient parallel sequencing of thousands of epialleles from bulk DNA samples. Thus, if single-cell epigenetic analyses identify loci where DNA methylation status is associated with clinically relevant phenotypes (e.g., therapy-resistance), amplicon bisulphite sequencing could be used to estimate the proportion of cells with that phenotype (). Although not currently in routine clinical use, amplicon bisulphite sequencing is transferrable to existing infrastructure, cost-efficient and relatively simple to analyse. Other epigenetic features such as chromatin accessibility and histone modifications are less amenable to such approaches because they are studied using count-based methods that select for a certain modification status, meaning that only some cells are represented in the data.
Cellular heterogeneity in cancer is associated with clinical challenges such as metastasis, therapeutic response and relapse. Single-cell technologies will identify target cell populations (by dimensional reduction methods) that could not be distinguished using traditional techniques. Single-cell epigenomics will identify modifications specific to those cells. For example, DNA methylation at a certain allele may be observed only in therapy resistant cells (red flag). Based on this information, simplified tests will be developed for clinical use. For example, amplicon bisulphite sequencing of specific loci may be used to advise clinicians of the proportion of cells in a sample with therapeutic resistance (each line indicates a sequencing read, black and white circles represent methylated and unmethylated cytosines respectively, the methylation pattern associated with therapy resistance is highlighted in red).
Conclusion
Single-cell epigenomic analyses hold much potential to uncover clinically relevant aspects of cancer biology. Initial studies have already applied these methodologies to the cellular heterogeneity of cancer, CTCs and therapeutic response. The clinical translation of biological insights requires careful consideration but may be achieved through either direct implementation of single-cell sequencing technologies or adaptation of existing infrastructure. In either case, development of cost-efficient high throughput platforms, appropriate standards and robust analytical pipelines, will be required for these technologies to reach their full potential. During the next decade of research, we can expect to see exciting technological, biological and clinical advances as these novel methods move toward their ultimate goal of delivering meaningful benefit to patients.
Cancer as an epigenomic disease
Cancer is associated with wide-spread disruption of the epigenome.
Epigenetic modulators are frequently mutated in several cancer types.
Clinical relevance of cancer epigenetics
Epigenomic information can be used to predict patient outcome in cancer.
Epigenetic therapies are used to treat several cancer types.
Single-cell epigenomics
Various epigenetic features can be assessed using single-cell sequencing techniques, allowing analysis of heterogeneous and rare cell populations.
These methods are developing improved through-put and cost-efficiency.
Clinical relevance of single-cell epigenetics
Single-cell analyses can reveal how certain cancer cells drive disease progression.
Emerging single-cell epigenomic studies are investigating genomic regulation in intra-tumoral heterogeneity, metastasis and therapy response.
Toward clinical applications of single-cell epigenetics
To implement single-cell epigenomic technologies in the clinic, further improvements in cost-efficiency, through-put, standardization and data analysis are required.
Alternatively, simplified clinical tests could be developed based on biological discoveries made using single-cell epigenomics.
Supplemental Table 1
Download MS Word (77.6 KB)Acknowledgments
HJ Lee thanks C Gedye and R Scott for helpful discussions regarding the challenges of cancer treatment and clinical implementation of new management strategies.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2217/epi-2020-0046
Financial & competing interests disclosure
HJ Lee has received research funding from: The Cancer Institute NSW, Australia (ECF171145); The National Health and Medical Research Council, Australia (GNT1143614, GNT1143614); The Ian Potter Foundation, Australia (20180029); The National Stem Cell Foundation of Australia (2018 Metcalf Prize); The Australian Research Council (DP200102903). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Meissner A . Epigenetic modifications in pluripotent and differentiated cells. Nature Biotechnol.28(10), 1079–1088 (2010).
- Baylin SB , JonesPA. A decade of exploring the cancer epigenome - biological and translational implications. Nat. Rev. Cancer11(10), 726–734 (2011).
- Jones PA . Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet.13(7), 484–492 (2012).
- Zhao Z , ShilatifardA. Epigenetic modifications of histones in cancer. Genome Biol.20(1), 245 (2019).
- Biswas S , RaoCM. Epigenetic tools (the writers, the readers and the erasers) and their implications in cancer therapy. Eur. J. Pharmacol.837, 8–24 (2018).
- Schubeler D . Function and information content of DNA methylation. Nature517(7534), 321–326 (2015).
- Okano M , XieS , LiE. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet.19(3), 219–220 (1998).
- Wu H , ZhangY. Reversing DNA methylation: mechanisms, genomics and biological functions. Cell156(1-2), 45–68 (2014).
- Feng Q , ZhangY. The MeCP1 complex represses transcription through preferential binding, remodeling and deacetylating methylated nucleosomes. Genes Dev.15(7), 827–832 (2001).
- Leighton G , WilliamsDC. The methyl-CpG–binding domain 2 and 3 proteins and formation of the nucleosome remodeling and deacetylase complex. J. Mol. Biol.432(6), 1624–1639 (2019).
- Itzen F , GreifenbergAK , BöskenCA , GeyerM. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res.42(12), 7577–7590 (2014).
- Jones PA , IssaJP , BaylinS. Targeting the cancer epigenome for therapy. Nat. Rev. Genet.17(10), 630–641 (2016).
- Du Q , BertSA , ArmstrongNJet al. Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat. Commun.10(1), 416 (2019).
- Thienpont B , SteinbacherJ , ZhaoHet al. tumor hypoxia causes DNA hypermethylation by reducing TET activity. Nature537(7618), 63–68 (2016).
- Ng JM , YuJ. Promoter hypermethylation of tumor suppressor genes as potential biomarkers in colorectal cancer. Int. J. Mol. Sci.16(2), 2472–2496 (2015).
- Bhatia V , GoelMM , MakkerAet al. Promoter region hypermethylation and mRNA expression of MGMT and p16 genes in tissue and blood samples of human premalignant oral lesions and oral squamous cell carcinoma. Biomed. Res. Int.2014, 248419–248419 (2014).
- Liyanage C , WathupolaA , MuraleetharanS , PereraK , PunyadeeraC , UdagamaP. Promoter hypermethylation of tumor-suppressor genes p16(INK4a), RASSF1A, TIMP3 and PCQAP/MED15 in salivary DNA as a quadruple biomarker panel for early detection of oral and oropharyngeal cancers. Biomolecules9(4), 148 (2019).
- Lv X , YeG , ZhangX , HuangT. p16 Methylation was associated with the development, age, hepatic viruses infection of hepatocellular carcinoma and p16 expression had a poor survival: a systematic meta-analysis (PRISMA). Medicine96(38), e8106 (2017).
- Liu Y , LanQ , SiegfriedJM , LuketichJD , KeohavongP. Aberrant promoter methylation of p16 and MGMT genes in lung tumors from smoking and never-smoking lung cancer patients. Neoplasia8(1), 46–51 (2006).
- Bilgrami SM , QureshiSA , PervezS , AbbasF. Promoter hypermethylation of tumor suppressor genes correlates with tumor grade and invasiveness in patients with urothelial bladder cancer. Springerplus3, 178 (2014).
- Fiolka R , ZuborP , JanusicovaVet al. Promoter hypermethylation of the tumor-suppressor genes RASSF1A, GSTP1 and CDH1 in endometrial cancer. Oncol. Rep.30(6), 2878–2886 (2013).
- Kang JH , KimSJ , NohDYet al. Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: correlation between CpG methylation in the p53 promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab. Invest.81(4), 573–579 (2001).
- Zhang L , LongX. Association of BRCA1 promoter methylation with sporadic breast cancers: evidence from 40 studies. Sci. Rep.5, 17869–17869 (2015).
- Kondo Y , ShenL , IssaJP. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol. Cell. Biol.23(1), 206–215 (2003).
- Di Cerbo V , SchneiderR. Cancers with wrong HATs: the impact of acetylation. Brief Funct. Genomics12(3), 231–243 (2013).
- Glozak MA , SetoE. Histone deacetylases and cancer. Oncogene26(37), 5420–5432 (2007).
- Kandoth C , MclellanMD , VandinFet al. Mutational landscape and significance across 12 major cancer types. Nature502(7471), 333–339 (2013).
- Shlush LI , ZandiS , MitchellAet al. Identification of pre-leukaemic haematopoietic stem cells in acute leukemia. Nature506(7488), 328–333 (2014).
- Jan M , SnyderTM , Corces-ZimmermanMRet al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med.4, 149ra118 (2012).
- Abelson S , CollordG , NgSWKet al. Prediction of acute myeloid leukemia risk in healthy individuals. Nature559(7714), 400–404 (2018).
- Desai P , Mencia-TrinchantN , SavenkovOet al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med.24(7), 1015–1023 (2018).
- The Cancer Genome Atlas Research Network . Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med.368(22), 2059–2074 (2013).
- Spencer DH , Russler-GermainDA , KetkarSet al. CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell168(5), 801–816e813 (2017).
- Rasmussen KD , JiaG , JohansenJVet al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev.29(9), 910–922 (2015).
- Brocks D , AssenovY , MinnerSet al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep.8(3), 798–806 (2014).
- Li S , Garrett-BakelmanFE , ChungSSet al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med.22(7), 792–799 (2016).
- Pan H , JiangY , BoiMet al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat. Commun.6, 6921 (2015).
- Gaiti F , ChaligneR , GuHet al. Epigenetic evolution and lineage histories of chronic lymphocytic leukemia. Nature569(7757), 576–580 (2019).
- Bian S , HouY , ZhouXet al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science362(6418), 1060–1063 (2018).
- Song L , LiY. Progress on the clinical application of the SEPT9 gene methylation assay in the past 5 years. Biomark. Med.11(6), 415–418 (2017).
- Chan KCA , JiangP , ChanCWMet al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl Acad. Sci. USA110(47), 18761–18768 (2013).
- Shen SY , SinghaniaR , FehringerGet al. Sensitive tumor detection and classification using plasma cell-free DNA methylomes. Nature563(7732), 579–583 (2018).
- Pixberg CF , RabaK , MullerFet al. Analysis of DNA methylation in single circulating tumor cells. Oncogene363223–3231 (2017).
- Pixberg CF , SchulzWA , StoeckleinNH , NevesRPL. Characterization of DNA methylation in circulating tumor cells. Genes6(4), 1053–1075 (2015).
- Stirzaker C , ZotenkoE , SongJZet al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun.6, 5899 (2015).
- Moran S , Martínez-CardúsA , SayolsSet al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol.17(10), 1386–1395 (2016).
- Wick W , WellerM , VanDen Bent Met al. MGMT testing – the challenges for biomarker-based glioma treatment. Nat. Rev. Neurol.10, 372 (2014).
- Cheng Y , HeC , WangMet al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct. Target. Ther.4(1), 62 (2019).
- Fenaux P , MuftiGJ , Hellstrom-LindbergEet al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol.28(4), 562–569 (2010).
- Fennell KA , BellCC , DawsonMA. Epigenetic therapies in acute myeloid leukemia: where to from here?Blood134(22), 1891–1901 (2019).
- Alqahtani A , ChoucairK , AshrafMet al. Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci. OA5(3), Fso372 (2019).
- Mohammad HP , BarbashO , CreasyCL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat. Med.25(3), 403–418 (2019).
- Jones PA , OhtaniH , ChakravarthyA , DeCarvalho DD. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer19(3), 151–161 (2019).
- Clark SJ , LeeHJ , SmallwoodSA , KelseyG , ReikW. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol.17, 72 (2016).
- Kelsey G , StegleO , ReikW. Single-cell epigenomics: recording the past and predicting the future. Science358(6359), 69–75 (2017).
- Schwartzman O , TanayA. Single-cell epigenomics: techniques and emerging applications. Nat. Rev. Genet.16, 716 (2015).
- Knapp DJHF , HammondCA , HuiTet al. Single-cell analysis identifies a CD33+ subset of human cord blood cells with high regenerative potential. Nat. Cell Biol.20(6), 710–720 (2018).
- Angermueller C , ClarkSJ , LeeHJet al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods13(3), 229–232 (2016).
- Argelaguet R , ClarkSJ , MohammedHet al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature576(7787), 487–491 (2019).
- Lareau CA , DuarteFM , ChewJGet al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nature Biotechnol.37(8), 916–924 (2019).
- Zhu C , PreisslS , RenB. Single-cell multimodal omics: the power of many. Nature methods17(1), 11–14 (2020).
- Satpathy AT , GranjaJM , YostKEet al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nature Biotechnol.37(8), 925–936 (2019).
- Hemberger M , DeanW , ReikW. Epigenetic dynamics of stem cells and cell lineage commitment. Digging Waddington’s canal. Nat. Rev. Mol. Cell Biol.10, 526–537 (2009).
- Lee HJ , HoreTA , ReikW. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell14(6), 710–719 (2014).
- Giraldo NA , Sanchez-SalasR , PeskeJDet al. The clinical role of the TME in solid cancer. Br. J. Cancer120(1), 45–53 (2019).
- Gustavson MD , Bourke-MartinB , ReillyDet al. Standardization of HER2 immunohistochemistry in breast cancer by automated quantitative analysis. Arch. Pathol. Lab. Med.133(9), 1413–1419 (2009).
- Suvà ML , TiroshI. Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol. Cell75(1), 7–12 (2019).
- Gao R , DavisA , McdonaldTOet al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet.48(10), 1119–1130 (2016).
- Tirosh I , VenteicherAS , HebertCet al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature539(7628), 309–313 (2016).
- Van Galen P , HovestadtV , WadsworthIi MHet al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell176(6), 1265–1281.e1224 (2019).
- Buenrostro JD , WuB , LitzenburgerUMet al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature523(7561), 486–490 (2015).
- Farlik M , SheffieldNC , NuzzoA , DatlingerP , SchöneggerA , KlughammerJ. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep.10(8), 1386–1397 (2015).
- Litzenburger UM , BuenrostroJD , WuBet al. Single-cell epigenomic variability reveals functional cancer heterogeneity. Genome Biol.18(1), 15 (2017).
- Corces MR , BuenrostroJD , WuBet al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet.48(10), 1193–1203 (2016).
- American Cancer Society . Cancer facts & figures 2020. (2020). www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf
- Lambert AW , PattabiramanDR , WeinbergRA. Emerging biological principles of metastasis. Cell168(4), 670–691 (2017).
- Fares J , FaresMY , KhachfeHH , SalhabHA , FaresY. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct. Target. Ther.5(1), 28 (2020).
- Chatterjee A , RodgerEJ , EcclesMR. Epigenetic drivers of tumorigenesis and cancer metastasis. Semin. Cancer Biol.51, 149–159 (2018).
- Tam WL , WeinbergRA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med.19, 1438 (2013).
- Chaffer Christine l , MarjanovicNemanja d , LeeTet al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell154(1), 61–74 (2013).
- Ramskold D , LuoS , WangY-Cet al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotech.30(8), 777–782 (2012).
- Pastushenko I , BrisebarreA , SifrimAet al. Identification of the tumor transition states occurring during EMT. Nature556(7702), 463–468 (2018).
- Ting David t , WittnerBen s , LigorioMet al. Single-Cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep.8(6), 1905–1918 (2014).
- Miyamoto DT , ZhengY , WittnerBSet al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science349(6254), 1351–1356 (2015).
- Lawson DA , BhaktaNR , KessenbrockK , PrummelKD , YuY , TakaiK. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature526 (2015).
- Gkountela S , Castro-GinerF , SzczerbaBMet al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell176(1), 98–112.e114 (2019).
- Iorgulescu JB , HararyM , ZoggCKet al. Improved risk-adjusted survival for melanoma brain metastases in the era of checkpoint blockade immunotherapies: results from a national cohort. Cancer Immunol. Res.6(9), 1039–1045 (2018).
- Sambi M , BagheriL , SzewczukMR. Current challenges in cancer immunotherapy: multimodal approaches to improve efficacy and patient response rates. J. Oncol.2019, 4508794 (2019).
- Jerby-Arnon L , ShahP , CuocoMSet al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell175(4), 984–997.e924 (2018).
- Duchmann M , ItzyksonR. Clinical update on hypomethylating agents. Int. J. Hematol.110(2), 161–169 (2019).
- Kim D , KobayashiT , VoisinBet al. Targeted therapy guided by single-cell transcriptomic analysis in drug-induced hypersensitivity syndrome: a case report. Nat. Med.26(2), 236–243 (2020).
- Flores-Montero J , Sanoja-FloresL , PaivaBet al. Next generation flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia31(10), 2094–2103 (2017).
- Theunissen P , MejstrikovaE , SedekLet al. Standardized flow cytometry for highly sensitive MRD measurements in B-cell acute lymphoblastic leukemia. Blood129(3), 347–357 (2017).
- Ossenkoppele G , SchuurhuisGJ. MRD in AML: does it already guide therapy decision-making?Hematology2016(1), 356–365 (2016).
- Izevbaye I , LiangLY , MatherC , El-HallaniS , MaglantayRJr , SainiL. Clinical validation of a myeloid next-generations sequencing panel for single-nucleotide variants, insertions/deletions and fusion genes. J. Mol. Diagn.22(2), 208–219 (2020).
- Sabour L , SabourM , GhorbianS. Clinical applications of next-generation sequencing in cancer diagnosis. Pathol. Oncol. Res.23(2), 225–234 (2017).
- Kim T , MoonJH , AhnJ-Set al. Next-generation sequencing–based posttransplant monitoring of acute myeloid leukemia identifies patients at high risk of relapse. Blood132(15), 1604–1613 (2018).
- Jongen-Lavrencic M , GrobT , HanekampDet al. Molecular minimal residual disease in acute myeloid leukemia. N. Engl. J. Med.378(13), 1189–1199 (2018).
- Enjeti AK , ChapmanK , TaylorPJ , MeldrumC. Congenital late onset thrombotic thrombocytopenic purpura: a diagnostic challenge. Pathology47(6), 585–586 (2015).
- Salomon R , KaczorowskiD , Valdes-MoraFet al. Droplet-based single cell RNAseq tools: a practical guide. Lab Chip19(10), 1706–1727 (2019).