
Abstract
Aim: Identification of aberrant hypermethylation in promoter regions of candidate genes to discover potential biomarkers for colorectal cancer. Materials & Methods: Genes BMP2, IRF4, KCNA1, LRRC7, NRG3, SLC27A6 and UNC5D were pre-selected in a bioinformatics study for their hypermethylation status in colorectal cancer. Methylation analysis was performed on 202 cancer tissue specimens to validate candidate genes. Results: Genes KCNA1 and UNC5D displayed methylation in 95.3 and 99.7% of The Cancer Genome Atlas dataset samples and in 96 and 98% of our experimentally tested samples, respectively. Conclusion:KCNA1 and UNC5D promoter hypermethylation holds diagnostic biomarker potential in patients with early colorectal cancer.
DNA methylation is a major epigenetic mechanism influencing gene regulation whereby modifications often coincide with disease phenotypes [Citation1]. In mammals, DNA methylation predominantly occurs by DNA methyltrasferases covalently modifying cytosine (C) residues into 5-methylcytosine (5-mC) at cytosine-phosphate-guanine (CpG) dinucleotides in a process called de novo methylation [Citation1,Citation2]. The majority of CpG sequences in mammalian somatic cells is methylated (70–80%), with the exception of CpG islands (CGIs) in gene promoter regions [Citation1]. Normal cells possess mechanisms to protect CGIs from methylation because hypermethylation of these regions often leads to transcriptional silencing [Citation3]. Together with direct inactivation of tumor suppressor genes [Citation3], hypermethylation can indirectly silence other classes of genes, including genes involved in metabolism, genes responsible for tumor cell invasion and DNA repair genes [Citation2,Citation4], resulting in cancerogenesis. Because promoter CGI hypermethylation was shown to occur in early colorectal cancer (CRC) [Citation5,Citation6], this event could serve as a prognostic or diagnostic evaluator.
Even though the urgency of early diagnosis is evident, it is difficult to find an entirely satisfactory method to clinically stage CRC. Conventional methods for CRC screening tend to be either ineffective or too invasive [Citation7]. The so-called tumor, node, metastases (TNM) staging system is commonly used in the clinical setting as a tool for treatment and prognosis. Despite some shortcomings and inconsistencies [Citation8], staging of cancer is a useful guide; however, limitations highlight the demand for new biomarker options. Even though methylation markers show an immense prognostic potential, they still have a limited availability in practice, and the need for sophisticated and effective methylation-based CRC biomarkers as an additional diagnostic or prognostic tool remains unmet. The urgency of early detection of CRC, for which the 5-year survival rate can be as high as 90% for early-stage patients but decreases to 10% for patients with distant metastases [Citation8], is enhanced by the fact that prognosis is closely related to the stage at diagnosis [Citation9,Citation10].
CRC is one of the most common cancers worldwide [Citation10], responsible for 9% of all cancer deaths. The quantity and diversity of molecular changes in an average CRC genome is large [Citation11,Citation12], and CRC encompasses multiple molecular subtypes of genomic instability [Citation13]. Although hypermethylation can be observed in all of these subtypes, an exceptionally extensive frequency of hypermethylation in CGI was observed in the CpG island methylator phenotype (CIMP) subgroup [Citation14,Citation15], which is believed to be radically different from other colon cancers. However, whether CIMP is simply at the far end of a continuous distribution of a biased selection of methylated genes or a group of truly distinctive cancers remains controversial [Citation15]. It is also not yet known whether the CIMP group emerges from a unique underlying cause in the process of cancerogenesis. A more consistent characterization of CIMP with specifically hypermethylated loci is a major challenge in CRC research.
Considering that epigenetic changes outnumber genetic mutations [Citation3], the detection of changes in gene promoter methylome holds great potential as an early CRC indicator, regardless of the biological consequence. Aberrantly methylated promoter sequences associated with CGIs can be used as biomarkers for cancer diagnosis, prognosis and prediction of responsiveness to therapy [Citation13,Citation16], because methylation modifications have been shown to reflect not only the presence but also the stage of the cancerogenic process [Citation9,Citation17]. In this study, DNA methylation profiling was performed to identify aberrantly methylated CpG islands within the promoter regions of candidate genes selected on the basis of a previous study by Hauptman et al. [Citation11]. As a tool for comparison, we used The Cancer Genome Atlas (TCGA) database, which offers genetic and epigenetic data from 381 CRC tissue samples. To identify new aberrantly methylated gene promoter sequences within the candidate genes BMP2, IRF4, KCNA1, LRRC7, NRG3, SLC27A6 and UNC5D, we applied methylation-specific high-resolution melting analysis (MS-HRM) to bisulfite converted DNA. The MS-HRM method discriminates between 5-mC containing and non-5-mC-containing DNA sequences, based on the difference in the melting curves of methylated and unmethylated templates. The analysis was performed on a large cohort of cancer tissue samples (n = 202). Because a multitude of factors can affect cancer initiation, progression and survival rate, survival analysis was performed on the clinical data collected over 5 years. The influence of clinical, pathological and genetic factors was graded separately and in different combinations to discover their influence on overall survival. Better understanding of the DNA methylation landscape in CRC could offer a starting point for novel diagnostic markers for timely and accurate detection of early CRC.
Materials & methods
Study design
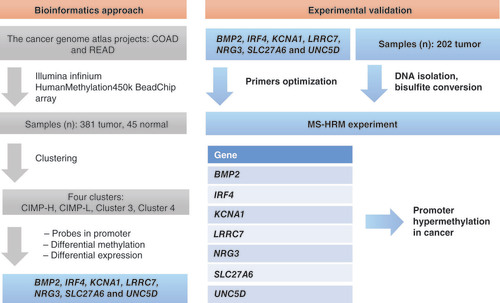
The study comprised three phases: identifying the candidate genes based on existing DNA methylation and gene expression data from Hauptman et al. [Citation11], experimentally validating the CRC tissue specimens and providing a data comparison for optimal CRC biomarker discovery. As presented in , methylation data were collected with Illumina Infinium HumanMethylation450 BeadChip (450k) array, which contained more than 450k methylation probes for each sample from projects colon adenocarcinoma (COAD) and rectum adenocarcinoma (READ), retrieved from TCGA (https://portal.gdc.cancer.gov/). For experimental validation, the seven most prominent genes were chosen to be analyzed on 202 cancer tissue samples. In the third phase, methylation status was associated to clinicopathological characteristics of patients, and Cox proportional hazards model was used to assess the influences on overall survival of CRC patients.
Unsupervised clustering of tumor samples resulted in four clusters. After literature mining and gene ontology, BMP2, IRF4, KCNA1, LRRC7, NRG3, SLC27A6 and UNC5D were chosen for further experimental validation on 202 samples. After DNA isolation, DNA was bisulfite converted and used in methylation-sensitive high-resolution melt experiment (MS-HRM) to test for promoter hypermethylation in cancer samples.
COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; CIMP: CpG island methylator phenotype; CIMP-L: CpG island methylator phenotype–low; CIMP-H: CpG island methylator phenotype–high.
Sample collection
A total of 202 cancer tissue specimens were used for the assessment of DNA methylation in CRC, of which 158 were fresh frozen tissue samples, and 44 tissue samples were stabilized in RNAlater solution (Invitrogen, Thermo Fisher Scientific, MA, USA). The samples () originated from patients diagnosed with CRC who had been treated with surgical resection. Tissue samples for this study were selected from the tissue bank of the Institute of Pathology, Faculty of Medicine, Ljubljana, Slovenia. The National Medical Ethics Committee of the Republic of Slovenia approved this research (0120-463/2019/6). There were 15 paired normal samples used for comparison in methylation data, and these healthy colon mucosa samples were collected at least 20 cm away from the tumor site. Both the average and the median age of patients were 64 years, with an age range between 31 and 89. There were 90 female patients (44.6%) and 112 male patients (55.4%) in our cohort. The occurrence of CRC in either colon or rectum was approximately the same, with 103 cases of colon (51.0%) and 99 cases of rectum (49.0%) cancer; 41 cases were located in the sigmoid colon (20.3%), followed by 39 cases in the ascending colon (19.3%), 17 cases in the transverse colon (8.4%) and 6 cases in the descending colon (3.0%). Tumors were classified as T1 in 27 (13.4%), T2 in 63 (31.2%), T3 in 88 (43.6%) and T4 in 24 (11.9%) patients. Regional lymph node metastases (N) were not present in 103 (51.0%) patients and were present in 98 (48.5%) patients; involvement of regional nodes was undetermined for one patient. Distant metastases (M) were present in 22 (10.9%) patients, absent in 166 (82.2%) patients and undetermined in 14 (6.9%) patients.
Table 1. Clinical features of 202 patients with colorectal cancer, whose samples were used for experimental validation of DNA methylation in gene promoter regions.
DNA extraction & bisulfite modification
DNA from tissue samples was extracted with an AllPrep® DNA/RNA/miRNA Universal Kit (Qiagen, Hilden, Germany), according to the manufacturer’s recommendations. DNA was qualified and quantified spectrophotometrically by NanoDrop ND-1000 (Thermo Fisher Scientific) and subjected to bisulfite conversion. Samples with a total of 5000 ng of DNA were bisulfite converted with an innuCONVERT Bisulfite Basic Kit (Analytik Jena AG, Jena, Germany), according to the manufacturer’s recommendations.
Methylation-specific high-resolution melting
Optimized MS-HRM reactions were performed using the following protocol: 1.00 μl of bisulfite converted DNA, 1.00 μl of each primer (forward and reverse), 0.50 μl of dNTP, 1.00 μl of HotStarTaq Plus Buffer (10x), 0.05 of μl HotStarTaq Plus Polymerase (5 U/μl) and 0.3 of μl of DNA-intercalating dye Syto®9 (Invitrogen, Thermo Fisher Scientific), diluted with nuclease-free water to obtain a total PCR reaction volume of 10 μl. Amplification of both methylated and unmethylated DNA was performed with candidate gene primers for MS-HRM, which were predesigned in Methyl Primer Express Software v1.0 (Thermo Fisher Scientific). Fully methylated and fully unmethylated commercially available bisulfite converted DNA (EpiTect PCR Control DNA Set, Qiagen) were used in each MS-HRM run to help with differential assessment of the methylation status of the samples. As a nontemplate control, 1.00 μl of ddH2O was used. An optimized cycling protocol for MS-HRM analysis on a Rotor-Gene Q (Qiagen) was performed, including initial denaturation at 95°C for 5 min; 45 cycles at 94°C for 15 s, annealing temperature () for 30 s and extension at 72°C for 30 s. MS-HRM analysis was performed immediately after PCR at 60–99°C with a 0.1°C ramp rate. Normalization of PCR product melting curves was adjusted using Rotor-Gene Q provided software (Qiagen) by taking into account normalization regions before and after the major drop in fluorescence.
Table 2. Primer sequences used in experiment.
Statistical analysis
The analysis of the association between methylation of selected genes and clinicopathological features, as well as survival analyses using the Cox proportional hazards model for univariate and multivariate analyses of clinical and methylation data, were done using statistical software IBM SPSS Statistics for Windows, version 24.0 (IBM Corp., NY, USA). For the purpose of association study between promoter CpG sites hypermethylation and standard clinicopathological features, binominal logistic regression approach was used. Multivariate analysis using the Cox regression model was performed using stepwise backward selection by starting with all explanatory variables and removing the least useful (the largest p-value) predictors gradually, until all variables have been added for the optimal model.
Results
Selection of candidate genes
DNA methylation analysis was performed to identify the methylation status of CpG sites within promoter regions of candidate genes. Genes were chosen on the basis of previous bioinformatics analysis based on DNA methylation and gene expression data, which was performed on samples from projects on COAD and READ. Briefly, methylation data were used in unsupervised clustering, which resulted in four clusters. The probes were subsequently subjected to the following conditions:
Probes had to be located in promoter regions of genes according to Ensembl regulatory build;
Probes had to be differentially methylated in each of all four clusters compared with normal samples;
Genes to which these probes were mapped had to be differentially expressed in each of the four clusters compared to normal samples.
Overlap of these conditions resulted in a list of genes with differentially methylated probes in promoter regions. On the basis of the preceding conditions, seven genes were chosen for experimental validation: BMP2, IRF4, KCNA1, LRRC7, NRG3, SLC27A6 and UNC5D.
Methylation analysis by high-resolution melting
On the basis of the preliminary bioinformatics analysis results, DNA methylation of specific promoter regions of the seven selected genes was validated on 202 CRC patient samples. Each gene was run separately with optimal parameters for each individual set of primers. In each MS-HRM run, two controls of 0% and 100% methylated bisulfite converted DNA were used to assess the methylation level of each sample.
We used Rotor-Gene Q dedicated software to obtain graphs of normalized melting curve peaks and melting curves to assess the methylation status of all samples. The results of the MS-HRM analysis are presented in . For comparison, we also added the methylation data of CRC samples from TCGA dataset. The results revealed that the gene with the most potential in both datasets was UNC5D (probe cg13561879), which was aberrantly methylated in 98.0% of our samples and in 99.7% of samples from TCGA. The degree of hypermethylation frequency for KCNA1 marker (probe cg03929977) was also notably high, being methylated in 96.0% of the samples in our set and 95.3% in TCGA dataset. Taking this into consideration, the high rate of methylation in the promoter regions of genes KCNA1 and UNC5D reveals their potential for a role in early diagnostics of CRC. Genes IRF4, LRRC7, NRG3 and SLC27A6 had more than 90% of samples methylated in TCGA dataset, whereas in our set, between 80 and 90% of samples were methylated. The least favorable methylation marker was the BMP2 gene, with 67.2% of samples methylated in TCGA dataset and 68.8% of samples methylated in our data set. The paired healthy mucosa samples showed unmethylated status when tested for hypermethylation in CpG sites of KCNA1 and UNC5D gene promoters compared with cancer samples as expected.
Table 3. The number of methylated and unmethylated probes for the TCGA samples (n = 381) and the study sample (n = 202).
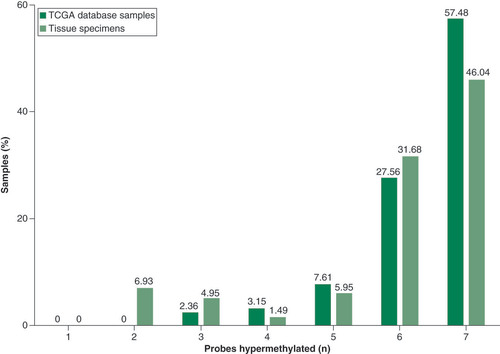
depicts a quantitative comparison of hypermethylated probes between TCGA database and the experimental results of methylation analysis. The majority of samples from both TCGA (n = 381) and experimental validation results (n = 202) showed hypermethylation in either six or seven of the probes tested. The remaining samples show hypermethylation in five, four, three or two probes; no samples had fewer than two probes aberrantly methylated.
The values are represented in a column plot with the number of probes hypermethylated on the x-axis (minimum zero, maximum seven probes) and the percentage of samples for each value on the y-axis. The representation shows the majority of samples having either six or seven methylated probes, from both TCGA database and our dataset.
TCGA: The Cancer Genome Atlas.
Associations between gene promoter methylation & clinicopathological features
Hypermethylation in the BMP2 gene promoter was associated with tumor location in colon (p = 0.006), whereas the unmethylated promoters of genes BMP2, IRF4 and LRRC7 were associated with cancer progression (p < 0.05). Methylation in the promoter of KCNA1 gene was associated with patients age greater than 60 (p = 0.039). Methylation of the LRRC7 gene promoter was associated with lack of metastases (p = 0.046), early disease stage (p = 0.007), tumor size T1 or T2 (p = 0.006) and the absence of chemotherapy (0.008). The associations for each gene promoter methylation status with clinicopathological characteristics are presented in .
Table 4. The association of methylation status of selected gene promoters to clinicopathological features.
Survival analysis
Univariate Cox regression analysis of the pathological and molecular data revealed that age greater than 60 years, location, lymph node infiltration, presence of metastases, tumor stage, tumor size and cancer progression affected patients’ survival (). Multivariate Cox regression analysis of the same initial pathological and molecular data showed that age greater than 60, location, presence of metastases, tumor stage, radiotherapy, cancer progression and status of methylation in the promoter region of IRF4 were factors independently affecting patients’ survival ().
Table 5. Cox regression analysis for overall survival.
Discussion
The traditional view of the genetic code being the only mode of cellular memory and the sole source of pathological phenotypes has been challenged by recently acquired knowledge on epigenetic landscape of various diseases, including CRC. CRC is a heterogeneous disease with distinct genetic and epigenetic alterations [Citation18]. Early detection of CRC lesions is currently the most effective approach for successful reduction of morbidity and mortality [Citation19]. However, patients with early CRC often lack symptoms, resulting in a delayed diagnosis and subsequently increased morbidity and mortality [Citation9,Citation10]. Identifying CRC-specific methylation markers offers a tool for unravelling the methylation pattern change in the cancerogenic process and could help tackle the issue of CRC heterogeneity recognition. Because DNA methylation is cell-type-specific, of binary character and stable through sample handling [Citation20], DNA methylation biomarkers have several advantages over genetic biomarkers for the use in in vitro diagnostics.
Although general genome-wide hypomethylation has been described as a clear sign of an altered homeostasis in tumorigenesis [Citation21], the present study focused more on specific hypermethylation of CpG nucleotides in promoter regions, which occurs through direct mutagenesis of 5 mC-containing sequences by deamination [Citation21]. CpG promoter hypermethylation has been described to occur frequently during carcinogenesis [Citation22] and indicates altered transcriptional regulation [Citation23]. However, with the biological function of methylation having no relevance, defining the methylation pattern change in cancer is highly beneficial in a diagnostic setting since it provides a targeted approach for the increasing demand of molecular cancer profiling options [Citation24].
Here, we identified and assessed new genes with aberrantly methylated probes in gene promoter regions for the purpose of novel biomarker discovery. Using TCGA database, analysis led to the identification of specific promoter CpG sites, located within BMP2, IRF4, KCNA1, LRRC7, NRG3, SLC27A6 and UNC5D. The BMP2 gene was identified to be in close proximity to a low-penetrance signal associated with CRC risk. The bone morphogenetic signaling pathway, regulated by BMP2, is reported to play a role in CRC development [Citation25]. A recent study found that methylation of BMP2 is a poor prognostic factor for stage III CRC [Citation26]. Silencing of gene IRF4 by methylation has been observed in the tissue of gastric cancer patients. The study also showed high methylation levels in IRF4 in noncancerous gastric mucosa from gastric cancer patients, especially in those with multiple cancers [Citation27]. LRRC7 is one of the driver genes in cancer [Citation28]. A recent study illustrated how the NRG3 product, when bound to a specific transcription factor in the extracellular space, can activate oncogenic pathways and promote the formation of colon tumors [Citation29]. The SLC27A6 gene encodes the long-chain fatty acid transport protein 6 and has not previously been described as showing promoter hypermethylation, although we were able to validate it in a high number samples. The UNC5D gene has been previously described as a possible biomarker target [Citation30]. Protein UNC5B is a transmembrane receptor, which induces apoptosis in the absence of ligand; cancer cells with reduced expression are thus unable to induce apoptosis, which is a selective advantage for tumor progression [Citation30]. Downregulation of gene expression has been shown in 27% of colorectal tumors [Citation31]. Additionally, increased mRNA levels were detected after demethylation treatment, suggesting that promoter hypermethylation was the reason for downregulation of UNC5D [Citation30]. This result is in line with our study, in which 99.7% of TCGA dataset and 98.0% of experimentally validated samples showed hypermethylation in the promoter regions. This supports the common conclusion that the UNC5D gene shows prominent biomarker qualities. The KCNA1 gene is a component of type A potassium channels, and its silencing has been related to the suppression of cervical cancer [Citation32]; however, its regulation was shown to be connected to mitochondrial dysfunction and did not show any relation to methylation levels. To the best of our knowledge, the present study is the first to associate the KCNA1 gene with high-frequency promoter hypermethylation in a large cohort of CRC samples.
DNA methylome profiling was performed to quantify the CGI methylation status within selected genes, using probes associated with the gene promoter regions. The vast majority of samples from both TCGA database and our experimental dataset showed hypermethylation in either six or seven probes tested, indicating high correlation between the two cohorts, and could provide the basis for the concept of a targeted multi-biomarker diagnostic approach. Additionally, two of the chosen probes, g03929977 and cg13561879 of the KCNA1 and UNC5D genes, respectively, could be confirmed as prominent hypermethylation markers because they displayed methylation in 95.3% and 99.7% of TCGA dataset samples and in 96.0% and 98.0% of our experimentally tested samples, respectively. Methylation in CpG promoter regions was experimentally validated on a large cohort of 202 CRC samples. Further quantification of CGI hypermethylation on an even larger number of samples could enable not only an enhanced understanding of cancer evolution but could also be applied in a clinical setting for early CRC detection.
Although research presented here offers a preliminary glimpse of possible roles of KCNA1 and UNC5D genes as diagnostic tools, the validity and applicability will need to be a subject of further work with the emphasis on increasing the patient samples numbers, using additional detection methods and ultimately performing a population study. Furthermore, although Cox regression survival analysis did indicate how multiple clinicopathological features can affect patients’ survival rates differently, KCNA1 and UNC5D should not be regarded as significant parameters for overall survival despite their potential diagnostic value. Even though the evident need for further validation limits the applicable scope of the study, undeniable potential for discovering novel biomarkers is in sight.
Conclusion
The present study identified novel candidates for CRC markers using aberrant promoter region hypermethylation detection. Seven genes with promoter hypermethylation were chosen based on bioinformatics data and validated using MS-HRM on a large cohort of CRC patient samples. Two genes out of this panel, KCNA1 and UNC5D, showed a very high frequency of methylation in both TCGA dataset and our experimental cohort of CRC samples. The bioinformatics approach for selecting prominent biomarker genes correlated well with experimental results, suggesting that an interconnected approach of combining preliminary bioinformatics methods with experimental validation could offer a powerful tool for the discovery of prominent biomarker genes. Furthermore, this result could indicate a future avenue of including the probes examined here in a targeted multi-biomarker approach for the crucial early detection of CRC.
Future perspective
As methylated DNA presented to be a stable biomarker that can be used in a non-invasive matter and is amenable to reliable analysis, it could serve in developing patient stratification before drug treatment and while monitoring patients’ response. To further define the suitability of KCNA1 and UNC5D as prominent molecular biomarkers for CRC, additional methods should be applied for comparison on an ever larger cohort of samples.
Furthermore, as described by Grady in 2007 [Citation15], the molecular features of CIMP tumors are at present characterized in a loose and inconsistent manner due to the complexity of CRC sorting. Although defining aberrant CGI is of particular interest when unravelling the initiation or CRC, the mechanism(s) are not yet clearly defined. Regardless of the subtyping challenges due to heterogeneity of colorectal tumors, determining CIMP status would provide useful insights into the role of aberrant hypermethylation in CRC.
Development of noninvasive, specific and selective biomarkers continues to represent an important research avenue, especially for enabling timely intervention in early disease stages. Analysis of aberrant methylation at specific locations in the genome is already entering a clinical setting for cancer and may become a standard diagnostic tool for diverse diseases with a diagnostic, prognostic and predictive aptitude. Nevertheless, one of the major challenges in the discovery of novel epigenetic biomarkers in the future could be the shortage of quantitative and standardized methodology given that high sensitivity and accuracy are of immense importance. Comprehensive analysis of bioinformatics data might offer a bridge for this limitation, along with properly assessed molecular features and well-characterized clinical information, leading to useful insights into disease presence or progression. In this way, routine genetic and DNA panel testing could replace selective biomarker testing, offering an enhancive holistic approach. Consequently, the importance of proper conversion of molecular results into clinically useful information could become more evident than ever before.
Genes BMP2, IRF4, KCNA1, LRRC7, NRG3, SLC27A6 and UNC5D were selected based on a bioinformatics study.
Analyses were performed on 381 colorectal cancer (CRC) samples from The Cancer Genome Atlas (TGCA) and on our own 202 CRC samples.
Methylation results were consistent in both cohorts.
Hypermethylation in either six of seven or seven of seven probes was observed in the majority of both TCGA and experimental samples.
Genes KCNA1 and UNC5D displayed methylation in 95.3% and 99.7% of the TCGA dataset samples and in 96% and 98% of our experimentally tested samples, respectively.
KCNA1 and UNC5D promoter hypermethylation could be used as new diagnostic biomarkers in patients with early colorectal cancer.
Methylation status of IRF4 gene is one of the factors independently affecting patients’ survival.
Our study is the first to associate the KCNA1 gene with a high-frequency promoter hypermethylation in a large cohort of CRC samples.
Ethical conduct of research
The authors state that they have obtained National Medical Ethics Committee Republic of Slovenia approval.
Financial & competing interests disclosure
The study was supported by the Slovenian Research Agency (research core funding no. P3-0054 and project J3-1754). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the paper apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Li E , ZhangY. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol.6(5), a019133 (2014).
- Sharma S , KellyTK , JonesPA. Epigenetics in cancer. Carcinogenesis31(1), 27–36 (2010).
- Shen H , LairdPW. Interplay between the cancer genome and epigenome. Cell153(1), 38–55 (2013).
- Yang M , ParkJY. DNA methylation in promoter region as biomarkers in prostate cancer. Methods Mol. Biol.863, 67–109 (2012).
- Chan AO , BroaddusRR , HoulihanPSet al. CpG island methylation in aberrant crypt foci of the colorectum. Am. J. Pathol.160(5), 1823–1830 (2002).
- Rashid A , ShenL , MorrisJSet al. CpG island methylation in colorectal adenomas. Am. J. Pathol.159(3), 1129–1135 (2001).
- Toth K , SiposF , KalmarAet al. Detection of methylated SEPT9 in plasma is a reliable screening method for both left- and right-sided colon cancers. PLoS One7(9), e46000 (2012).
- O’connell JB , MaggardMA , KoCY. Colon cancer survival rates with the New American Joint Committee on Cancer sixth edition staging. J. Natl. Cancer Inst.96(19), 1420–1425 (2004).
- Ashktorab H , BrimH. DNA methylation and colorectal cancer. Curr. Colorectal Cancer Rep.10(4), 425–430 (2014).
- Wojdacz TK , DobrovicA. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res.35(6), e41 (2007).
- Hauptman N , JevsinekSkok D , SpasovskaE , BostjancicE , GlavacD. Genes CEP55, FOXD3, FOXF2, GNAO1, GRIA4, and KCNA5 as potential diagnostic biomarkers in colorectal cancer. BMC Med. Genom.12(1), 54 (2019).
- Nazemalhosseini Mojarad E , KuppenPJet al. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed. Bench6(3), 120–128 (2013).
- Lao VV , GradyWM. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol.8(12), 686–700 (2011).
- Toyota M , AhujaN , Ohe-ToyotaMet al. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA96(15), 8681–8686 (1999).
- Grady WM . CIMP and colon cancer gets more complicated. Gut56(11), 1498–1500 (2007).
- Kim H , WangX , JinP. Developing DNA methylation-based diagnostic biomarkers. J. Genet. Genomics45(2), 87–97 (2018).
- Leygo C , WilliamsM , JinHCet al. DNA methylation as a noninvasive epigenetic biomarker for the detection of cancer. Dis. Markers2017, 3726595 (2017).
- Wood LD , ParsonsDW , JonesSet al. The genomic landscapes of human breast and colorectal cancers. Science318(5853), 1108–1113 (2007).
- Simon K . Colorectal cancer development and advances in screening. Clin. Interv. Aging.11, 967–976 (2016).
- Bock C , HalbritterF , CarmonaFJet al. BLUEPRINT consortium. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat. Biotechnol.34(7), 726–737 (2016).
- Jin B , LiY , RobertsonKD. DNA methylation: superior or subordinate in the epigenetic hierarchy?Genes Cancer2(6), 607–617 (2011).
- Kalari S , PfeiferGP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv. Genet.70, 277–308 (2010).
- Kok-Sin T , MokhtarNM , AliHassan NZet al. Identification of diagnostic markers in colorectal cancer via integrative epigenomics and genomics data. Oncol. Rep.34(1), 22–32 (2015).
- Campregher PV , HamerschlakN. Novel prognostic gene mutations identified in chronic lymphocytic leukemia and their impact on clinical practice. Clin. Lymphoma Myeloma Leuk.14(4), 271–276 (2014).
- Fernandez-Rozadilla C , PallesC , Carvajal-CarmonaLet al. BMP2/BMP4 colorectal cancer susceptibility loci in northern and southern European populations. Carcinogenesis34(2), 314–318 (2013).
- Miura T , IshiguroM , IshikawaTet al. Methylation of bone morphogenetic protein 2 is associated with poor prognosis in colorectal cancer. Oncol. Lett.19(1), 229–238 (2020).
- Yamashita M , ToyotaM , SuzukiHet al. DNA methylation of interferon regulatory factors in gastric cancer and noncancerous gastric mucosae. Cancer. Sci.101(7), 1708–1716 (2010).
- Palaniappan A , RamarK , RamalingamS. Computational Identification of novel stage-specific biomarkers in colorectal cancer progression. PloS One11(5), e0156665–e0156665 (2016).
- Liu Y , WangS-Q , LongY-Het al. KRASG12 mutant induces the release of the WSTF/NRG3 complex, and contributes to an oncogenic paracrine signaling pathway. Oncotarget7(33), 53153–53164 (2016).
- Okazaki S , IshikawaT , IidaSet al. Clinical significance of UNC5B expression in colorectal cancer. Int. J. Oncol.40(1), 209–216 (2012).
- Thiebault K , MazelinL , PaysLet al. The netrin-1 receptors UNC5H are putative tumor suppressors controlling cell death commitment. Proc. Natl. Acad. Sci. USA100(7), 4173–4178 (2003).
- Liu L , ChenY , ZhangQ , LiC. Silencing of KCNA1 suppresses the cervical cancer development via mitochondria damage. Channels13(1), 321–330 (2019).