
Abstract
Aims & objectives: To investigate DNA methylation patterns in asthenozoospermic and normozoospermic sperm and to explore the potential roles of differential methylations in the etiology of the disease. Materials & methods: The authors performed whole-genome bisulfite sequencing analysis between normozoospermic controls and asthenozoospermic individuals. Results: The authors identified 238 significant differentially methylated regions. These differentially methylated regions were annotated to 114 protein-coding genes, with many genes showing associations with spermatogenesis, sperm motility etc. Conclusion: There are plenty of genomic regions exhibiting altered DNA methylation in asthenozoospermia, a number of which are located within or adjacent to sperm-related genes, suggesting novel methylation markers of asthenozoospermia and potential epigenetic regulation mechanisms through DNA methylation in the disease.
Infertility or subfertility has aroused wide social concerns in recent decades, and around half of the subfertile couples are affected by male factor infertility [Citation1]. Around 80% of male infertility cases are related to asthenozoospermia with variable degrees of severity [Citation2]. Asthenozoospermia is absent or reduced sperm motility in the fresh ejaculate, which specifically means that the proportion of progressively motile spermatozoa is below the lower limit (32%) [Citation3]. Asthenozoospermia itself is a complex and multifactorial disease that may result from genetic, epigenetic or environmental factors. Multiple genetic factors contributing to asthenozoospermia have been identified and summarized [Citation4–6]. However, a considerable proportion of the cases were still diagnosed with unexplained causes. Apart from genetic factors, epigenetic mechanisms, including DNA methylation, may represent an attractive explanation for the etiology of asthenozoospermia.
A widely accepted definition of epigenetics is the heritable changes of gene expression without a change of DNA sequence, of which DNA methylation is the most prominent and extensively studied epigenetic mechanism [Citation7,Citation8]. Methylation of the fifth atomic position of cytosines on genomic DNA is the form of DNA methylation that has been most commonly studied so far. In humans or other mammals, methylation of the fifth atomic position of cytosines primarily occurs at CpG dinucleotides, although methylation of non-CpG cytosines (CHG or CHH when H stands for A, G or T) also exists in some cases [Citation9,Citation10]. CpGs are functionally most important due to their symmetrical structures, which makes them peculiar epigenetic marks that can be passed on to daughter cells after DNA replication [Citation11–14]. Through interplay with other epigenetic mechanisms, including histone modifications and transcription factors, DNA methylation pattern acts as another layer of regulation of gene expression and securer of genome stability [Citation15–18]. In principle, methylated CpGs at regulatory genomic regions are negatively associated with gene expression, although increasing evidence in recent studies is also suggesting a positive relationship in some situations depending on their relative locations on the genome [Citation19–22]. Importantly, DNA methylation has been reported to play vital roles in crucial development and normal physiology, including spermatogenesis and embryogenesis [Citation23–25].
The impacts of DNA methylation on sperm development can be manifested in different aspects. Two major DNA methylation reprogramming events take place during the human developmental process, with the first wave occurring in primordial germ cells during early embryogenesis and the second in the preimplantation embryo following fertilization. Genomewide DNA methylation patterns are erased and later on re-established in both periods, which are windows of susceptibility that may introduce epigenetic errors and thus affect fertility [Citation26]. During the long and continuous process of human sperm production, impairment of spermatogenesis or abnormal semen parameters have been reported to be associated with toxins, smoking, diet or other environmental factors [Citation27–31]. It has been reported that some influences may last for generations, probably associated with altered DNA methylation marks in spermatozoa [Citation32–34]. In the past 20 years, growing evidence has supported the idea that DNA methylation may also contribute to male infertility, which profits from the wide application of microarray-based technologies and next-generation sequencing. Aberrant DNA methylation has been reported to be associated with male subfertility and abnormal semen parameters, and plenty of differential methylation sites or regions have been identified in several disease states, including teratozoospermia and oligozoospermia [Citation35–44]. For instance, several candidate gene studies of DNA methylation have found an association of aberrant methylation of H19 with male infertility, including teratozoospermia and oligozoospermia [Citation45,Citation46]. A methylation study on the spermatogenesis-associated genes (SPATA4, SPATA5 and SPATA6) showed that hypermethylation of these genes correlated with oligozoospermic infertility [Citation47]. Another study demonstrated increased defective methylation of DAZL promoter CpG islands in oligoasthenoteratozoospermia patients [Citation48]. However, only very few genes have been investigated multiple times and there has been little overlap of results from different genomewide DNA methylation studies. Moreover, many studies have used testis or blood to investigate fertility issues and only a limited number of studies have directly performed methylation profiling of human sperm samples. In studies that investigated reduced fertility, different types of sperm abnormalities may not have been distinguished, which may have led to failure in capturing subtle but functional changes in DNA methylation patterns of a certain type of abnormality. Besides, most of the previous studies used array-based or targeted-sequencing methods and thus were restricted to only a part of the human genome.
In this study, the authors performed the gold-standard whole-genome bisulfite sequencing (WGBS) on normozoospermic and asthenozoospermic sperm samples to specifically investigate whether DNA methylation is involved in the etiology of asthenozoospermia and to build an atlas of asthenozoospermia that provides a valuable reference and resources for future studies.
Materials & methods
Ethics approval & sample acquisition
This research was approved by all respective institutional research and ethics boards. The participants in this study were selected from volunteers or patients seeking fertility treatment. Informed written consent was obtained from each participant. Participant information was de-identified before analysis. Semen samples were collected after at least 3 days of abstinence. After liquefaction for 20–30 min at 37°C, semen analyses of all samples were performed according to the standard WHO recommendations [Citation3]. To reduce the contamination of DNA from nonsperm cells during the sample collection, the authors excluded the semen samples for which the nonsperm cell concentration exceeded 1%. The authors further microscopically examined all semen samples. Consequently, nonsperm cells were far fewer than the spermatozoa, and the spermatozoa/nonsperm cell ratios of our samples ranged from about 150 to 1000. Additionally, a recent study showed that seminal cfDNA (e.g., from broken cells) has little contaminating impact on sperm DNA methylation analysis [Citation49]. Thus, the methylation marks that were extracted from the seminal samples should represent the true epigenetic feature of spermatozoa. In total, 12 semen samples were collected and were divided into two groups: normozoospermic control samples (n = 5) and asthenozoospermic samples (n = 7). The semen parameters of the different study groups are summarized in .
Table 1. Semen parameters of different study groups.
Sperm DNA isolation
For bisulfite sequencing, the total genomic DNA of all samples was isolated. Precipitated clean sperm samples were lysed with a lysis buffer containing a final concentration of 100 mM Tris·Cl (pH 8.0), 10 mM EDTA, 500 mM NaCl, 1% SDS and 2% ß-mercaptoethanol and were incubated together with proteinase K at 55°C for 2 h. DNA was then isolated using the QIAamp® DNA Mini Kit (Qiagen, MD, USA) according to the manufacturer’s protocol. Inspection of DNA degradation or contamination was performed by running agarose gels. The purity of DNA was checked using the NanoPhotometer® spectrophotometer (Implen, CA, USA), and concentration was measured by using Qubit® DNA Assay Kit in Qubit® 2.0 fluorometer (Life Technologies, CA, USA).
Library construction & WGBS
A total amount of 100 ng genomic DNA spiked with 0.5 ng lambda DNA were fragmented by sonication to 200–300 bp with the Covaris S220 (Covaris LLC, MA, USA). The DNA fragments then underwent bisulfite treatment with the commercial EZ DNA Methylation-Gold™ Kit (Zymo Research, CA, USA) before library construction. The concentration and quality of the library were assessed on the Agilent Bioanalyzer 2100 system. Pair-end sequencing of samples was performed on the Illumina NovaSeq platform (Illumina, CA, USA) and 150 bp reads were generated. Image analysis and base calling were performed with the Illumina CASAVA pipeline.
WGBS data analysis
FastQC v0.11.8 was used to check the sequence quality and generate quality reports. Low-quality bases and the adaptor sequences were trimmed with Trim Galore v0.6.6 [Citation50], and the cleaned data with high-quality reads for each sample were used for the next alignment step. Cleaned reads were mapped to the human reference genome (GRCh38) using Bismark v0.23.0 with the following alignment parameters: “bismark -X 600 -1 ${READ1} -2 ${READ2}” [Citation51]. Duplicates were removed using sambamba v0.8.0 [Citation52]. Methylation information was extracted using bismark_methylation_extractor from Bismark v0.23.0 for downstream analysis.
Differentially methylated region calling, gene annotation & gene ontology analysis
The DMRichR v1.7.2 package with R software v4.1.1 was used to identify differentially methylated regions (DMRs) and to annotate DMR-related genes [Citation53–55]. CpG sites with at least 5× sequencing coverage across all samples were retained for DMR calling. The minimum number of CpGs required for a DMR was set to five. Statistical algorithms from Bioconductor R package “dmrseq” were leveraged and DMRs were identified in a two-step approach [Citation55], which accounts for both interindividual and inter-CpG variability across the entire genome. Methylation data were first smoothed and candidate DMRs with a difference were assembled; then a regional statistic was estimated for each DMR and permutation analysis of a pooled null distribution was performed to identify significant DMRs. The absolute value for the cutoff of the single CpG coefficient was set at 0.1 for candidate region discovery. After computation, p-values, methylation differences and other statistics were assigned to each DMR. DMRs that passed the p < 0.05 threshold were selected as significant DMRs. The authors performed gene ontology (GO) analysis of biological process, cellular component and molecular function using the Bioconductor R package ‘rGREAT’ [Citation56]. Two statistical tests, the binomial test over genomic regions and the hypergeometric test over genes, were conducted in GREAT analysis. These two statistical tests would compensate for each other, thus providing an accurate picture of annotation enrichments.
Results
Global DNA methylation profiles in normozoospermic & asthenozoospermic sperm
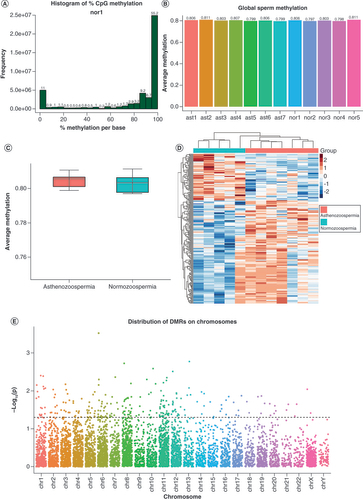
To obtain the DNA methylation profiling of both normozoospermic and asthenozoospermic human sperm and compare their methylomes, the authors performed WGBS on five normozoospermic and seven asthenozoospermic samples. Semen parameters of the samples are summarized in . The percentages of progressively motile sperm in the asthenozoospermic group were evidently below the normal threshold (<32%). For each sample, about 330 million paired-end reads were obtained and some basic statistics of the WGBS experiment are summarized in Supplementary Table 1. The majority of cytosine methylations occurred in the CpG context, while non-CpG cytosine methylation (CHG or CHH) was observed at a very low level (less than 1%). In this case, the authors focused only on CpG cytosine methylation across the whole genome in later analysis. To further inspect the quality of the WGBS experiment process in our study, we calculated and visualized the distribution of methylation values for all CpGs of each sample, where a bimodal distribution would suggest good experimental quality. A shows the distribution of methylation values of one normal control sample, and such distributions of all other samples were also examined and are shown in Supplementary Figure 1. Upon extraction of methylation information, only CpGs that were covered by at least five reads in all 12 samples were retained for further analysis. A total of 23,725,450 CpG sites were retained and used for global methylation analysis as well as the regional methylation analysis. Then, the authors first explored the global level of DNA methylation in the two study groups. For each sample, the average CpG methylation values were computed and plotted, as in B. The global methylation differences between the groups were then tested through one-way analysis of variance, and no significant differences were observed in terms of global DNA methylation between the groups (p > 0.05; C). Nevertheless, since this measure of global methylation refers to a composite average derived from all the detected CpG sites across the entire genome, potential alterations in certain areas may have been covered up. Therefore, the authors performed a further investigation to identify regional methylation changes across the whole genome.
(A) Histogram of methylation values for all CpGs in one representative sample. As expected, it showed bimodal distributions, suggesting a high experimental quality. (B) Global methylation levels of all samples. The ast1-7 and nor1-5 are sample IDs of asthenozoospermia and normozoospermia, respectively. (C) No significant global DNA methylation differences were found between groups (p > 0.05). (D) Heatmap of methylation data z-scores of 238 significantly differentially methylated regions. (E) Distribution of candidate DMRs across chromosomes, where the dotted line represents a threshold of p < 0.05, and dots above the line were chosen as significant DMRs.
DMR: Differentially methylated region.
Identification of asthenozoospermia-associated DMRs
Despite the lack of global methylation difference between the two groups, the regional analysis suggested that there are plenty of DMRs across the genome. With a rigorous permutation-based algorithm from ‘dmrseq’ the authors successfully identified 238 significant DMRs between the normozoospermic and asthenozoospermic groups (D & E). This methodology enables the inference of DMRs with good specificity and sensitivity. In this smoothing-based approach, CpGs with higher coverage are given a higher weight and are used to infer the methylation level of adjacent CpGs that have lower coverage. In addition, this approach performs statistical testing on the DMRs themselves with a region statistic for each DMR rather than testing for the differences in a single CpG, which are then assembled into DMRs. Thus, this analysis offers accurate false discovery rate control. For the samples, the absolute value for the cutoff of the single CpG coefficient that was used to discover candidate regions was set to 0.1, with a minimum of five CpGs required for a region. The maximum distance between two neighboring CpGs that were included in the same DMR was no more than 1000 bp. A heatmap of methylation data z-scores of the 238 DMRs is illustrated in D. In the heatmap, each row represents a DMR and each column represents a sample. Overall, the authors identified 6726 candidate DMRs across the genome and then plotted the chromosome distribution of all candidate DMRs using a Manhattan plot, as shown in E. The 238 significant DMRs above the dotted line (p < 0.05) from the Manhattan plot were chosen for further analyses. Methylation differences and genomic annotations of all candidate DMRs were also plotted and were collected in Supplementary Figure 2.
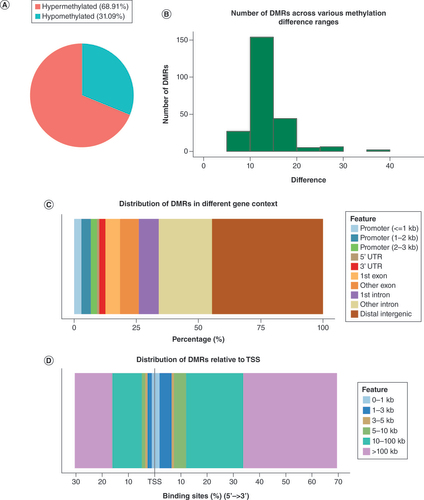
The authors then defined hypermethylation and hypomethylation as increased or decreased methylation levels in asthenozoospermic sperm compared with normozoospermic sperm, respectively. Overall, they identified 69% hypermethylated DMRs (n = 164) and 31% hypomethylated DMRs (n = 74; A). They further grouped the DMRs into distinct levels of methylation difference to look into the degree of alteration (B). Most of the DMRs fell into a difference of 10–20 (83.2% of the total DMRs), while around 5.5% of the DMRs showed a higher extent of more than 20 methylation difference.
(A) Count of hypermethylated DMRs and hypomethylated DMRs. (B) Histogram showing the distribution of degrees of methylation difference. (C) Percentage of DMRs in distinct genomic contexts. (D) Relative distances of DMRs to the TSS.
DMR: Differentially methylated region; TSS: Transcription start site.
Genomic locations of asthenozoospermia-associated DMRs
Since DNA methylation plays a crucial role in the regulation of gene expression in a genomic context-dependent manner, the authors then investigated the locations of DMRs relative to distinct genomic features (promoter, 5′UTR, 3′UTR, exon etc.). As shown in C, differential methylations were most frequent in the distal intergenic regions (44.54%), followed by introns (29.41%) and exons (13.44%). Also, about 9.24% of the DMRs were located near promoters, suggesting potential regulation of gene expression. Less frequent locations were 3′UTR (2.52%) and 5′UTR (0.84%). In addition, the distances of DMRs relative to the transcription start site are shown in D, and a small number of DMRs were located quite near the transcription start site (<3 kb).
Annotation of DMRs & functional-based analysis of DMR-associated genes
To further explore the biological significance of these methylation changes between groups, the authors next annotated the DMRs to find out whether they were associated with functional genes. Collectively, these 238 DMRs were annotated to 187 genes, with 27 genes harboring two or more DMRs, including 114 protein-coding genes, 65 ncRNA genes and eight pseudogenes. They first took a closer look at the protein-coding genes. As methylations of CpGs in promoter regions of coding genes are commonly associated with gene transcription, the authors summarized those protein-coding genes associated with DMR(s) located in their promoter regions (. As shown in , there were significant methylation differences in promoter regions of 17 protein-coding genes, suggesting that expression of these genes in human sperm may be associated with DNA methylation. Strikingly, the majority of these genes have been shown to play important roles in the mitotic cell cycle, transcriptional regulation and sperm functions, indicating potential involvement in sperm production. In addition, some of the genes, including IK, MAP1S, XRRA1 and AKIRIN1, also showed high expression in testis, according to previous genomewide studies [Citation57,Citation58]. Importantly, TDRP, which encodes the testis development-related protein, has been reported to function in sperm motility and may play a role in spermatogenesis [Citation59,Citation60]. Since low motility is the main manifestation of asthenozoospermia, TDRP may be an interesting candidate gene that is worth further investigation.
Table 2. Summary of genes harboring differentially methylated regions in their promoter regions.
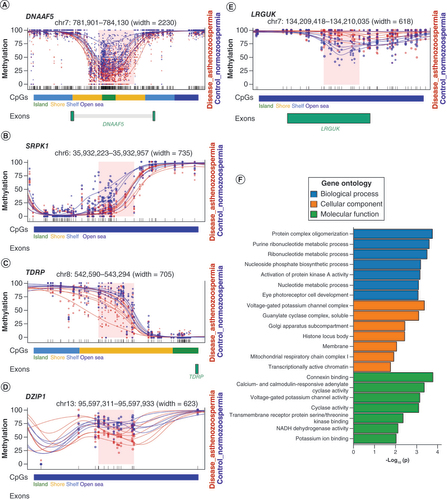
In addition, increasing evidence is supporting the roles of DNA methylation even when they do not show in promoter regions. For instance, it has been reported that some highly expressed genes also show high DNA methylation levels in their gene bodies. In this case, attention should also be paid to those DMRs that locate in gene bodies or intergenic regions. The authors then did a thorough search of all the protein-coding genes in the literature and databases and found that a number of those genes (i.e., LRGUK, DZIP1, DNAAF5, SRPK1 and TDRP) had been directly involved in spermatogenesis or in maintaining sperm functions. For instance, DZIP1 encodes for a centrosome-related protein and its mutation is related to morphological abnormalities of the sperm flagella, which affects the motility of sperm [Citation61]. DNAAF5 is involved in the assembly or stability of axonemal dynein arms, which is also related to the functions of the sperm flagella [Citation62]. These genes are summarized in , and the DMRs regarding these genes are illustrated in A–E.
Table 3. Differentially methylated region(s)-harboring genes involved in sperm functions or spermatogenesis.
(A–E) Plots of five selected DMRs with regard to genes functioning in sperm motility or spermatogenesis. Each dot represents the methylation level of an individual CpG in a single sample, and the size of the dot is representative of coverage. The lines represent smoothed methylation levels for each sample, either normozoospermic controls (blue) or asthenozoospermic samples (red). CpG annotations are shown below the plot. If the DMR is located in the gene body of a gene, the exon of the gene is also shown below. (F) Gene ontology analysis of biological process, cellular component and molecular function of DMR-annotated genes.
DMR: Differentially methylated region.
GO analysis of DMR-associated genes
Apart from those spermatogenesis- or motility-related genes, more than 60 of the DMR-associated genes have been reported to show altered expression in the testis of azoospermic or teratozoospermia patient samples, or to have restricted expression toward testis, suggesting that these genes may also indirectly function in male fertility [Citation57,Citation58]. Some of these genes are involved in transcription regulation, cell proliferation and microtubules, such as MAF and TUBA3E [Citation63–65]. To further investigate all the DMR-associated genes, the authors then performed GO analysis of the biological process, cellular component and molecular function (F). For biological process, the differentially methylated genes were mostly clustered to genes related to protein complex oligomerization, purine ribonucleotide metabolic process, ribonucleotide metabolic process, activation of protein kinase A activity etc. For cellular component, these genes were mostly enriched in genes involved in voltage-gated potassium channel complex. For molecular function, the top three related functions were connexin binding, calcium- and calmodulin-responsive adenylate cyclase activity and voltage-gated potassium channel activity. Notably, cAMP/protein kinase A and calcium pathways are shown to be two general sperm motility-regulating signaling pathways [Citation66,Citation67]. The results of the present study suggest that DNA methylation may also be involved in sperm motility through epigenetic regulation of genes in these two signaling pathways.
Discussion
Asthenozoospermia contributes to a large proportion of subfertility cases and thus affects many couples [Citation1]. While some genetic factors have already been discovered in asthenozoospermia and its resulting male infertility, there are still many pieces missing from the puzzle regarding its etiology [Citation2]. The emerging picture is that DNA methylation may play an important role in human developmental processes relevant to male fertility or spermatogenesis [Citation35,Citation36,Citation39,Citation41]. Several studies examined sperm DNA methylation profiles of specific genes or genomic regions from males with unexplained infertility, varicocele, oligozoospermia and many other types of abnormal sperm conditions [Citation35,Citation39,Citation40,Citation68]. However, these studies only targeted limited gene loci or obtained information from a restricted number of CpG sites on the human genome [Citation69], and few studies have directly investigated sperm DNA methylation in asthenozoospermia. In this study, the authors performed WGBS of sperm from asthenozoospermia patients as well as normozoospermic controls to get a comprehensive map of DNA methylation patterns in both diseased and healthy states. With the single-base resolution method, the authors were able to identify asthenozoospermia-related DMRs and their associated genes. To their knowledge, this is the first study to directly compare sperm DNA methylation patterns from asthenozoospermia and normozoospermic controls and report regional specific changes at a genomewide level.
Consistent with previous studies regarding sperm DNA methylations [Citation35,Citation36,Citation39,Citation44,Citation70], the findings of the present study strengthen the association between DNA methylation and male fertility. Although the global levels of DNA methylation in the two study groups showed no significant difference, the authors were able to identify 238 significant DMRs from the detected methylated CpG sites, indicating the presence of differential DNA methylation in the genomic region from asthenozoospermia patients. Given that clustered CpGs are more likely functionally important in impacting the gene activity during development [Citation71], the present novel analysis thus focused on the genomic regional analysis of showing clustered differential methylation instead of single CpG sites. The data indicated that these DMRs are located most frequently in distal intergenic regions and gene bodies (introns and exons), though nearly 9% of the DMRs were also located near promoters.
These promoter-located DMRs could be further annotated to 17 protein-coding genes, including several cell cycle- or mitosis-related genes such as IK, MAP1S, CHAMP1, TUBGCP5 and PSTPIP1, which is reasonable, considering the importance of mitosis during sperm production. Notably, the gene PSTPIP1 was also reported to overlap differentially methylated CpGs in a previous study comparing sperm methylomes of fertile men and in vitro fertilization patients [Citation36], which coincides with the present study’s results. Further screening and validation of these genes may be promising and rewarding in the field of reproductive research. On the other hand, while these DMRs in promoter regions most likely suggest potential roles in the regulation of relevant gene expression, the DMRs in gene bodies or distal intergenic regions cannot be overlooked, given that increasing evidence is supporting the idea that DNA methylation can function distinctly in different genomic contexts [Citation20].
The authors annotated hundreds of protein-coding genes associated with these asthenozoospermia-related DMRs. Particularly, several sperm-related genes, such as LRGUK, DZIP1, DNAAF5, SRPK1 and TDRP, have been identified [Citation59,Citation61,Citation62,Citation72,Citation73], indicating that expression levels of these genes in sperm may be under the regulation of DNA methylation-dependent mechanisms. For example, TDRP was reported to be expressed in spermatogenic cells and its expression was lower in the testis tissues of azoospermic men [Citation59,Citation60]; DZIP1 had a crucial function in sperm motility and its mutation can lead to morphological abnormalities of the sperm flagella and may induce asthenozoospermia [Citation61]; DNAAF5 also encodes for a protein involved in the function of sperm flagella and thus may affect sperm motility [Citation62]. Apart from these genes with known direct or indirect functions in sperm, there were also a large number of DMR-associated genes showing restricted expression in the testis or altered expression in the testis of patients with abnormal sperm, suggesting that these genes may also be related to spermatozoon development and male fertility [Citation57]. Notably, GO analysis of the DMR-related genes has shown an enrichment in the activation of PKA activity, which is consistent with the previous evidence that the cAMP/protein kinase A pathway is important in regulating sperm motility [Citation66,Citation67]. Taken together, the current findings provide promising targets for further investigation of asthenozoospermia and male infertility.
Most of the current body of research on human spermatozoal DNA methylations is limited, either focusing on semen parameters or simply using array-based assays [Citation35,Citation40]. In terms of asthenozoospermia, only one promoter-targeted sequencing study has identified a few CpGs/DMRs in promoter regions, thus providing only limited information [Citation69]. In addition, the regional analysis of CpGs may provide more prominent functional insights into related genes, compared with a single CpG variation analysis. The present study shows that there are plenty of unique DMRs in asthenozoospermia and that DNA methylation alterations are multifacetedly associated with the abnormality, which may be primarily through the regulation of crucial genes involved in sperm production, either directly or indirectly. These sperm-related genes may be promising candidate targets for further investigation of the etiology of asthenozoospermia, and experimental validation of expressions of these genes in sperm with low motility can provide more solid evidence of these results.
Nevertheless, there are also a few limitations of this study. First, the sample size was relatively small. Practically, collecting sperm samples and extracting DNA are rather difficult to achieve in a short period of time. Above all, it usually takes a much longer time to select and collect suitable samples due to the specialty of semen samples, especially for those asthenozoospermia cases. In addition, the isolation of genomic DNA of mature sperm remains a challenge, since DNA is very tightly packed in sperm and there is no perfect experimental protocol to address this issue. Therefore, considering the accessibility of sperm samples and the loss rate of sperm sample DNA, the authors maximized the utilization of a sperm sample as much as possible to achieve the current results. Certainly, the validation of DMRs may be an indispensable step, which is supposed to take at least a few more months, given the challenges mentioned above. In fact, a large-scale multi-omics (i.e., bisulfite sequencing, RNA sequencing and assay for transposase-accessible chromatin using sequencing) cohort analysis as well as loci-specific methylation status validation experiments on those selected promising target genes will be implemented in the next step of the authors’ research plan.
Second, considering the crosstalk between different layers of epigenetic marks, such as histone modifications, a combination of multi-omics that includes both evidence from gene expression level and other epigenetic levels may provide more detailed insights into the whole picture. Finally, the heterogeneity of individual sperm may cover up some methylation changes that the study failed to detect, since motilities of sperm even from the same individual can vary from each other, which may represent different fates during spermatogenesis. It will be a good idea to separate different fractions of sperm with varied motilities and perform genomewide methylation analysis in future studies.
Conclusion
Our study has provided a novel and detailed analysis of the DNA methylation patterns in asthenozoospermia and normal controls and has identified methylation alterations associated with genes and pathways that may contribute to the abnormal phenotypes as well as infertility. This is the first study to implement whole-genomewide profiling of DNA methylation in asthenozoospermia, as previous studies used either array-based methods or targeted-sequencing methods. Although our current results still lack evidence for direct causation of the disease, the identification of asthenozoospermic-related DMRs and their associated genes is intriguing, and these genomic regions and genes provide promising candidate sites or genes for further investigations of the etiology behind asthenozoospermia and male infertility. Our study has further consolidated DNA methylation as a crucial participant in asthenozoospermia and male infertility. We have successfully identified novel asthenozoospermic-related DMRs and their associated genes, providing promising targets for further investigation of this disease. Importantly, a larger scale, multi-omics study with integrated analysis of both methylation and gene expression data, as well as experimental validation for promising candidate genes, will be performed to investigate the etiology in detail. With the rapid progress of advanced technologies such as scRNA sequencing and single-cell bisulfite sequencing, inspiring findings in this area will surely flourish in the near future and great contributions will also be made to health and medical areas.
There was no significant difference in terms of global DNA methylation levels between normozoospermic and asthenozoospermic sperm.
Regional analysis identified 238 asthenozoospermia-associated differentially methylated regions (DMRs), of which 164 DMRs were hypermethylated and 74 DMRs were hypomethylated.
The 238 DMRs were annotated to 187 genes, which included 114 protein-coding genes, 65 ncRNA genes and eight pseudogenes.
Gene ontology analysis showed clusters of DMR-associated genes related to biological pathways, including protein complex oligomerization, purine ribonucleotide metabolic process, ribonucleotide metabolic process and activation of protein kinase A activity.
Seventeen DMRs were located near promoter regions of corresponding protein-coding genes, suggesting potential regulation of gene expression.
Five DMR-related genes (LRGUK, DZIP1, DNAAF5, SRPK1 and TDRP) have been reported to be directly involved in spermatogenesis or the maintenance of sperm functions, indicating that these genes may be interesting candidates for further investigation.
Author contributions
C Fan, F Li and B Shen conceived and supervised the study. J Li performed DNA extraction, library construction and bisulfite sequencing. J Xu, T Yang and F Li performed sperm collection and semen analysis. J Li, J Chen and C Fan performed data and statistical analyses. J Li and C Fan wrote the manuscript. All authors contributed to and approved the final version of the manuscript.
Ethical conduct of research
The authors state that this work has been approved by all respective institutional research and ethics boards (WHO International Clinical Trials Registry ChiCTR2200056153). The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Supplemental Pdf
Download PDF (1.8 MB)Supplemental Table 1
Download MS Excel (12.2 KB)Supplemental Table 2
Download MS Excel (45.5 KB)Acknowledgments
The authors thank the participants of the study who made this work possible, the medical staff of West China Second University Hospital of Sichuan University and the members of Institutes for Systems Genetics at West China Hospital, Sichuan University.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2217/epi-2022-0122
Financial & competing interests disclosure
This work was supported and funded by the International Cooperation Initiative grant (no. 139190042) from West China Hospital, Sichuan University, Chengdu, China. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was used in the creation of this manuscript.
Additional information
Funding
References
- Agarwal A , BaskaranS , ParekhNet al. Male infertility. Lancet397(10271), 319–333 (2021).
- Tu C , WangW , HuT , LuG , LinG , TanYQ. Genetic underpinnings of asthenozoospermia. Best Pract. Res. Clin. Endocrinol. Metab.34(6), 101472 (2020).
- Cooper TG , NoonanE , Von EckardsteinSet al. World Health Organization reference values for human semen characteristics. Hum. Reprod. Update16(3), 231–245 (2010).
- Toure A , MartinezG , KherrafZEet al. The genetic architecture of morphological abnormalities of the sperm tail. Hum. Genet.140(1), 21–42 (2021).
- Li WN , ZhuL , JiaMM , YinSL , LuGX , LiuG. Missense mutation in DNAJB13 gene correlated with male fertility in asthenozoospermia. Andrology8(2), 299–306 (2020).
- Akbari A , PipitoneGB , AnvarZet al. ADCY10 frameshift variant leading to severe recessive asthenozoospermia and segregating with absorptive hypercalciuria. Hum. Reprod.34(6), 1155–1164 (2019).
- Jaenisch R , BirdA. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet.33 Suppl., 245–254 (2003).
- Bird A . DNA methylation patterns and epigenetic memory. Genes Dev.16(1), 6–21 (2002).
- Yu B , DongX , GravinaSet al. Genome-wide, single-cell DNA methylomics reveals increased non-CpG methylation during human oocyte maturation. Stem Cell Rep.9(1), 397–407 (2017).
- Ehrlich M , Gama-SosaMA , HuangLHet al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res.10(8), 2709–2721 (1982).
- Robert MF , MorinS , BeaulieuNet al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet.33(1), 61–65 (2003).
- Rhee I , BachmanKE , ParkBHet al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature416(6880), 552–556 (2002).
- Kim GD , NiJ , KelesogluN , RobertsRJ , PradhanS. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J.21(15), 4183–4195 (2002).
- Okano M , BellDW , HaberDA , LiE. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell99(3), 247–257 (1999).
- Chodavarapu RK , FengS , BernatavichuteYVet al. Relationship between nucleosome positioning and DNA methylation. Nature466(7304), 388–392 (2010).
- Robertson KD , Ait-Si-AliS , YokochiT , WadePA , JonesPL , WolffeAP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet.25(3), 338–342 (2000).
- Razin A . CpG methylation, chromatin structure and gene silencing – a three-way connection. EMBO J.17(17), 4905–4908 (1998).
- Henckel A , NakabayashiK , SanzLA , FeilR , HataK , ArnaudP. Histone methylation is mechanistically linked to DNA methylation at imprinting control regions in mammals. Hum. Mol. Genet.18(18), 3375–3383 (2009).
- Luo C , HajkovaP , EckerJR. Dynamic DNA methylation: in the right place at the right time. Science361(6409), 1336–1340 (2018).
- Moen EL , MarianiCJ , ZullowHet al. New themes in the biological functions of 5-methylcytosine and 5-hydroxymethylcytosine. Immunol. Rev.263(1), 36–49 (2015).
- Yang X , HanH , DeCarvalho DD , LayFD , JonesPA , LiangG. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell26(4), 577–590 (2014).
- Maunakea AK , NagarajanRP , BilenkyMet al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature466(7303), 253–257 (2010).
- Smith ZD , MeissnerA. DNA methylation: roles in mammalian development. Nat. Rev. Genet.14(3), 204–220 (2013).
- Meissner A , MikkelsenTS , GuHet al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature454(7205), 766–770 (2008).
- Oakes CC , LaSalle S , SmiragliaDJ , RobaireB , TraslerJM. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev. Biol.307(2), 368–379 (2007).
- Smallwood SA , KelseyG. De novo DNA methylation: a germ cell perspective. Trends Genet.28(1), 33–42 (2012).
- Schisterman EF , SjaardaLA , ClemonsTet al. Effect of folic acid and zinc supplementation in men on semen quality and live birth among couples undergoing infertility treatment: a randomized clinical trial. JAMA323(1), 35–48 (2020).
- Veron GL , TisseraAD , BelloRet al. Impact of age, clinical conditions, and lifestyle on routine semen parameters and sperm kinematics. Fertil. Steril.110(1), 68–75 e64 (2018).
- Salas-Huetos A , BulloM , Salas-SalvadoJ. Dietary patterns, foods and nutrients in male fertility parameters and fecundability: a systematic review of observational studies. Hum. Reprod. Update23(4), 371–389 (2017).
- Sharma R , HarlevA , AgarwalA , EstevesSC. Cigarette smoking and semen quality: a new meta-analysis examining the effect of the 2010 World Health Organization laboratory methods for the examination of human semen. Eur. Urol.70(4), 635–645 (2016).
- Evans HJ , FletcherJ , TorranceM , HargreaveTB. Sperm abnormalities and cigarette smoking. Lancet1(8221), 627–629 (1981).
- Senaldi L , Smith-RaskaM. Evidence for germline non-genetic inheritance of human phenotypes and diseases. Clinical Epigenetics12(1), 136 (2020).
- Weigmann K . Lifestyle in the sperm: there is growing evidence that epigenetic marks can be inherited. But what is the nature of the information they store and over how many generations do they prevail?EMBO Reports15(12), 1233–1237 (2014).
- Siddeek B , MauduitC , SimeoniU , BenahmedM. Sperm epigenome as a marker of environmental exposure and lifestyle, at the origin of diseases inheritance. Mutat. Res. Rev. Mutat. Res.778, 38–44 (2018).
- Urdinguio RG , BayónGF , DmitrijevaMet al. Aberrant DNA methylation patterns of spermatozoa in men with unexplained infertility. Hum. Reprod.30(5), 1014–1028 (2015).
- Aston KI , UrenPJ , JenkinsTGet al. Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertil. Steril.104(6), 1388–1397.E5 (2015).
- Jenkins TG , AstonKI , PfluegerC , CairnsBR , CarrellDT. Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLOS Genetics10(7), e1004458 (2014).
- Jenkins TG , JamesER , AlonsoDFet al. Cigarette smoking significantly alters sperm DNA methylation patterns. Andrology5(6), 1089–1099 (2017).
- Jenkins TG , AstonKI , MeyerTDet al. Decreased fecundity and sperm DNA methylation patterns. Fertil. Steril.105(1), 51–57e51–e53 (2016).
- Santana VP , JamesER , Miranda-FurtadoCLet al. Differential DNA methylation pattern and sperm quality in men with varicocele. Fertil. Steril.114(4), 770–778 (2020).
- Marques PI , FernandesS , CarvalhoF , BarrosA , SousaM , MarquesCJ. DNA methylation imprinting errors in spermatogenic cells from maturation arrest azoospermic patients. Andrology5(3), 451–459 (2017).
- Louie K , MinorA , NgR , PoonK , ChowV , MaS. Evaluation of DNA methylation at imprinted DMRs in the spermatozoa of oligozoospermic men in association with MTHFR C677T genotype. Andrology4(5), 825–831 (2016).
- Wu X , LuoC , HuLet al. Unraveling epigenomic abnormality in azoospermic human males by WGBS, RNA-Seq, and transcriptome profiling analyses. J. Assist. Reprod. Genet.37(4), 789–802 (2020).
- Laqqan M , TierlingS , AlkhaledY , LoPorto C , SolomayerEF , HammadehM. Spermatozoa from males with reduced fecundity exhibit differential DNA methylation patterns. Andrology5(5), 971–978 (2017).
- Boissonnas CC , AbdalaouiHE , HaelewynVet al. Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile men. Eur. J. Hum. Genet.18(1), 73–80 (2010).
- He W , SunY , ZhangSet al. Profiling the DNA methylation patterns of imprinted genes in abnormal semen samples by next-generation bisulfite sequencing. J. Assist. Reprod. Genet.37(9), 2211–2221 (2020).
- Sujit KM , SinghV , TrivediS , SinghK , GuptaG , RajenderS. Increased DNA methylation in the spermatogenesis-associated (SPATA) genes correlates with infertility. Andrology8(3), 602–609 (2020).
- Navarro-Costa P , NogueiraP , CarvalhoMet al. Incorrect DNA methylation of the DAZL promoter CpG island associates with defective human sperm. Hum. Reprod.25(10), 2647–2654 (2010).
- Barney R , StalkerK , LutesA , BaylesA , AstonK , JenkinsT. Assessment of seminal cell-free DNA as a potential contaminate in studies of human sperm DNA methylation. Andrology10(4), 702–709 (2022).
- Krueger F . Trim Galore. https://github.com/FelixKrueger/TrimGalore
- Krueger F , AndrewsSR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics (Oxford, England)27(11), 1571–1572 (2011).
- Tarasov A , VilellaAJ , CuppenE , NijmanIJ , PrinsP. Sambamba: fast processing of NGS alignment formats. Bioinformatics (Oxford, England)31(12), 2032–2034 (2015).
- Hansen KD , LangmeadB , IrizarryRA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome biology13(10), R83 (2012).
- Yu G , WangLG , HeQY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics (Oxford, England)31(14), 2382–2383 (2015).
- Korthauer K , ChakrabortyS , BenjaminiY , IrizarryRA. Detection and accurate false discovery rate control of differentially methylated regions from whole genome bisulfite sequencing. Biostatistics (Oxford, England)20(3), 367–383 (2019).
- Gu Z . rGREAT: Client for GREAT Analysis (2021). https://github.com/jokergoo/rGREAT, http://great.stanford.edu/public/html/
- Spiess AN , FeigC , SchulzeWet al. Cross-platform gene expression signature of human spermatogenic failure reveals inflammatory-like response. Hum. Reprod.22(11), 2936–2946 (2007).
- Platts AE , DixDJ , ChemesHEet al. Success and failure in human spermatogenesis as revealed by teratozoospermic RNAs. Hum. Mol. Genet.16(7), 763–773 (2007).
- Mao S , WuF , CaoXet al. TDRP deficiency contributes to low sperm motility and is a potential risk factor for male infertility. Am. J. Transl. Res.8(1), 177–187 (2016).
- Wang X , JiangH , ZhouWet al. Molecular cloning of a novel nuclear factor, TDRP1, in spermatogenic cells of testis and its relationship with spermatogenesis. Biochem. Biophys. Res. Commun.394(1), 29–35 (2010).
- Lv M , LiuW , ChiWet al. Homozygous mutations in DZIP1 can induce asthenoteratospermia with severe MMAF. J. Med. Genet.57(7), 445–453 (2020).
- Zariwala MA , KnowlesMR , LeighMW. Primary ciliary dyskinesia. In: GeneReviews.AdamMP, ArdingerHH, PagonRAet al.et al. ( Eds). University of Washington, WA, USA (1993).
- Zhu X , DuY , LiDet al. Aberrant TGF-beta1 signaling activation by MAF underlies pathological lens growth in high myopia. Nat. Commun.12(1), 2102 (2021).
- Hutchins JR , ToyodaY , HegemannBet al. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science328(5978), 593–599 (2010).
- Hurt EM , WiestnerA , RosenwaldAet al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell5(2), 191–199 (2004).
- Finkelstein M , EtkovitzN , BreitbartH. Ca(2+) signaling in mammalian spermatozoa. Mol. Cell. Endocrinol.516, 110953 (2020).
- Martinez-Leon E , Osycka-SalutC , SignorelliJet al. Fibronectin stimulates human sperm capacitation through the cyclic AMP/protein kinase A pathway. Hum. Reprod.30(9), 2138–2151 (2015).
- Jenkins TG , AstonKI , HotalingJM , ShamsiMB , SimonL , CarrellDT. Teratozoospermia and asthenozoospermia are associated with specific epigenetic signatures. Andrology4(5), 843–849 (2016).
- Du Y , LiM , ChenJet al. Promoter targeted bisulfite sequencing reveals DNA methylation profiles associated with low sperm motility in asthenozoospermia. Hum. Reprod.31(1), 24–33 (2016).
- Camprubí C , Salas-HuetosA , Aiese-CiglianoRet al. Spermatozoa from infertile patients exhibit differences of DNA methylation associated with spermatogenesis-related processes: an array-based analysis. Reprod. Biomed. Online33(6), 709–719 (2016).
- Jones PA . Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Rev. Genetics13(7), 484–492 (2012).
- Liu Y , DeboerK , DeKretser DMet al. LRGUK-1 is required for basal body and manchette function during spermatogenesis and male fertility. PLOS Genetics11(3), e1005090 (2015).
- Papoutsopoulou S , NikolakakiE , ChalepakisG , KruftV , ChevaillierP , GiannakourosT. SR protein-specific kinase 1 is highly expressed in testis and phosphorylates protamine 1. Nucleic Acids Res.27(14), 2972–2980 (1999).