
Abstract
Aim: Nonhuman primates are essential for research on many human diseases. The Infinium Human Methylation450/EPIC BeadChips are popular tools for the study of the methylation state across the human genome at affordable cost. Methods: We performed a precise evaluation and re-annotation of the BeadChip probes for the analysis of genome-wide DNA methylation patterns in rhesus macaques and African green monkeys through in silico analyses combined with functional validation by pyrosequencing. Results: Up to 165,847 of the 450K and 261,545 probes of the EPIC BeadChip can be reliably used. The annotation files are provided in a format compatible with a variety of standard bioinformatic pipelines. Conclusion: Our study will facilitate high-throughput DNA methylation analyses in Macaca mulatta and Chlorocebus sabaeus.
Background
DNA methylation is an epigenetic mark associated with gene regulation. It impacts a number of key biological processes including genomic imprinting, X-chromosome inactivation, repression of transposable elements, aging, carcinogenesis and immunity against infectious diseases [Citation1]. In mammalian genomes, DNA methylation occurs mainly as the methylation of a cytosine (5-methylcytosine) immediately followed by a guanine. Methylated cytosines are more susceptible to deamination yielding thymines [Citation2–4]. Due to their increased mutation rate, CpG dinucleotides have been depleted during evolution and are thus under-represented in the genome [Citation5]. However, a higher density of mostly unmethylated CpG dinucleotides is found in CpG islands, generally localized in the first exon and intron or in the promoter region of genes [Citation6,Citation7].
CpG methylation can be measured in humans using the Infinium Human Methylation450 BeadChip Array (Infinium 450K), which measures methylation levels at more than 450,000 CpGs across the human genome [Citation8]. This array has been replaced by the Infinium Human Methylation EPIC BeadChip Array, which adds about 350,000 CpGs localized in enhancers [Citation9]. Due to its accuracy and capacity to analyze large cohorts at an affordable cost, the Infinium microarrays have been used in a wide range of epigenome-wide association studies in humans (see review [Citation10]).
Only few specifically dedicated genome-wide microarray tools are currently available for the analysis of nonhuman primate models. As these species have a close phylogenic proximity with humans and a high percentage of DNA identity and gene homology, the use of human microarrays in studies in nonhuman primate models could be possible. Indeed, human gene expression microarrays have been used in various studies on monkeys, from Hepatitis C Virus and simian immunodeficiency virus (SIV) infection [Citation11–13] to asthma [Citation14] or glaucoma [Citation15]. The human Affymetrix HG-U133 Plus 2.0 GeneChip has also been used for a gene expression study in the phylogenetically more distant lemur Microcebus murinus, which has been shown to be an excellent model for Alzheimer’s disease ([Citation16]; Pichon et al., unpublished data). In the latter study, human microarrays detected about 20% of lemur transcripts, corresponding to the expected rate due to the divergence of both species.
Similarly, the human Infinium 450K has been used for DNA methylation studies in great apes [Citation17]. Seventy-three percent of the probes designed to the human genome mapped to the bonobo genome, 72–77% to chimpanzee, 61% to gorilla and 44% to orangutan genomes [Citation17,Citation18]. Studies were also performed in monkeys of the cynomolgus macaque species (Macaca fascicularis) [Citation19], rhesus macaque (Macaca mulata, MM) [Citation18] and baboon [Citation20]. For example, using different selection criteria for probes yielding reliable signals, 61% of human probes were mapped and annotated to the Macaca fascicularis genome and subsequently used to study the impact of birth weight on gene methylation and expression in Macaca fascicularis [Citation21]. Microarrays initially designed for the interrogation of the human genome have thus been used to study gene expression or DNA methylation of nonhuman primate samples. This can be successfully done under the condition that a thorough evaluation of reliable CpG-targeting probes is performed for the species of interest.
Several monkey species are widely used models to study complex human diseases contributing to unraveling the mechanisms or evaluating treatment options of human and animal diseases. Rhesus macaques are for instance frequently used for the development of vaccines against infectious diseases, including Sars-CoV-2, HIV, influenza and ebola virus [Citation22–26]. They are also widely used in various studies ranging from development and imprinting, to addiction and social cognition [Citation27–32]. In parallel, the Chlorocebus genus, among which figure prominently African green monkeys (AGMs), have been a gold standard model for investigation of several infectious diseases, such as yellow fever, trypanosoma and plague in the past [Citation33,Citation34]. AGMs, in particular Chlorocebus sabaeus (CS), are included in studies of neurological disorders, in pharmacological trials [Citation35] and more recently also for the identification of mechanisms of protection against HIV/AIDS, MERS-CoV and SARS-CoV2 [Citation26,Citation36–40].
MM and CS have therefore, together with baboons and cynomolgus macaques, become reference animal models for preclinical research. Nonetheless, no commercial off-the-shelf DNA methylation microarrays are available for these species. In the present study, we evaluated the use of the Infinium 450K and Infinium EPIC BeadChips for genome-wide DNA methylation analyses in samples from CS and MM and conducted an in-depth analysis of the reliable probes for these two old world monkey genomes. Results show that about one third of the Infinium 450K or EPIC human-designed probes can be reliably used to study DNA methylation in these Cercopithecidae and that the majority map to gene features. We provide for each species and each microarray a list of annotated probes using the latest genome and annotation builds that can be used by the scientific community for genome-wide DNA methylation studies in CS or MM. These detailed data on probe behavior were obtained with stringent criteria. They provide the flexibility to the research community to focus their analysis only to those probes identified as reliable.
Methods
Study approval
Animals were housed at the IDMIT center of the Commissariat à l’Energie Atomique (CEA, Fontenay-aux-Roses, France, permit number: A 92–032-02). The CEA complies with Standards for Human Care and Use of Laboratory of the Office for Laboratory Animal Welfare (OLAW, USA) under OLAW Assurance number #A5826-01. The Central Committee for Animals at Institut Pasteur or Ethical Committee of Animal Experimentation (CETEA-DSV, IDF, France) approved all animal experimental procedures (Notification numbers: 10-051b, 12-006b). The studies were conducted in strict accordance with the international European guidelines 2010/63/UE on protection of animals used for experimentation and other scientific purposes (French decree 2013–118).
Samples
Blood was collected from 13 AGM (Chlorocebus sabaeus) and 17 rhesus macaques (Macaca mulatta) by venipuncture on EDTA tube, which were part of a study on DNA methylation changes upon SIV infection [Citation68]. Several blood samples taken at different timepoints during the disease course were available for some of the animals and in total 21 CS and 25 MM samples were included in the study. However, as samples were taken at different timepoints of the infection, and since the infection modified the DNA methylation pattern, we treated the samples from distinct time points as independent samples and avoided to use them to test for reproducibility. CD4+ peripheral blood mononuclear cells were purified, as described previously using magnetic anti-CD4 beads (Miltenyi Biotec, Bergisch Gladbach, Germany) [Citation13]. CD4+ T cell purity after isolation was confirmed using flow cytometry (median 97%, IQR 93%–98%). DNA was extracted from CD4+ T cells using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany), according to manufacturer’s protocol.
Infinium 450K analysis
One μg of DNA was bisulfite-treated using the EpiTect® 96 Bisulfite Kit (Qiagen) and analysed using the Infinium Human Methylation 450K BeadChips (Illumina, CA, USA) according to the manufacturer’s protocol.
Mapping of 50 bp probe sequences from Infinium 450K & EPIC
Reference genomes used in this work were downloaded from Ensembl: MMUL10.0 for MM and ChlSab1.1 for CS. Infinium 450K microarrays provide DNA methylation measures for 482,421 CpG sites across the human genome (135,476 Infinium I and 346,945 Infinium II probes). In the same way, Infinium EPIC (Version 2020 B5) allows measuring of 862,927 CpGs (142,137 Infinium I and 720,790 Infinium II probes).
Because of the two different chemistries and the different targets of the single base extension, the unconverted 50 bp probe design sequences targeting CpG positions in the human genome from the Infinium 450K and EPIC arrays provided in the BeadChip manifests were used. Thus, all sequences ended or started with a CpG dinucleotide. We did not use directly the sequences of the Infinium I of Infinium II probes, but rather the sequences that were used to create these probes. Sequences were mapped to the CS and MM genomes, using Bowtie [Citation41], allowing only a unique position on the respective genome and up to three mismatches. Sequences were thus classified as ‘perfect match,’ ‘1 mismatch,’ ‘2 mismatches,’ ‘3 mismatches’ or ‘unmapped/nontargeting’ depending on the number of mismatches attributed by Bowtie. The number and position of mismatches are provided in the manifest files in the Supplementary Material and the numbering refers to the original design sequence. Nontargeting probes included the probes with either more than three mismatches or probes that did not map to a unique location. Because of the necessity to only keep probe sequences that can reliably hybridize on simian genomes and inform on methylation state of the CpG sites, we removed probes containing mismatches at the CpG site, which were annotated as ‘CS-nontargeting’ or ‘MM-nontargeting’. Sequences with intact CpG sites were qualified as ‘CS-functional’ or ‘MM-functional.’ Among the CS-functional or MM-functional probes, we also determined the exact position of the closest mismatch, if any, relative to the CpG. If the mismatch was three or more bases away from the CpG, probes were retained in the final selection of valid probes as detailed in the results section.
Annotation of Infinium 450K & EPIC probes in MM & CS
To annotate probes, the annotations files for MM and CS were retrieved from Ensembl (Version 101), containing 64,228 and 28,078 transcripts for 35,432 and 27,982 genes (coding, noncoding and pseudogenes), respectively. In accordance with the human annotation file provided by Illumina for the arrays we divided transcripts into six different categories: promoter region ranging from 1 to 200 bp upstream of the TSS (TSS200), promoter region ranging from 201 to 1500 bp upstream of the TSS (TSS1500), 5′UTR, first exon (1stExon), 3′UTR and gene bodies, excluding the 5′ and 3′ UTRs and first exons (Body). Similarly, CpG islands prediction files were downloaded from the University of California Santa Cruz (UCSC) genome browser (containing respectively 28,580 and 26,663 CpG islands for MM and CS) for the annotation relative to CpG islands following again the annotation criteria used by Illumina for the human genome. UCSC islands predictions are based on the following parameters: CpG obs/exp ratio >0.6, CG content >50%, length >200 bp. Shores are defined as island-flanking regions ranging to up to 2000 bp and shelves are defined as island-flanking regions ranging from 2,001 bp to 4,000 bp. Northern and southern shores and shelves (noted N_ and S_) are respectively defined as the upstream and downstream shores or shelves according to chromosomal coordinates.
Preprocessing & correction for Infinium I/II shift of 450K data for monkeys
To normalize signals from the probes in the two monkey species, a refined version of the subset quantile normalization (SQN) pipeline [Citation42], which performs the SQN at the level of each individual sample prior to a between-sample quantile normalization, was used to correct for the difference in the performance and dynamic range of Infinium I and Infinium II probes. The original Illumina annotation file used in the pipeline were replaced by the ones created for MM and CS. Due to the stringent criteria in the probe selection process, no further filtering for example, nonspecific probes, was required.
Pyrosequencing analysis
Quantitative DNA methylation analysis for validation was performed by pyrosequencing of bisulfite-treated DNA [Citation43]. Six regions of interest for validation were amplified using 30 ng of bisulfite-treated human genomic DNA and 5 to 7.5 pmol of forward and reverse primer, one of them being biotinylated. Sequences for oligonucleotides for PCR amplification and pyrosequencing are shown in Supplementary Table 1. Reaction conditions were either 1X HotStar Taq buffer supplemented with 1.6 mM MgCl2, 100 μM dNTPs, 5 pM of each primer and 2.0 U HotStar Taq polymerase (Qiagen, Courtaboeuf, France) in a 25 μl volume or 1X Phusion U Hotstart (Thermo Fisher Scientific, Les Ulis, France) with 5 pM of each primer. The PCR program consisted of a denaturing step of 15 min at 95°C followed by 50 cycles of 30 s at 95°C, 30 s at the respective annealing temperature and 20 s at 72°C, with a final extension of 5 min at 72°C. 10 μl of PCR product were rendered single-stranded as previously described [Citation43] and 4 pmol of the respective sequencing primer were used for analysis. Quantitative DNA methylation analysis was carried out on a PSQ 96MD system with the PyroGold SQA Reagent Kit (Qiagen) and results were analyzed using the PyroMark CpG software (V.1.0.11.14, Qiagen).
Results
Throughout the manuscript, we use a nomenclature of ‘targeting’ probes, referring to probes that map and potentially target a CpG dinucleotide in the respective simian genomes allowing still for mismatches at any place in the probe including the CpG site, and ‘functional probes,’ which target an intact CpG site in the simian genomes, as well as ‘valid probes,’ which is the final selection of probes based on the selection criteria described below in the results section.
First, we mapped the human probes to the simian genomes. All 50 bp probe sequences designed to target a CpG in the human genome, were extracted from Illumina’s manifest files for each microarray (482,421 sequences for the Infinium 450K and 862,927 for the Infinium EPIC).
Mapping probes of the Infinium 450K & EPIC BeadChip on the CS genome
From the 482,421 Infinium 450K CpG probes designed for the human genome, 231,217 (47.9%) mapped to CS genome according to our parameters, of which 36,439 (15.8%) were perfect matches, 57,785 (25.0%) had one mismatch, 69,312 (30.0%) had two and 67,681 (29.3%) had three mismatches ( & ). Among the 251,204 nontargeting probes, 4539 (1.8%) were removed due to nonunique mapping. We investigated in detail the distribution of all the substitutions at the mismatches from the entire mapped probe set (Supplementary Figure 1A & Supplementary Table 2). A nucleotide located in a CpG site included in the probe sequence had a higher chance (around 5% in the middle of the probe sequence and 8–12% at their extremities) to be substituted than a base located outside the targeted CpG site (about 3%). Moreover, about 70% of these substitutions were T (CS)→C (human) or, respectively A→G nucleotide substitutions in CpG sites, thus replacing the cytosine or the guanine of the CpG site, respectively. These substitutions with a weak to strong (W→S) nucleotide substitutions were less frequent along the entire 50 bp probe sequence, where they represented around 40%. Frequencies of substitutions were higher at the extremities of the mappable, in other words, targeting probes (Supplementary Figure 1A & Supplementary Table 2), where 25.8% (61,413 probes) presented a mismatch at a CpG dinucleotide and 85% of them were weak to strong (W→S) nucleotide substitutions between CS and human (Supplementary Table 3). Among those ∼71% were T→C or, respectively, A→G nucleotide substitutions of the cytosine or the guanine of the CpG site, respectively (Supplementary Table 3).
Proportion of probes of the Infinium 450K and the EPIC BeadChip mapping perfectly (targeting, functional perfect matched probes), with mismatches (targeting, functional mismatch probes), with a mutation at the CpG nucleotide (targeting, nonfunctional mismatch probes) or with more than three mismatches (nontargeting) to the CS and MM genome.
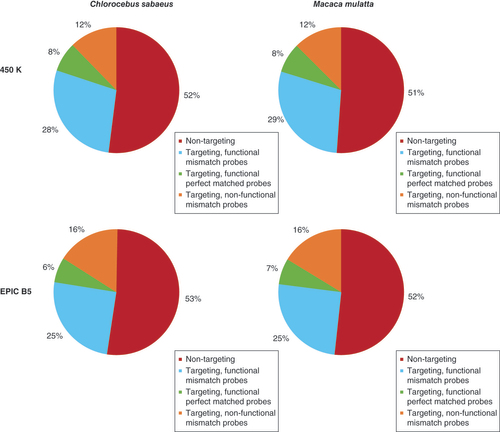
Table 1. Mapping of the 50 bp probe design sequences from the Infinium 450K and EPIC microarrays on the two simian genomes.
We then performed a more careful study of mismatches at the targeted CpG position, which showed that 59,305 (25.6%) of the targeting probe sequences had at least one mismatch at the CpG site targeted by the probe and were thus identified as CS-nonfunctional probes. Out of the 231,217 targeting probes, 171,912 (74.4%) probe sequences were targeting a CpG dinucleotide in the CS genome and were thus identified as CS-functional (). From these CS-functional probes, 21.2% had no mismatch at all, 51.8% had the mismatch at more than 10 bp from the CpG and 46,457 (27%) presented at least one mismatch at 10 bp or less from the CpG (Supplementary Table 4). The latter were further analyzed in more detail as described below.
Results from the Infinium EPIC BeadChip were very similar to those from Infinium 450K, with 408,578 (47.4%) probes designed for the human genome mapping to the Chlorocebus sabaeus genome. Of these, 55,433 (13.6%) were perfect matches, 97,588 (23.9%) had one mismatch, 126,777 (31.0%) had two mismatches and 128,780 (31.5%) had three mismatches. Among the 454,349 nontargeting probes, 6807 (1.5%) were removed due to nonunique mapping. From these targeting probes, 270,694 (66.3%, vs 74.4% in Infinium 450K) were identified as CS-functional and 137,884 were identified as CS-nonfunctional (). From these CS-functional probes, 73,793 presented at least one mismatch at 10 bp or less from the CpG (Supplementary Table 4) and were further investigated as described below.
As for Infinium 450K, we analyzed the distribution of all the substitutions from the whole mapped probe set (Supplementary Table 5). From this distribution, it appeared that a base located in a CpG site had a higher chance to be substituted on the EPIC BeadChip (around 17%) than on Infinium 450K (around 13%, hypergeometric test p < 0.001). In CpG sites, W→S nucleotide substitutions represented around 65%, while this type of substitutions represented around 20% along the entire 50 bp probe sequences.
Altogether, for their use in DNA methylation analysis using the two human BeadChips, 31.4% (EPIC) and 35.6% (450K) of the probes could be classified as functional for CS.
Mapping probes of the Infinium 450K & EPIC BeadChip on the MM genome
Similarly to CS, we found that of the 482,421 Infinium 450K CpG probes designed for the human genome, 235,585 (48.8%) mapped to the MM genome. Of these, 37,547 (15.9%) were perfect matches, 59,669 (25.3%) had one mismatch, 70,625 (30.0%) had two and 67,744 (28.8%) had three mismatches. Among the 246,836 nontargeting probes, 5835 (2.4%) were removed due to nonunique mapping. Investigating the position of the respective mismatches showed that 59,904 (25.4%) probe sequences designed to the human genome had at least one mismatch at the CpG site and were thus identified as MM-nonfunctional. Among substitutions at the CpG sites ∼71% were again W→S substitutions (Supplementary Figure 1B & Supplementary Table 3). 175,681 (74.6%) probe sequences designed to the human genome were identified as MM-functional (). From these MM-functional probes, 47,230 presented at least one mismatch at 10 bp or less from the CpG (Supplementary Table 4) requiring further evaluation as described below.
From the 862,927 Infinium EPIC CpG probes designed for the human genome, 416,382 (48.3%) mapped to the MM genome according to our parameters, from which 57,426 (13.8%) were perfect matches, 101,042 (24.3%) had one mismatch, 128,651 (30.9%) had two mismatches and 129,263 (31.0%) had three mismatches. Among the 446,545 nontargeting probes, 8827 (2.0%) were removed due to nonunique mapping. Of the targeting probes, 277,020 probes (66.5%, vs 74.6% in Infinium 450K) were identified as MM-functional and 139,362 were identified as MM-nonfunctional (). Of these MM-functional probes, 74,928 presented at least one mismatch at 10 bp or less from the CpG (Supplementary Table 4).
As above, we studied more carefully the distribution of all the substitutions from the whole mapped probes set (Supplementary Tables 6 & 7) and found a very similar distribution in MM as in CS.
Altogether, for their use in DNA methylation analysis using the two human BeadChips, 32.1% (EPIC) and 36.4% (450K) of the probes could be classified as functional for MM.
The results were similar for the two species and when allowing up to three mismatches for the 50 base pair probes potentially about a third of the probes on the respective arrays were targeting CpG positions in the two simian genomes. There was a strong overlap between the two simian species with 145,639 (about 63%) and 224,882 (about 55%) mapped and functional probes in common between the two species for the Infinium 450K and Infinium EPIC microarrays, respectively.
This probe set contained mismatches within some of the probes (outside the CpG position), which could influence probe behavior, a point which has been neglected in most studies so far. Therefore, final validation and refinement of the reliable probe sets required experimental analysis of samples from the respective species.
Validation & refinement of our selection criteria for the Infinium 450K BeadChip
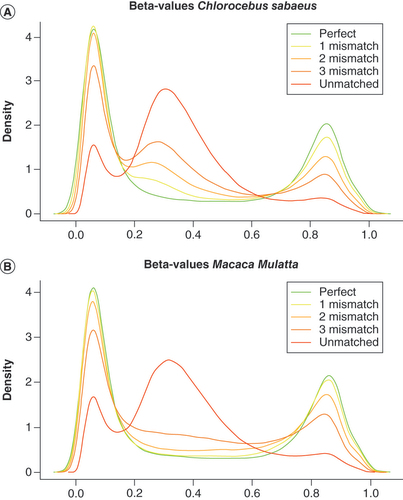
To determine to which extent CpG probes designed for the human genome and mapping to simian genomes could efficiently be used to detect methylation in these two simian models, we analyzed 25 CD4+ T cell samples from MM and 21 from CS on the Infinium 450K array (). The density distribution of beta-values of the probes identified as perfectly matching were very similar to the bimodal beta-value density distribution commonly observed for human samples [Citation42]. Beta-value density distribution of probes containing a single mismatch remained close to beta-value density distribution of perfectly matched probes, and this density distribution was still similar for probes with two mismatches. However, probes containing three mismatches showed a density distribution of beta-values started to deviate from the expected bimodal distribution, supporting the restriction to a maximum of three mismatches. These results were similar for both species.
Mapped probes for Cholocebus sabaeus(A) and Macaca muletta (B) are classified into categories representing probes with a perfect match, 1, 2 or 3 mismatches. For probes with three mismatches (dark orange) the beta-value distribution start to deviate from the bimodal distribution observed for probes with fewer or no mismatches. Unmapped probes (in red) have an aberrant beta-value distribution. For reasons of clarity only the average density curve derived from 21 Chlorocebus sabaeus and 25 Macaca mulatta samples is shown.
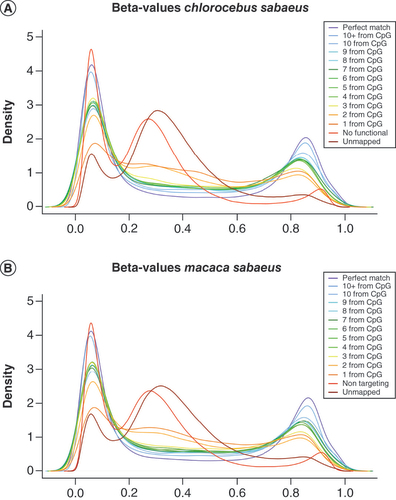
When analyzing the beta-value density distribution of probes with regard to the position of the mismatch, we observed that, in both species, the beta-value distribution was closely related to the mismatch position (). Thus, probes with a mismatch localized at 1 or 2 bp from a CpG site presented an aberrant density distribution of beta-value among valid probes whereas probes containing a mismatch at 3–4 bp or more away from the CpG were similar to probes without mismatches independent of the number of mismatches present (one to three). We thus chose to remove probes presenting a mismatch at 1 or 2 bp from the CpG from the list of functional Infinium probes on simian models for both 450K and EPIC arrays. With this last filter, 162,053 CS-valid and 165,847 MM-valid probes remained for Infinium 450K (i.e., 33% of the Infinium 450K CpG probes), among which 136,702 (∼83%) were common to both species. For the Infinium EPIC array, 255,227 CS-valid and 261,545 MM-valid probes remained after filtering out probes presenting a mismatch at 1 or 2 bp from the CpG (i.e. 29% of the Infinium EPIC CpG probes), 211,065 (∼82%) of them were common to both species.
Mapped probes for Chlorocebus sabaeus(A) and Macaca mulatta(B) are classified into categories representing probes with a perfect match and according to the mismatch position localized from 1 bp to 10 bp or more from the CpG dinucleotide. Probes containing a mismatch localized at 1 or 2 bp (dark orange and orange) do no longer show the expected beta-value density distribution compared with the other mapped probes (yellow, greens, blues and purple). Nontargeting probes (red) and unmapped probes (dark red) have an aberrant beta-value distribution. For reasons of clarity only the average density curve derived from 21 Chlorocebus sabaeus and 25 Macaca mulatta samples is shown.
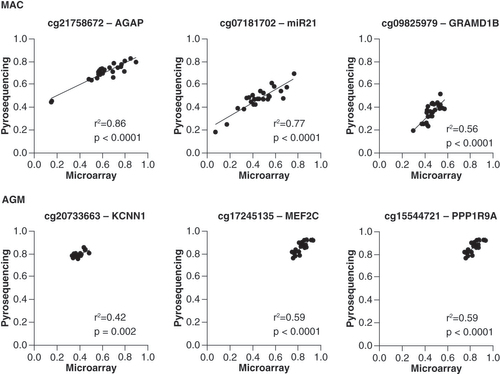
Furthermore, using pyrosequencing, which as a sequencing-by-synthesis method is not dependent on human probes but uses species-specific amplification and sequencing primers, we validated DNA methylation levels measured by the respective Infinium probes at three CpG positions in each species showing a high correlation between the two orthogonal technologies and validating our approach of selecting reliable probes (). The selected probes had either no, one or two mismatches (cg07181702, cg20733663, cg17245135 (n = 0); cg21758672, cg15544721 (n = 1); cg09825979 (n = 2)). The Infinium data correlated well with the pyrosequencing data and this independently of the presence, number or the position of the mismatch.
Top row: Macaca mulatta, bottom row: Chlorocebus sabaeus.
Annotation of valid probes
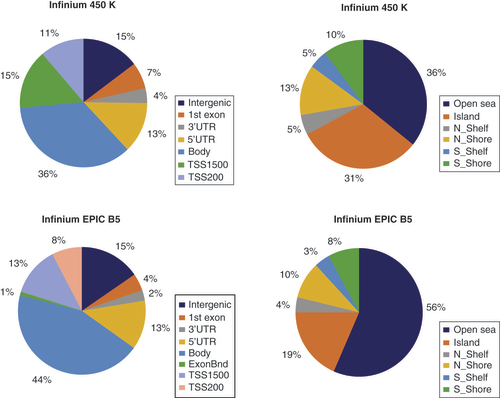
The annotation of the probes to the Homo sapiens genome (GRCh37) as described in the manifest files for both microarrays showed that the additional content of the Infinium EPIC BeadChip compared with the Infinium 450K array was mainly located in gene bodies, and in the open sea using the gene feature and CpG island feature annotation, respectively (). To provide the user with similar information as contained in the Illumina manifest for the human genome, we annotated the location of the CpGs with matching probes according to the Ensembl gene annotation (version 101).
All CpG probes are valid for human (482,421 probes in Infinium 450K and 862,927 in Infinium EPIC), which represent 802,912 and 1,605,008 gene features annotations in total for Infinium 450K and Infinium EPIC, respectively. Note that one probe can be attributed to different transcripts and thus gene features, whereas one probe is attributed to only one island feature.
Chlorocebus sabaeus
Among the 162,053 CS-valid probes on the Infinium 450K BeadChip, 102,674 were annotated for a gene feature (63%, representing 16,259 genes) and 107,906 for a CpG island feature (66%) using the CS genome. 74,858 CS-valid probes were annotated for both a gene and an island feature. CS-valid probe sequences principally targeted CpGs located around transcription start sites and gene bodies (about 65% in total) and around 35% of the CS-valid probes target CpGs in intergenic regions (vs 14% in human). For the probes annotated to a gene in the CS genome, the fraction of probes located in the TSS500, the 5′UTR or the first exon decreased, while more probes were located in gene bodies and upstream regions (TSS1500). According to UCSC islands prediction (see Materials and Methods section), 38% of human-designed CS-valid probes target CpG probes located in CpG islands, which is similar to the proportions and distribution for the human genome ().
We defined 162,053 Chlorocebus sabaeus-valid probes for Infinium 450K and 255,227 for Infinium EPIC, representing 171,608 and 267,827 gene feature annotations in total for Infinium 450K and Infinium EPIC, respectively. Note that one probe can be attributed to different transcripts and thus gene features, whereas one probe is attributed to only one island feature. The stacked barplots show the gene feature and CpG island annotation in comparison to the same probes annotated to the human arrays, as given in the BeadChip manifest files.
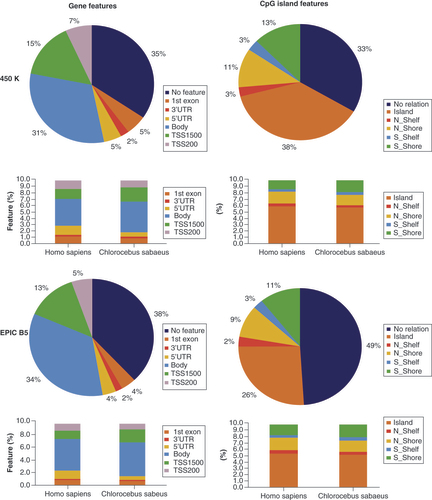
Among the 255,227 CS-valid probes on the Infinium EPIC BeadChip, 153,430 were annotated for a gene feature (60%, representing 17,701 genes) and 129,928 (51%) for a CpG island feature. 90,093 probes were annotated for both a gene and an island feature. The proportion of the CpG islands categories followed the trend observed for the human genome with an increased proportion in intergenic regions and gene bodies on the EPIC arrays. At the same time, as for the 450K BeadChip, but even more pronounced, probes were depleted for the 5′UTR category and slightly increased for upstream regions ().
Macaca mulatta
Among the 165,847 MM-valid probes on the Infinium 450K array, 132,952 were annotated to a gene feature (88%, representing 18,667 genes) and 111,148 (72%) to a CpG island feature (). A total of 96,944 MM-valid probes were annotated to both a gene and an island feature. As for CS-valid probes, but much more pronounced, MM-valid probe sequences principally targeted CpGs located in TSS regions and gene bodies (about 91% in total, 9% in intergenic regions). For the probes annotated to a gene in the MM genome, the fraction of probes located in the TSS500, the first exon and particularly the 5′UTR decreased, while many more probes were located upstream regions (TSS1500) and particularly in gene bodies. The distributions among island features according to the UCSC CpG island prediction was more similar between species ( & ).
We defined 165,847 Macaca mulatta-valid probes for Infinium 450K and 261,545 for Infinium EPIC, representing 382,780 and 610,010 gene feature annotations in total for Infinium 450K and Infinium EPIC, respectively. Note that one probe can be attributed to different transcripts and thus gene features, whereas one probe is attributed to only one island feature. The stacked barplots show the gene feature and CpG island annotation in comparison to the same probes annotated to the human arrays, as given in the BeadChip manifest files.
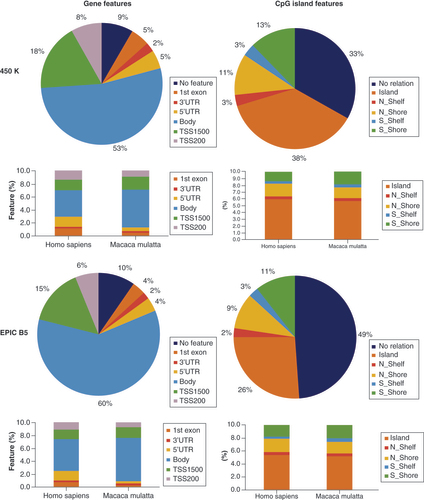
Among the 261,545 MM-valid probes from Infinium EPIC mapped on the MM genome, 200,125 were annotated for a gene feature (77%, representing 20,539 genes) and 134,091 (51%) for a CpG island feature. 117,045 MM-valid probes were annotated for both a gene and an island feature. Similar to the results for the 450K BeadChip, the fraction of probes located in the TSS500, the first exon and particularly the 5′UTR decreased, while many more probes were located upstream regions (TSS1500) and particularly in gene bodies.
Compared with Homo sapiens, reliable Infinium 450K and EPIC probes followed the overall distribution of probes when using the CpG island feature annotation, while for the gene feature annotation the proportion of probes in intergenic regions was increased especially in CS (, & ). Overall 50-60% of all simian genes were covered by at least one valid probe.
450K & EPIC manifest files for CS & MM
We provide the scientific community with new manifest files for Infinium 450K and EPIC BeadChips, adapted for genome-wide DNA methylation studies in CS or MM (Supplementary Material). We provide for each microarray and each species two files: one containing the whole set of CS-valid or MM-valid probes (filtered for probes with a mismatch at 1 or 2 bp prior to the CpG site) and another file containing only perfectly matched probes. These annotation files retain the format of the original Illumina manifest and can thus be used without further modifications in analysis pipelines for BeadChips such as SQN [Citation42] or the widely used ChAMP pipeline [Citation44]. All columns of the respective manifest files are described in detail in the supporting file (manifest header descriptions file included in the compressed Supplementary Material). The last column details the number of mismatches as well as the string for the position of the mismatch returned by Bowtie. The position of a mismatch is given relative to the base of the core probe sequence included in the manifest files. The interested user can thus select a subset of probes based on more stringent parameters, if needed. Of note, the string for the mismatch of the positions should be reversed for probe sequences mapping on the reverse strand of the monkey genomes.
Discussion
Epigenome-wide association studies (EWAS) have recently been performed on many phenotypes, traits and diseases including cancer, immune, neurodegenerative and infectious diseases, with now more than 500 EWAS published [Citation45]. Additionally, many more large-scale studies are likely to be conducted in the near future, linking complex diseases and traits with changes in the epigenome. Furthermore, DNA methylation holds the promise to explain at least a part of the influences the environment has on phenotype [Citation46]. Cell lines or blood cells do in most cases not appropriately recapitulate the phenotype of complex diseases, requiring the use of tissue or animal models to further complement our understanding of disease etiology and evaluate potential future treatments. CS and MM represent reference models in biomedical research and due to the recently realized importance of epigenetics for human disease, it would be of importance to include DNA methylation analysis in comprehensive multilevel-omics analyses. DNA methylation varies more across tissues than between distinct primate species [Citation47]. However, few tools are currently available for nonhuman primates.
In the presented work we determined, which probes of the most widely used DNA methylation arrays can be used to reliably analyze CpGs in the genomes of CS and MM.
The homology between human and MM at the nucleotide level has been reported to be on average 93.5 and 97.5% in genes [Citation48]. Considering the gene-centered approach for the design of the 450K array, the average rate of nucleotide substitution of 3.4% along the 50 bp probe sequences (Supplementary Table 6) is very similar to the reported rates. Furthermore, consistent with a higher nucleotide substitution rate in intergenic regions, these are increased to 3.6% on the EPIC array, on which mainly sequences in intergenic regions were added. The valid probes were very similar between CS and MM, with over 80% of mapped and valid CpG probe sequences in common between the two species. This is in agreement with the phylogenic proximity of these two Cercopithecidae [Citation49,Citation50]. Thus, similar mapping rates for both CS and MM are expected and found in our study (30–34% for EPIC and 450K BeadChips, respectively). Annotation rates were nonetheless much lower for AGM compared with MM, probably because the current annotation dates back 5 years for AGM and significantly smaller numbers of species-specific sequences and RNASeq data for Vervet-AGM are available leading to a lower quality of the annotation file. The available gene set comprises models based on orthologous proteins as well as longest translations of some human gene models, while for MM more RNA-seq data are available together with a more recent annotation explaining the higher quality of the annotation for MM compared to CS.
MM and Homo sapiens have diverged about 25 million years ago and their sequence identity is about 93% [Citation48]. At random, we should therefore expect about 86.5% of CpG sites in common between MM and humans. Nevertheless, only 66.5% of mapped probe sequences were identified as MM-targeting. The percentage of probes that can be reliably used identified in this study differed from the one identified in a previous study conducted on M. fascicularis [Citation19], a species closely related to M. mulatta, in which 61% of human probes mapped to the simian genome in contrast to the 48.8% identified here for M. mulatta. This difference can be explained by the more stringent parameters for mapping probe sequences in our study rather than differences between the two simian genomes. For instance, we allowed only three mismatches instead of the four mismatches allowed in the study by Ong et al. [Citation19] to align Infinium 450K probe sequences on the simian genomes as we observed that beta-value densities showed aberrant distributions for probes with more than three mismatches. The same behavior was observed for probes with fewer mismatches close to the targeted CpG. This feature has not been considered in previous studies, but had clearly strong influence on the measured DNA methylation levels (). Furthermore, in contrast to Ong et al. [Citation19], we only kept uniquely matched sequences on simian genomes because probes measuring DNA methylation levels at CpGs in multiple genomic regions are difficult to interpret and thus are not informative, as has been demonstrated for human samples on the arrays [Citation51]. Indeed, overall we have annotated 88% (MM) of aligned probes sequences, close to the 94% reported in Ong et al. [Citation19]. The same filtering approach as proposed by Ong et al. [Citation19] was subsequently also used for a study on osteoarthritis using baboon samples which concluded that a total of 44% of probes on the 450K BeadChip could be reliably used [Citation20], similar to the levels found here.
A similar approach allowing up to four mismatches was also proposed for the analysis of DNA methylation and hydroxymethylation in MM using the 450K BeadChip [Citation18]. When comparing our list of valid probes with the ones identified by Chopra et al., slightly more probes with a perfect match were identified in our study (37,547 vs 35,901, of which 33,001 were in common) and 165,847 probes with mismatches (vs 154,030, with 129,340 probes in common). Looking into more detail in the perfectly matched probes only identified by Chopra et al. (n = 2900), 773 probes were also among our valid probes, but had one to three mismatches, while 2127 had more than three mismatches and were thus in our nontargeting category. There several possible reasons for this discrepancy, including the use of different genome builds (MM1.0 vs MM10) as well as differences in the parameters to map the sequences to the MM genome.
Recently, human DNA methylation capture panels were adapted to the analysis of DNA methylation of nonhuman primates and notably AGM. While this approach allows capturing a larger proportion of regulatory elements in the simian genomes [Citation52], it comes at a higher price and is thus less well suited for projects with larger number of samples. Furthermore, in quantitative comparisons the Infinium BeadChips outperformed the sequencing-based approach in terms of quantitative precision even when pooling CpG levels of closely neighboring CpGs in the sequencing approach [Citation53]. These results were obtained in human samples, it can thus be expected that the quantitative ratios will deviate even stronger in the presence of sequence mismatches between the capture probes and the simian genomes. Reduced representation bisulfite sequencing (RRBS) [Citation54] has been shown to correlate well with the results obtained from 450K arrays in Macaca fascicularis [Citation19] and will cover more CpGs than BeadChips. However, the coverage will in general be lower, leading to a reduction in the ability to precisely quantify to assess DNA methylation levels at CpGs [Citation55]. Furthermore, RRBS requires the use of restriction enzymes, which will – dependent on their recognition site – select genomic regions with a certain CpG density, making this approach less universal compared with BeadChips, which cover diverse regions in terms of CpG density and genic annotation ( & ).
Of note, W→S nucleotide substitutions represented ∼40% of mismatches and around 85% when looking at CpG sites in agreement with the weak-to-strong bias observed during recent human evolution [Citation56–58]. The same results were obtained for CS and similar results were found with the Infinium EPIC. For the latter, we observed a slightly increased substitution rate at CpG sites concomitant with a slightly lower number of valid probes compared with Infinium 450K for both species. This observation might be explained by the localization of the 350,000 additional probes in enhancers on the EPIC arrays as regulatory regions have been suggested to be fast evolving regions during human divergence from other primates [Citation59,Citation60]. As the overall percentage of reliable probes between the different generations of BeadChips remains similar, it can be expected that future human generations of human Infinium DNA methylation microarrays, will also improve the coverage of the simian genomes.
Annotation of these valid probes for gene features showed higher proportion of probes targeting CpGs located in intergenic regions compared with CpGs in promoter or gene regions. This tendency was increased in CS compared with MM. In contrast, the frequency of location of valid probes within CpG island features were similar between the two species. It is important to point out the different annotation levels between these distinct species, which impacts the number of identified/predicted genes and transcripts in each species. Indeed, the human annotation (version 101 from Ensembl) contains 60,671 genes and pseudogenes for 229,487 transcripts plus thousands of alternate sequences, whereas 35,432 genes are annotated in MM for 64,228 transcripts and 27,982 genes are annotated in CS for only 28,072 transcripts [Citation61,Citation62]. CpG island feature annotations, in counterpart, are directly predicted from reference sequence. This might explain the observed differences between the species depending on the genomic region. It is indeed important to keep in mind that simian annotations generally are still less precise and documented than human annotation and this lack of information may contribute to the observed differences.
Nonetheless, we provide to the research community a list of 162,053 annotated CS-valid probes corresponding to 16,259 genes for CS and a list of 165,847 annotated MM-valid probes corresponding to 18,667 genes for MM for the Infinium 450K array, as well as a list of 255,227 annotated CS-valid probes corresponding to 17,701 genes for CS and a list of 261,545 annotated MM-valid probes corresponding to 20,539 genes for MM for the Infinium EPIC array. While previous studies identified list of probes, they did not provide a species-specific annotation file. Furthermore, we annotated probes according to Illumina gene and CpG island features and built annotation files following the Illumina manifest file format. We kept the human GRCh37 annotation for comparison purpose and our annotation files can directly be used with a number of processing tools. Although the proportions of the different gene features are altered compared with the distribution for the human genome with a reduction in gene bodies and around the TSS, the current generation of the methylation microarray cover around 60% of all known genes in the two species and these genes are covered by two to six probes allowing multiple reliable measurements of DNA methylation to increase confidence in the obtained data. This observation will also be reinforced by the fact that human arrays contain proportionally more probes in the promoter regions (often associated with CpG islands) ensuring that promoter associated CpG-rich regions remain sufficiently covered. While genes of special interest for a research group might be missing from these lists, the arrays provide the currently most comprehensive tool for DNA methylation analysis at a reasonable price. The number of available reliable probes exceeds by several orders of magnitude the number of probes available on the first generation of the Infinium DNA methylation BeadChips, the 27K array, which led to the discovery of DNA methylation changes following environmental exposure such as tobacco smoking [Citation63] or disease-associated changes [Citation64,Citation65], and whose success led to the development of the higher-density DNA methylation arrays evaluated in this study. Monkey-specific microarrays would constitute ideal tools. However, commercial off-the-shelf DNA methylation microarrays for model organisms have been awaited in vain. On the other hand, comprehensive whole genome bisulfite sequencing projects remain for most laboratories prohibitively expensive when performed at coverage allowing reliable methylation calls.
In summary, our approach using standard bioinformatic tools and validated quality criteria using orthogonal analysis technology concerning notably the importance of the number and position of any mismatches should be applicable to other simian and perhaps even other mammalian species. Our approach allows a rapid selection of reliable probes for any organism with an annotated genome sequence. Of course, a more divergent genome sequence would lead to fewer probes that can be used with high confidence and for some organisms this might eventually represent an unfavorable cost/output balance, such as mice where only 10–12 k probes for the 450K BeadChip and 13K for the EPIC array were found reliable [Citation18,Citation66,Citation67]. For these organisms sequencing-based approaches such as RRBS or capture-based selection might be more appropriate.
Conclusion
The presented work investigated in depth the suitability of human DNA methylation Infinium BeadChips for the use in two widely used simian model species. The annotation files are provided in a format compatible with a variety of preprocessing, normalization and analytical pipelines designed for data analysis from 450K/EPIC arrays facilitating high-throughput DNA methylation analyses in MM and CS for many questions of biomedical relevance. Especially, for the current and potential future pandemics, nonhuman primate models will be important for the development of efficient treatments and vaccines and this might require the understanding of the disease process. The tools described in this work provide a straightforward approach enabling the inclusion of DNA methylation analysis in current analyses.
Future perspective
Technological advances in microarray technology have allowed commercializing microarrays with an ever-increasing density and with the recognition of the contribution of epigenetic changes to many human complex diseases, it can be anticipated that methylation BeadChips with an increased number of probes will become available for the analysis of the human genome. This would in turn allow for a more comprehensive analysis of the DNA methylation landscape in simian samples. Furthermore, while it is rather unlikely that commercial off-the-shelf arrays will become available for specific animal models, the identification of reliable probes, might allow to subset available human probes and create arrays with this subset of probes. The reduced required space on the BeadChip surface might enable to analyze a larger number of animals on a single array and thereby increase sample size of studies and their biological or clinical relevance.
The Infinium Human Methylation 450K and Methylation EPIC BeadChips are useful tools for the study of the methylation state of hundreds of thousands of CpGs across the human genome at affordable cost.
Due to their close developmental, immunological and neurological proximity with humans, nonhuman primates are essential for studies and preclinical research of many human diseases.
Rhesus macaques (Macaca mulatta) and African green monkeys (Chlorocebus sabaeus) are among the major nonhuman primate models utilized in biomedical research and of importance for treatment and prevention of infectious diseases such as Covid-19.
In this study we demonstrate through in silico analyses using stringent criteria combined with functional validation by pyrosequencing, that up to 165,000 of the 450K and 261,000 probes of the EPIC BeadChip can be reliably used in M. mulatta or C. sabaeus.
We provide species-specific annotation files in a format compatible with a variety of preprocessing, normalization and analytical pipelines designed for data analysis from 450K/EPIC arrays, facilitating high-throughput DNA methylation analyses in M. mulatta and C. sabaeus.
Author contributions
F Pichon, M Müller-Trutwin and J Tost designed the study. F Busato and SP Jochems conducted experiments. F Pichon, Y Shen, F Busato and SP Jochems performed the bioinformatic analyses. R Le Grand and JF Deleuze provided infrastructure and financial support. B Jacquelin and R Le Grand coordinated the animal follow-up and provided samples. F Pichon and J Tost wrote the manuscript. All authors read the final manuscript.
Ethical conduct of research
Animals were housed at the IDMIT center of the Commissariat à l’Energie Atomique (CEA, Fontenay-aux-Roses, France, permit number: A 92–032-02). The CEA complies with Standards for Human Care and Use of Laboratory of the Office for Laboratory Animal Welfare (OLAW, USA) under OLAW Assurance number #A5826-01. The Central Committee for Animals at Institut Pasteur or Ethical Committee of Animal Experimentation (CETEA-DSV, IDF, France) approved all animal experimental procedures (Notification numbers: 10-051b, 12-006b). The studies were conducted in strict accordance with the international European guidelines 2010/63/UE on protection of animals used for experimentation and other scientific purposes (French decree 2013–118).
Supplemental Document
Download MS Excel (14.7 KB)Supplemental Document
Download Comma-Separated Values File (14.5 MB)Supplemental Document
Download Comma-Separated Values File (64.6 MB)Supplemental Document
Download Comma-Separated Values File (16.6 MB)Supplemental Document
Download Comma-Separated Values File (73.1 MB)Supplemental Material
Download Zip (78.3 MB)Supplemental Document
Download Comma-Separated Values File (23.1 MB)Supplemental Document
Download Comma-Separated Values File (26.5 MB)Supplemental Document
Download MS Word (10.5 MB)Supplemental Document
Download MS Word (88.8 KB)Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2217/epi-2020-0200
Financial & competing interests disclosure
The authors would like to acknowledge grant support from ANRS and the institutional budget from the CNRGH. SPJ was recipient of a PhD fellowship from the University Paris Diderot, Sorbonne Paris Cité. The Infectious Disease Models and Innovative Therapies (IDMIT) center in Fontenay-aux-Roses, France, is funded by the French government’s Investissements d’Avenir program for infrastructures (PIA) under grant ANR-11-INBS-0008 and the PIA grant ANR-10-EQPX-02-01. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Andersen GB , TostJ. A Summary of the Biological Processes, Disease-Associated Changes, and Clinical Applications of DNA Methylation.In: DNA Methylation Protocols: Methods in Molecular Biology.TostJ (Eds).Humana Press, NY, USA, 1708, 3–30 (2018).
- Ehrlich M , WangRY. 5-Methylcytosine in eukaryotic DNA. Science212(4501), 1350–1357 (1981).
- Hwang DG , GreenP. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl Acad. Sci. USA101(39), 13994–14001 (2004).
- Walser JC , PongerL , FuranoAV. CpG dinucleotides and the mutation rate of non-CpG DNA. Genome Res.18(9), 1403–1414 (2008).
- Duret L , GaltierN. The covariation between TpA deficiency, CpG deficiency, and G*C content of human isochores is due to a mathematical artifact. Mol. Biol. Evol.17(11), 1620–1625 (2000).
- Rollins RA , HaghighiF , EdwardsJRet al. Large-scale structure of genomic methylation patterns. Genome Res.16(2), 157–163 (2006).
- Saxonov S , BergP , BrutlagDL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl Acad. Sci. USA103(5), 1412–1417 (2006).
- Sandoval J , HeynH , MoranSet al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics6(6), 692–702 (2011).
- Moran S , ArribasC , EstellerM. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics8(3), 389–399 (2016).
- Flanagan JM . Epigenome-wide association studies (EWAS): past, present, and future. In: Cancer Epigenetics: Methods in Molecular Biology (Methods and Protocols, VermaM (Eds), Humana Press, New York, NY, 1238, 51–6310.1007/978-1-4939-1804-1_3 (2015).
- Lanford RE , BiggerC , BassettS , KlimpelG. The chimpanzee model of hepatitis C virus infections. ILAR J.42(2), 117–126 (2001).
- Bosinger SE , JacquelinB , BeneckeA , SilvestriG , Muller-TrutwinM. Systems biology of natural simian immunodeficiency virus infections. Curr. Opin. HIV AIDS7(1), 71–78 (2012).
- Jacquelin B , MayauV , TargatBet al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest.119(12), 3544–3555 (2009).
- Zou J , YoungS , ZhuFet al. Microarray profile of differentially expressed genes in a monkey model of allergic asthma. Genome Biol.3(5), R0020 (2002).
- Miyahara T , KikuchiT , AkimotoM , KurokawaT , ShibukiH , YoshimuraN. Gene microarray analysis of experimental glaucomatous retina from cynomologous monkey. Invest. Ophthalmol. Vis. Sci.44(10), 4347–4356 (2003).
- Abdel Rassoul R , AlvesS , PantescoVet al. Distinct transcriptome expression of the temporal cortex of the primate Microcebus murinus during brain aging versus Alzheimer’s disease-like pathology. PLoS One5(9), (2010).
- Hernando-Herraez I , Prado-MartinezJ , GargPet al. Dynamics of DNA methylation in recent human and great ape evolution. PLoS Genet.9(9), e1003763 (2013).
- Chopra P , PapaleLA , WhiteATet al. Array-based assay detects genome-wide 5-mC and 5-hmC in the brains of humans, non-human primates, and mice. BMC Genomics15, 131 (2014).
- Ong ML , TanPY , MacIsaacJLet al. Infinium monkeys: Infinium 450K array for the Cynomolgus macaque (Macaca fascicularis). G3 (Bethesda)4(7), 1227–1234 (2014).
- Housman G , HavillLM , QuillenEE , ComuzzieAG , StoneAC. Assessment of DNA methylation patterns in the bone and cartilage of a nonhuman primate model of osteoarthritis. Cartilage10(3), 335–345 (2019).
- Buschdorf JP , OngML , OngSXet al. Low birth weight associates with hippocampal gene expression. Neuroscience318, 190–205 (2016).
- Gaudinski MR , CoatesEE , NovikLet al. Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody mAb114 targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study. Lancet393(10174), 889–898 (2019).
- Rockx B , KuikenT , HerfstSet al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science368(6494) 1012–101510.1126/science.abb7314 (2020).
- de Wit E , FeldmannF , CroninJet al. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl Acad. Sci. USA117(12), 6771–6776 (2020).
- Maisonnasse P , GuedjJ , ContrerasVet al. Hydroxychloroquine use against SARS-CoV-2 infection in non-human primates. Nature585(7826), 584–587 (2020).
- Munoz-Fontela C , DowlingWE , FunnellSGPet al. Animal models for COVID-19. Nature586(7830), 509–51510.1038/s41586-020-2787-6 (2020).
- Sritanaudomchai H , MaH , ClepperLet al. Discovery of a novel imprinted gene by transcriptional analysis of parthenogenetic embryonic stem cells. Hum. Reprod.25(8), 1927–1941 (2010).
- Provencal N , SudermanMJ , GuilleminCet al. The signature of maternal rearing in the methylome in rhesus macaque prefrontal cortex and T cells. J. Neurosci.32(44), 15626–15642 (2012).
- Sayers K , EvansTA , MenzelE , SmithJD , BeranMJ. The misbehaviour of a metacognitive monkey. Behaviour152(6), 727–756 (2015).
- Brutcher RE , NaderSH , NaderMA. Evaluation of the reinforcing effect of quetiapine, alone and in combination with cocaine, in rhesus monkeys. J. Pharmacol. Exp. Ther.356(2), 244–250 (2016).
- Gao F , NiuY , SunYEet al. De novo DNA methylation during monkey pre-implantation embryogenesis. Cell Res.27(4), 526–539 (2017).
- Barrett RLC , DawsonM , DyrbyTBet al. Differences in frontal network anatomy across primate species. J. Neurosci.40(10), 2094–2107 (2020).
- Gichuki CW , NantulyaVM , SayerPD. Trypanosoma brucei rhodesiense: use of an antigen detection enzyme immunoassay for evaluation of response to chemotherapy in infected vervet monkeys (Cercopithecus aethiops). Trop. Med. Parasitol.45(3), 237–242 (1994).
- Bossart KN , RockxB , FeldmannFet al. A Hendra virus G glycoprotein subunit vaccine protects African green monkeys from Nipah virus challenge. Sci. Transl. Med.4(146), 146ra107 (2012).
- Jentsch JD , RedmondDEJr , ElsworthJD , TaylorJR , YoungrenKD , RothRH. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science277(5328), 953–955 (1997).
- Jasinska AJ , SchmittCA , ServiceSKet al. Systems biology of the vervet monkey. ILAR J.54(2), 122–143 (2013).
- Sodora DL , AllanJS , ApetreiCet al. Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts. Nat. Med.15(8), 861–865 (2009).
- Smits SL , vanden Brand JM , de LangAet al. Distinct severe acute respiratory syndrome coronavirus-induced acute lung injury pathways in two different nonhuman primate species. J. Virol.85(9), 4234–4245 (2011).
- Nalca A , ToturaA , LivingstonV , FrickO , DyerD. African green monkey model of Middle East respiratory syndrome coronavirus (MERS_CoV) infection. Int. J. Infect. Dis.79, 99–100 (2019).
- Cross RW , AgansKN , PrasadANet al. Intranasal exposure of African green monkeys to SARS-CoV-2 results in acute phase pneumonia with shedding and lung injury still present in the early convalescence phase. Virol. J.17(1), 125 (2020).
- Langmead B , TrapnellC , PopM , SalzbergSL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol.10(3), R25 (2009).
- Touleimat N , TostJ. Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics4(3), 325–341 (2012).
- Tost J , GutIG. DNA methylation analysis by pyrosequencing. Nat. Protoc.2(9), 2265–2275 (2007).
- Tian Y , MorrisTJ , WebsterAPet al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics33(24), 3982–3984 (2017).
- Xiong Z , LiM , YangFet al. EWAS Data Hub: a resource of DNA methylation array data and metadata. Nucleic Acids Res.48(D1), D890–D895 (2020).
- Chung FF , HercegZ. The promises and challenges of toxico-epigenomics: environmental chemicals and their impacts on the epigenome. Environ. Health Perspect.128(1), 15001 (2020).
- Pai AA , BellJT , MarioniJC , PritchardJK , GiladY. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet.7(2), e1001316 (2011).
- Rhesus Macaque Genome S , AnalysisC , GibbsRAet al. Evolutionary and biomedical insights from the rhesus macaque genome. Science316(5822), 222–234 (2007).
- Perelman P , JohnsonWE , RoosCet al. A molecular phylogeny of living primates. PLoS Genet.7(3), e1001342 (2011).
- Fomsgaard A , Muller-TrutwinMC , DiopOet al. Relation between phylogeny of African green monkey CD4 genes and their respective simian immunodeficiency virus genes. J. Med. Primatol.26(3), 120–128 (1997).
- Chen YA , LemireM , ChoufaniSet al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics8(2), 203–209 (2013).
- Lee JR , RyuDS , ParkSJet al. Successful application of human-based methyl capture sequencing for methylome analysis in non-human primate models. BMC Genomics19(1), 267 (2018).
- Heiss JA , BrennanKJ , BaccarelliAAet al. Battle of epigenetic proportions: comparing Illumina’s EPIC methylation microarrays and TruSeq targeted bisulfite sequencing. Epigenetics15(1–2), 174–182 (2020).
- Gu H , SmithZD , BockC , BoyleP , GnirkeA , MeissnerA. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc.6(4), 468–481 (2011).
- Carmona JJ , AccomandoWPJr , BinderAMet al. Empirical comparison of reduced representation bisulfite sequencing and Infinium BeadChip reproducibility and coverage of DNA methylation in humans. NPJ Genom Med.2, 13 (2017).
- Pollard KS , SalamaSR , LambertNet al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature443(7108), 167–172 (2006).
- Katzman S , CapraJA , HausslerD , PollardKS. Ongoing GC-biased evolution is widespread in the human genome and enriched near recombination hot spots. Genome Biol. Evol.3, 614–626 (2011).
- Gotea V , ElnitskiL. Ascertaining regions affected by GC-biased gene conversion through weak-to-strong mutational hotspots. Genomics103(5–6), 349–356 (2014).
- Pollard KS , SalamaSR , KingBet al. Forces shaping the fastest evolving regions in the human genome. PLoS Genet.2(10), e168 (2006).
- Shibata Y , SheffieldNC , FedrigoOet al. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet.8(6), e1002789 (2012).
- Svardal H , JasinskaAJ , ApetreiCet al. Ancient hybridization and strong adaptation to viruses across African vervet monkey populations. Nat. Genet.49(12), 1705–1713 (2017).
- Warren WC , JasinskaAJ , Garcia-PerezRet al. The genome of the vervet (Chlorocebus aethiops sabaeus). Genome Res.25(12), 1921–1933 (2015).
- Breitling LP , YangR , KornB , BurwinkelB , BrennerH. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet.88(4), 450–457 (2011).
- Halvorsen AR , HellandA , FleischerTet al. Differential DNA methylation analysis of breast cancer reveals the impact of immune signaling in radiation therapy. Int. J. Cancer135(9), 2085–2095 (2014).
- Hasler R , FengZ , BackdahlLet al. A functional methylome map of ulcerative colitis. Genome Res.22(11), 2130–2137 (2012).
- Wong NC , NgJ , HallNEet al. Exploring the utility of human DNA methylation arrays for profiling mouse genomic DNA. Genomics102(1), 38–46 (2013).
- Gujar H , LiangJW , WongNC , MozhuiK. Profiling DNA methylation differences between inbred mouse strains on the Illumina Human Infinium MethylationEPIC microarray. PLoS One13(3), e0193496 (2018).
- Jochems SP , JacquelinB , TchitchekNet al. DNA methylation changes in metabolic and immune-regulatory pathways in blood and lymph node CD4+ T cells in response to SIV infections. Clin. Epigenetics. 12(1), 18810.1186/s13148-020-00971-w.(2020).