
Abstract
As a link between a stable genome and a dynamic environment, epigenetics is a promising tool for mapping age-related changes in human DNA. Methylated cytosine changes at specific loci are generally less studied in sperm DNA than in somatic cell DNA. Age-related methylation changes can be connected to various reproductive health problems and multiple disorders in offspring. In addition, they can be helpful in forensic fields, where testing of specific loci in semen samples found at sexual assault crime scenes can predict a perpetrator’s age and narrow down the police investigation. This review focuses on age-related methylation changes in sperm. It covers the biological role of methylation, methylation testing techniques and the implications of methylation changes in forensics and clinical practice.
Plain language summary
DNA methylation is a biological process that can change the activity of a gene without changing its sequence. We do not know much about DNA methylation in sperm and what changes methylation undergoes during the lifespan. These changes can, however, be important both for health and solving crimes. Presperm cells renew themselves, which gives rise to new sperm cells, from youth to death, accumulating cell divisions prone to error. This is why sperm cells are affected by age more than nondividing eggs. Methylation is specific in different tissues of the body. The ratio between number of sperm cells, white blood cells, and other cell types is highly variable and hardly predicted, which may distort the results. Clinical studies have shown that older fathers have worse reproductive health. Their children can develop metabolic, neurological and behavioral disorders. This also applies to younger men whose DNA methylation pattern is similar to that of older men. Methylation changes allow us to build a model capable of predicting the age of an unknown person with a mean error of about 5 years. This can be helpful for police investigators in cases of sexual assault, when biological material is found but there is no match in the police database.
Tweetable abstract
A number of methyl groups on DNA changes with age. In sperm DNA, these changes can influence the successful conception and health of offspring or help police find an unknown criminal offender by predicting their age.
Previously, it was believed that sperm had little or no contribution to conception beyond the safe delivery of paternal nuclear DNA to the oocyte. Recently, evidence has arisen that the sperm epigenome plays an important role in embryonic development through gene imprinting and coding-specific RNA [Citation1,Citation2]. Also, it was found that an older age of the father negatively affects reproductive health outcomes that were previously attributed only to an older age of the mother [Citation3]. Although age-related changes in DNA methylation in somatic cells have been extensively studied, especially since the publication of “epigenetic clocks” by Hannum et al. [Citation4] and Horvath [Citation5], the impact of aging on the methylation profile of germ cells is much less known, and existing studies of germ cell methylation are mostly focused on known imprinted regions [Citation2,Citation3].
Although methylation of oocytes has only reproductive consequences, methylation of sperm can also be used forensically since ejaculate is the primary biological material used for perpetrator identification in sexual assault cases. In this review, we describe the biological processes behind DNA methylation in sperm cells in comparison with somatic cells as well as some state-of-the-art forensic applications of age estimation from sperm methylation profiles.
The role of DNA methylation
A correct methylation signal is crucial for maintaining proper gene expression control and is one of the tools for cell type specification [Citation6]. Methylation can silence gene expression in two ways: by physically preventing the binding of transcription factors to the promoter and by participating in shaping chromatin into a form such that the DNA is not open to transcription [Citation7]. Proper methylation and subsequent silencing of specific genes in embryonic totipotent stem cells are two of the factors sending individual cell types on their specific developmental pathways and restricting their proliferative potential [Citation8]. The dynamic character of DNA methylation helps facilitate cellular adaptation to a changing environment [Citation6].
Defects in proper methylation signals can lead to developmental defects and carcinogenesis, with global hypomethylation and local hypermethylation being two of the hallmarks of cancer [Citation7]. This can promote insertional mutagenesis by dysregulation of retrotransposons or tumorigenesis by the overexpression of oncogenes and silencing of tumor suppressors. In sperm cells, an aberrant DNA methylation signal can result in improper chromatin condensation in the sperm head and abnormal gene expression, leading to infertility [Citation9].
Development of sperm DNA methylation signals
Although oocytes are in a state of meiotic arrest, spermatogonial cells have a self-renewing potential and divide continuously, giving rise to differentiating daughter cells. Until the beginning of puberty, they undergo approximately 35 divisions, and this number increases roughly every 2 or 3 weeks to more than 800 replication cycles at the age of 50 years [Citation2,Citation10]. This could represent an evolutionary benefit, producing a more diverse population of germ cells, but each replication cycle brings a higher risk of error, not only during replication of the DNA sequence but also during the replication of epigenetic markers. Because replication mechanisms have an error rate at least one order of magnitude higher for epigenetic information compared with genetic information, spermatozoa from older men are burdened with more epigenetic than sequence changes. Epigenetic markers are known to be heritable, but the methylation profiles of the sperm fertilizing an egg and the mature sperm of this new individual are separated by several waves of extensive methylation reprogramming (), allowing the establishment of a specific male methylation profile [Citation11].
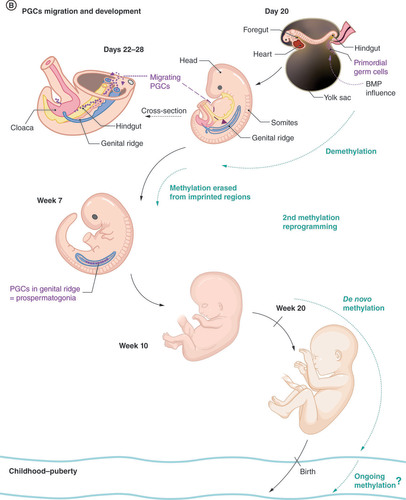
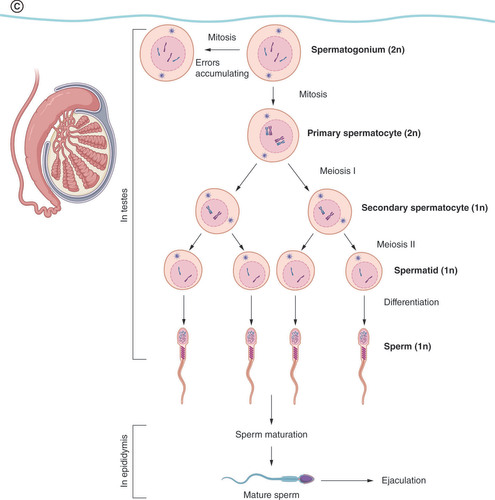
(A) Preimplantation and germ layer formation stage. In the Day 13, bilaminar disc formation is shown only in close-up for the sake of clarity. The rest of the structures are similar to the Day 12 stage. Ectodermal (red), mesodermal (orange), and entodermal (blue) structures are coded in the same color in each stage. (B) Second half of embryonic development. Embryo in the Days 22–28 stage is shown in the overview and cross-section to better demonstrate the migration route of PGCs as they pass the hindgut to the genital ridge. In the Week 7 stage, the digestive system is not shown for the sake of clarity. (C) Development of mature sperm cells in puberty. Prospermatogonia are called spermatogonia postnatally and have self-renewing function, providing a constant pool of precursor cells throughout the male lifetime.
BMP: Bone morphogenetic protein; PGC: Primordial germ cell.
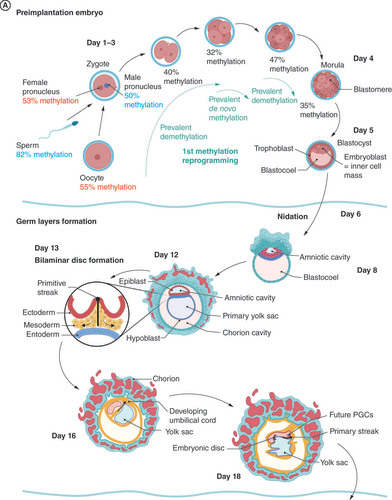
The first global remodeling of the methylation profile takes place 10–12 h after fertilization, and although it is mostly understood as demethylation, single-cell sequencing shows that it is a dynamic process characterized by massive global methylation erasure and local remethylation () [Citation2,Citation12,Citation13]. Demethylation goes from around 80% in the sperm to around 50% in the male pronucleus in the zygote, where enhancers and gene body regions, particularly, are demethylated [Citation12]. Methylation erasure continues during the first cell division and the formation of morula, when introns and short interspersed nuclear elements, especially Alu elements, are significantly enriched.
Additionally, de novo methylation starts during preimplantation development and is more prevalent than demethylation in the development of the eight-cell stage embryo. Potentially active repeat elements (i.e., short interspersed nuclear elements, long interspersed nuclear elements and long terminal repeats) are methylated to protect genome stability [Citation12].
The ratio between methylation decrease (blue) and methylation increase (red) taking place during development from one stage to another is shown in the bar graph. Numerical values correspond to the percentage of methylated genome, and values highlighted in orange and purple correspond to the female and male pronucleus, respectively.
Data taken from [Citation12].
![Figure 2. First wave of global epigenetic reprogramming. The ratio between methylation decrease (blue) and methylation increase (red) taking place during development from one stage to another is shown in the bar graph. Numerical values correspond to the percentage of methylated genome, and values highlighted in orange and purple correspond to the female and male pronucleus, respectively.Data taken from [Citation12].](/cms/asset/b9b6221d-53c3-48c2-b845-ce56311072d7/iepi_a_12324502_f0005.jpg)
Demethylation of the paternal genome is much faster and more profound than that of the maternal genome [Citation12]. Despite starting with higher methylation levels (82% methylation compared with 55% methylation of the oocyte), the average methylation of the paternal genome drops lower than that of the maternal genome in the two-cell stage of the embryo (15% of the paternal genome is methylated compared with 23% of the maternal genome). This difference also persists in the postimplantation embryo, although it is lower.
Analysis of differentially methylated regions (DMRs) – namely, analysis of genomic regions with different methylation statuses among a set of samples, such as different tissues, different individuals, different time points for an individual and diseased and healthy samples, depending on the context of the study – shows that methylation of the paternal genome is enriched in somatic cell-specific enhancers, whereas methylation of the maternal genome is enriched in CpG islands and gene promoters. In both genomes, short interspersed nuclear elements, especially Alu repeats, are methylated.
At the beginning of their development, primordial germ cells (PGCs) are epigenetically identical to other cells of the epiblast, showing stable values of DNA methylation and X-inactivation [Citation2]. Under the influence of bone morphogenetic protein, PGCs undergo a second wave of DNA methylation remodeling, with almost complete erasure of DNA methylation. During this process, PGCs start to migrate past the hindgut to the genital ridge, where DNA methylation is also erased from imprinting control regions and meiotic genes [Citation14]. High levels of DNA methylation are conserved in only several specific genomic regions (termed ‘escapees’) generally connected with genes expressed in the brain and associated with neurological and metabolic disorders, implying the possible transgenerational inheritance of epigenetic markers in these areas.
Under the influence of the SRY protein, somatic cells of the genital ridge undergo sex determination for the development of male gonads, differentiating into Sertoli cells. Subsequently, PGCs become developmentally restricted to the male sex, becoming gonocytes, also called prospermatogonia [Citation15]. Sex-specific subpopulations of cells can be detected from week 9 [Citation14]. By the seventh to tenth week of embryonic development, the global level of methylation in prospermatogonia is 5.0–8.0%, whereas somatic cells are mostly hypermethylated, with a mean methylation of 67.5–80.0% [Citation2,Citation11,Citation14,Citation16].
The window for extreme hypomethylation is quite long in human male germ cell precursors compared with, for example, those in mice. The process of de novo methylation starts in prospermatogonia in mitotic arrest around the 20th week, but because of developmental asynchrony, hypomethylated prospermatogonia may be found until the 26th week [Citation11,Citation14]. De novo methylation starts in the DNA repetition areas, especially retrotransposons and imprinted genes, creating a sex-specific methylation profile of germline cells [Citation17]. DNA transposons and long interspersed nuclear elements are significantly overrepresented among DMRs in younger and older men [Citation18]. It is unclear at which point we could consider the methylation signal established. Global methylation levels seem not to change significantly after birth, whereas spermatogonia remain in mitotic arrest [Citation17], but some studies [Citation11] suggest that this process continues after birth and is not completed until the start of puberty. More research is needed to investigate this postnatal phase of germ cell development.
Spermatogenesis itself starts in puberty, and prominent epigenetic changes include chromatin packaging and the exchange of the majority of histones for protamines. The number of nucleosomes bound to histones in humans is around 12.5% [Citation2,Citation19]. The areas conserved on histones are mostly CpG rich and contain promoters of developmentally important genes, imprinted areas and genes that code for miRNA [Citation19]. Spermatogonia can mitotically either self-renew or give rise to a primary spermatocyte entering meiosis, producing four spermatids from one primary spermatocyte [Citation20]. During the final stage of spermatogenesis (called spermiogenesis), the structure of the round spermatid polarizes, an acrosomal cap and a tail develop and an early sperm cell is oriented to the epididymis, where it undergoes a maturation process, finally acquiring the capacity to fertilize an egg.
Interesting question is whether there is a possibility of a third wave of methylation reprogramming during post-testicular sperm maturation, as the sperm enters caput epididymis. To further investigate this possibility, Galan et al. studied seven stages of developing sperm in mice and reported modest global hypomethylation in caput epididymis sperm, which was not observed in the preceding or succeeding developmental stages [Citation13]. Cell-free DNA, presumably derived from the somatic cells of the epididymis, was identified as the source of this shift in methylation profiles, emphasizing the differences between somatic and germline profiles. Interestingly, this cell-free material was found only in virgin mice and disappeared after mating.
Errors in the erasure or re-establishment of DNA methylation signals in individual PGCs may lead to the development of spermatogonia that produce, via clonal division, a subpopulation of sperm with defective methylation profiles [Citation11]. Incomplete methylation of retrotransposons may also lead to their uncontrolled propagation throughout the genome, cause insertional mutagenesis and promote carcinogenesis [Citation17]. Although the continuous division of spermatogonia creates and accumulates more epigenetic than sequencing errors, waves of reprogramming partly prevent their passing on to another generation.
A study by Jenkins et al. found no difference between patients with younger and older grandparents regarding levels of global methylation and only subtle changes in selected age-related regions, suggesting that age-related changes do not persist for two generations [Citation21]. This is consistent with a study by Leitão et al., who compared almost 100 patients with severely impaired spermatogenesis with healthy controls and detected no recurrent epigenetic mutations [Citation11]. By contrast, it has already been shown that epigenetic changes caused by other factors (e.g., exposure to pollutants such as the pesticide dichlorodiphenyltrichloroethane) obtain imprinting-like features, evading methylation erasure and creating an abnormal epigenome in embryonic stem cells, affecting all forming somatic cells and tissues and subsequently producing altered germline cells [Citation20].
Sperm cell sampling
Mature sperm cells capable of fertilization are stored in the epididymides. Their concentrated suspension is diluted by prostatic fluid, and subsequently seminal vesicle fluid in the urethra, to form the ejaculate during sexual activity [Citation22]. Considering this anatomical succession, sequential ejaculate fractions are not equal, and their composition is influenced by circumstances such as abstinence time before ejaculation as well as the level and/or duration of sexual arousal, which affects the smooth muscle cell contractions emptying glands and actively transporting spermatozoa. Samples collected in a container in a laboratory setting can thus have different compositions compared with the individual fractions ejaculated during sexual intercourse. depicts the proportional composition of human semen; however, the literature sources vary, so this information cannot be considered definitive.
Values are listed as percentages.
Data taken from [Citation23–25].
![Figure 3. Composition of human semen. Values are listed as percentages.Data taken from [Citation23–25].](/cms/asset/29ef112d-9d55-4e78-bc56-3805c18c7c8b/iepi_a_12324502_f0006.jpg)
Human semen consists of a 2–5% cellular fraction, whereas 95–98% is a seminal plasma rich in sugars, glycans, lipids, ions, metabolites, cell-free DNA, miRNAs, peptides and proteins secreted by the seminal vesicles, prostate, epididymides and bulbourethral glands [Citation23]. Of semen’s cellular fraction, Long et al. report that approximately 85% is allotted to sperm cells and 84% of the rest is progenitor sperm cells [Citation24]. Although the authors also report the proportion of white blood cells in semen to be around 13%, there is no official consensus defining the physiological ratio of leukocytes to other cell types in semen [Citation22]. During genitourinary tract or accessory gland infections such as prostatitis or other inflammatory processes, the proportion of leukocytes rises. A higher proportion of immature germ cells in semen might occur during various developmental disorders of the genitourinary tract, with a varicocele or as an adverse effect during cytotoxic treatments [Citation24]. The remaining portion of nonsperm cells consists of squamous epithelial cells from the excretory ducts, epithelial cells from the seminal vesicles and exfoliated Sertoli cells [Citation25]. In addition, the amount of these cells can rise during seminal vesiculitis.
Differences between somatic&germ cell methylation
Contamination of somatic DNA in sperm samples is described as one of the major confounding factors in sperm methylation studies [Citation26]. The set of 217 DMRs, potentially useful as diagnostic markers of male idiopathic infertility published by Luján et al. [Citation27], was unable to distinguish normozoospermic from oligozoospermic samples in independent testing by Leitão et al. [Citation11] but pinpointed samples containing somatic DNA among clean sperm samples. In the study by Leitão et al., 2462 genes showed differential expression between blood and sperm samples, with promoters of these genes being mostly methylated in blood and unmethylated in sperm.
Although the epigenetic clock by Horvath is described as a multitissue model [Citation5], it cannot be used for age prediction using testicular tissue and sperm cells [Citation1]. When tested on a small set of semen samples, the mean absolute error (MAE) of the prediction was 13.3 years compared with an MAE between 4 and 5 years for somatic tissues [Citation28]. Testing of another smaller three-CpG blood-based prediction model yielded an MAE of 37.3 years [Citation29]. This confirms that the loci used for age prediction in somatic cells have no predictive value in sperm [Citation30].
Global hypomethylation with regional gene-associated hypermethylation is considered an important feature of aging in somatic cells. In sperm DNA, the opposite situation was identified, with significant global hypermethylation and specific hypomethylated loci [Citation1,Citation19,Citation21]. Hypermethylation has been associated with CpG islands and hypomethylation with CpG shores in sperm DNA.
Frequently dividing cells are burdened with accumulating epigenetic mutations, or epigenetic drift [Citation31]. However, models that successfully predict age from various tissues, such as the clocks of Hannum et al. [Citation4] and Horvath [Citation5], confirm that epigenetic age does not measure proliferation rate, as the included tissues have variable – or even zero (e.g., neuronal cells) – proliferation rates [Citation32].
Although the average methylation change at a single CpG in somatic cells is 0.05–0.15% per year, Jenkins et al. reported a stronger effect of age on methylation in sperm cells, with an average change of >0.4% per year in 39 genomic regions [Citation1]. In these especially altered regions, age-related changes can be within the range of 10–12% during the male reproductive lifespan. By contrast, according to Heidegger et al., the predictive effect of an individual CpG in sperm cells is smaller than in somatic cells (R2 0.34–0.35 in sperm vs 0.66–0.68 in blood) [Citation33]. The diverse character of these studies (one worked with 1 kbp long genomic windows [Citation3], whereas the other used single CpGs [Citation30]) may contribute to these contradictory results, but further and more uniform research is needed.
The magnitude of DNA methylation changes in sperm DNA
In a study by Oluwayiose et al. [Citation3], 1698 CpGs were associated with age in tested sperm DNA, 91% of which showed an increase in methylation of 0.2–1.7% for every 5 years of age [Citation3]. Among this set of CpGs, none overlapped with the epigenetic clocks of Hannum et al. [Citation4] and Horvath [Citation5] based on somatic cells and only 30% overlapped with the model developed by Jenkins et al. [Citation50] for chronological age prediction from sperm DNA methylation.
When wider DMRs were considered, 1146 were detected by Oluwayiose et al. [Citation3]. In contrast to individual CpG methylation, only 57% DMRs showed hypermethylation, and 59% in another similarly designed study by Pisarek et al. [Citation34]. A study by Bernhardt et al. detected a similar number of DMRs (1565), but the ratio between hypermethylated and hypomethylated was reversed, with only 26% of DMRs hypermethylated [Citation10]. In that study, hypermethylated regions were mostly associated with biological processes connected to synapsis and abnormal behavior, whereas the majority of hypomethylated areas were enriched in male infertility phenotypes. This may be one of the reasons behind age-associated idiopathic male infertility. The results of these studies are summarized in .
Table 1. Number of CpGs and age-associated differentially methylated regions and percentage of loci hypermethylated with higher age in included studies.
Clinical impacts of age-related epigenetic sperm changes
Age-related changes in DNA methylation patterns are associated with three main reproductive health outcomes: lower probability of successful conception, both naturally and with assisted reproduction methods; higher risk of abnormal embryonic and fetal development; and development of various disorders in offspring [Citation3]. As these are all complex multifactorial processes that include genetic and epigenetic influences, we could expect that the influence of paternal age comprises a higher number of small-scale changes, rather than just one or two highly penetrating epigenetic variants [Citation10].
The study by Oluwayiose et al., which looked at 47 infertile couples, identified characteristic CpG hypermethylation patterns in the oldest fathers compared with the youngest [Citation3]. Results from two intermediate age groups suggested a dose-dependent impact of age-related methylation changes on successful conception. The linear relationship between age and methylation was confirmed in another study [Citation2].
In the study by Oluwayiose et al., successful live birth was reported in 57% of couples in the youngest fathers group, 35% of couples in the intermediate group and none of the couples in the oldest fathers group [Citation3]. Among the younger patients with methylation patterns corresponding to the older group’s pattern, there were also no successful live births. The probability of conception decreased by 4% each year. Pilsner et al. reported the same trend, with a 17% lower cumulative probability of achieving pregnancy for couples with a predicted older father, together with a longer time to achieving pregnancy and gestational age of the developing embryo [Citation30].
Age-related changes in sperm DNA methylation are located mostly in the area of embryonic development and neurological development genes [Citation3,Citation19,Citation35,Citation36]. The most enriched ontology terms include flavonoid glucuronidation, transmission on chemical synapses, histone ubiquitination [Citation30], opioid and neuronal nitric oxide synthase signaling pathways [Citation19], and DNA binding [Citation18]. Also, homeobox genes were significantly enriched, specifically between hypomethylated regions [Citation18].
Age-associated methylation changes were reported in genes connected to diabetes mellitus [Citation1], hypertension [Citation1], autism spectrum disorder [Citation1,Citation19], bipolar disorder [Citation1,Citation19], schizophrenia [Citation1,Citation19] and other neurological and developmental disorders, especially those affecting the cerebellum (e.g., ataxia and general learning disabilities) [Citation35]. The same effect was experimentally demonstrated in mice, where DNA methylation changes in the sperm of older males manifested at the level of gene expression in areas associated with schizophrenia and bipolar disorder as well as at the behavioral level [Citation3]. Fewer exploratory activities, lower response rates to acoustic startle signals and decreased cognitive functions were also observed in the offspring of older males [Citation35]. This effect was preserved even in the next generation, with hypomethylation persisting in 75% of the offspring of older grandfathers [Citation1].
Most of differentially methylated genes reported in several comparable studies are associated with embryonic neural development [Citation11]. However, there is a little overlap in tangible genes. Of the seven studies reviewed by Bernhardt et al., only three genes – namely, DLGAP2, PRDM16 and SLC22A18AS – were reported in at least five studies, with 90% of the reported genes included in only the original study [Citation10].
Age-induced DNA methylation changes could become a useful diagnostic tool. Array-based DNA methylation profiles from blood samples can highlight differences between infertile men and fertile controls [Citation17]. The previously mentioned studies have shown that the methylation of sperm DNA can be even more informative. Candidate genes, which can be considered epigenetic markers of lower fertility and higher risk of neurodevelopmental disorders in offspring, are starting to emerge.
Forensic applications of age-related epigenetic sperm changes
Age prediction from DNA is important in the field of forensics. The ability to predict an unknown person’s different phenotype traits based on their DNA could be an important forensic tool. This can provide information in cases where biological traces have been found but comparing DNA profiles with police databases or suspect profiles yields no match. Various visible traits, such as age, pigmentation, occurrence of freckles and premature hair graying or balding, together with lifestyle traits, such as diet, exercise, smoking and alcohol or drug consumption, can help to significantly narrow down the pool of potential suspects. Age prediction is currently one of the most pursued tools of DNA phenotyping [Citation37]. Forensic age prediction from blood samples has been deeply studied [Citation38,Citation39], but we lack a predictive model for other forensically relevant tissues, such as semen samples, which are often found after sexual assaults [Citation28,Citation34].
Age prediction models
The majority of studies researching age-related changes in DNA methylation of semen samples use whole genome sequencing or array-based approaches as the starting point ().
Table 2. Summary of methods used as a starting point in studies of age-related methylation changes in semen samples.
The consistency between chronological age and methylation in specific loci suggests the existence of regions more prone to epigenetic changes, but there is only a little overlap between the most informative and predictive loci in individual studies. Different predictive models with similar outcomes use different loci, making it impossible to identify a set of the most informative. Pilsner et al. built two independent age prediction models, one based on individual CpGs and the other based on the entire DMRs [Citation30]. Prediction errors were very similar, but of the 318 CpGs included in the DMR-based model, only ten (less than 1%) overlapped with the 120 CpGs included in the CpG-based model. From a practical point of view, the authors decided to use the CpG-based model for further clinical testing, as it requires fewer loci and minimizes the cost of the array, but it is possible that both models have an independent association with reproductive health outcomes.
For clinically focused prediction models, it can be advantageous to use a large number of markers to make the resulting model as precise as possible even if the effect of individual loci on the predicted age is low. However, these models are generally inapplicable for forensic practices, where the generally low quality and quantity of available DNA are the biggest limiting factors [Citation33,Citation34,Citation40].
Clinically and forensically focused models also selected their most informative CpGs based on different criteria. Although for medical purposes age acceleration or deceleration compared with the real chronological age is the key factor in predicting health-related outcomes [Citation1,Citation3], forensic predictors must be focused on chronological age itself, with a minimum influence of other lifestyle, environmental and health factors.
The first forensically focused prediction model using sperm DNA methylation was developed by Lee et al. using the three most informative CpGs identified with a 450K Infinium array (Illumina, CA, USA) in 12 semen samples [Citation28]. The model itself was constructed on a set of 68 samples (31 as the training set and 37 as the testing set, with an MAE of 4.2 and 5.4 years, respectively), and after retraining with the whole set, it reached an MAE of 4.7 years. These results were confirmed by an independent validation study, which found an MAE of 4.8 years for 12 volunteer samples and 5.2 years for 19 real-life casework samples, with various methods of sampling and a storage time from 3 months to 17 years [Citation41].
Although the prediction model developed by Lee et al. was based on the single-base extension method [Citation28] and a newer study by Pisarek et al. used next-generation sequencing [Citation34], the resulting performance of both models was similar, with an MAE of 4.3 and 5.1 years in the training and testing data sets, respectively. Lee´s prediction model applied to Pisarek´s samples resulted in an MAE of 5.7 years. This difference can be explained by the use of different technology as well as interpopulation variability in DNA methylation, considering that these studies were conducted in South Korea and Poland, respectively.
Despite Pisarek et al. testing three CpGs [Citation34] from the model developed by Lee et al. [Citation28], together with their top hits from the 850K EPIC array (Illumina, CA, USA), only one was included in the final six-CpG prediction model (NOX4, also known as FOLH1B) (). This final model, along with a wider panel comprising 13 loci and 36 CpGs, was validated by five laboratories from the VISAGE Consortium, which assessed its reproducibility and sensitivity [Citation33]. For two mock crime scene samples, the average difference between predicted and chronological age was 2.0 and 5.4 years.
Table 3. Markers used for individual prediction models.
A variety of blood-based age prediction studies describe the phenomenon of decreasing prediction sensitivity with increasing age [Citation42–44]. A validation study by Lee et al. confirmed this for sperm DNA as well [Citation41]. Although the general MAE was 4.8 years, the mean error increased from 2.9 years for individuals in their 20s to 7.2 years for individuals in their 50s. The results and parameters of the aforementioned studies are summarized in .
Table 4. Comparison of performance of various prediction models based on sperm DNA methylation changes.
The decreasing precision of the prediction with increasing age must also be taken into account when comparing prediction results from the different studies shown in . The model developed by Li et al. offers the lowest MAE but is built on samples from individuals aged 21–54 years and validated in individuals younger than 46 years [Citation45], whereas Pisarek et al. worked with individuals aged 26–57 years [Citation34] and Lee et al. included individuals up to the age of 73 years [Citation28]. Mutual comparability is also limited by the use of differential extraction by Li et al. [Citation45] and the swim-up sperm fraction by Laurentino et al. [Citation18] compared with other studies that worked with whole semen samples.
Limiting factors
Population sample size
The main limitation of age-related methylation studies in sperm is very small sample size. In most studies, the entire sample set is less than 100 samples, and even the largest use sets smaller than 400 samples (). Similar studies that have developed age prediction models for blood samples generally use at least 100 samples, with average sample sets consisting of 200–300 samples and the largest exceeding 1000 samples [Citation29,Citation46]. Apart from the fact that the population of potential sample donors is approximately halved by sex, semen is considered a much more private biological material than, for example, blood, and the number of volunteers willing to provide a sample for research is generally lower. Cooperation with assisted reproduction clinics may help to increase the total number of samples but burdens the sample set with potential selection bias, as most donors are looking for help with fertility issues. With the decline in sperm quality and quantity among the general population over the last few decades and many reproductive health problems passing undetected, we must choose between a specific known bias in samples from individuals with fertility problems and a bias we cannot fully define but that is representative of the general population in samples from healthy volunteers [Citation22]. Small sample sets like this tend to misrepresent biological variability in the population and lead to the overfitting of prediction models, which then perform significantly worse on independent samples.
Differentially methylated regions
Comparing the results of different studies can be complicated because of unclear definitions of the differentially methylated area in terms of length and difference thresholds. Leitão et al. defined the DMR as a region of at least ten CpGs with a methylation difference of at least 80% between two individuals of different ages [Citation11]. However, Oluwayiose et al., for example, used clustering algorithms starting with two CpGs in 1000 bp [Citation3].
Typing platform
Pyrosequencing is generally considered the gold standard, but many phenotyping studies use the single-base extension method SNaPshot (Applied Biosystems, MA, USA) or the mass spectrometry-based EpiTYPER (Agena Bioscience, CA, USA), and massively parallel sequencing is becoming more and more common. The use of different typing platforms can also be a source of inconsistent results, as loss of accuracy of methylation data has been reported during technology transfer among different methods [Citation46,Citation47].
Sample composition
The composition of the sample itself can significantly influence the entire study. As already mentioned, ejaculate is not created until it leaves the body, and its composition changes during ejaculation, between instances of sexual activity and depending on many individual physical and psychological factors [Citation22]. Because collecting the entire sample in a single container erases differences in individual fractions of the ejaculate, studies focused on reproduction lose specific information from the initial fraction of ejaculate, which is most effective for conception, whereas laboratory studies work with samples that do not correspond to forensic samples, such as individual semen stains and vaginal swabs. The ratio between individual cell types in the semen of an unknown person can hardly be estimated. The number of sperm per milliliter of semen is highly variable and a low sperm count can result in relative enrichment of nonspermatozoal cells; the number of white blood cells increases during infection; and atypical cells with aberrant methylation profiles can be present during cancer [Citation24,Citation25].
Purification methods
Considering the significantly different methylation profiles of somatic cells, most studies use some kind of purification of the semen so as to analyze only the sperm DNA. However, sample purity can itself be a confounding factor. Pilsner et al. [Citation30] detected a number of age-associated CpGs comparable to Oluwayiose et al. [Citation3] (2364 and 1698, respectively), but fewer of these regions were hypermethylated (44 and 91%, respectively). Although Oluwayiose et al. worked with samples that had been checked using microscopy for any somatic contamination, Pilsner et al. used whole semen samples after one step of 40% gradient centrifugation. It is possible that significant differences in obtained values are the result of DNA methylation of somatic cells.
There are a wide variety of methods applicable to the purification of semen samples. Swim-up, chemotactic methods and microfluidic chips are based on sperm motility; density gradient centrifugation, with a variable number of steps to select a fraction of the sample, is based on sedimentation; and advanced methods use the specific binding of sperm to, for example, hyaluronan. The effectiveness of these methods is variable, and there is no consensus regarding their priority for DNA methylation studies. Most of the aforementioned studies used one- or two-step gradient centrifugation [Citation3,Citation30], in some cases with swim-up enrichment of the motile sperm fraction [Citation10,Citation19].
The use of whole semen samples can increase predictive error as a result of the unpredictable ratio of sperm and nonsperm cells but may result in epigenetic profiles more applicable to real-life samples, especially in forensic settings, where semen stains from clothing or bedsheets are often analyzed. A whole semen prediction model could be more robust for this kind of situation than a model built on purified sperm cells. However, purified sperm DNA models have their place in forensics as well because when analyzing vaginal swabs, differential lysis must be performed to eliminate the genetic material of the woman so only sperm DNA remains. The forensically focused studies by Lee et al. [Citation28], Li et al. [Citation45], and Pisarek et al. [Citation34] refer to the same genetic loci and to each other despite the use of differential dithiothreitol-based extraction of sperm DNA by Li et al. and the use of whole semen samples by Lee et al. and Pisarek et al.
Biological factors
Lifestyle and environmental factors can be significant confounding factors in studies focusing on age-related changes in DNA methylation since they can influence epigenetic patterns as well [Citation3]. Although age is considered to be the most profound and predictable modifier of sperm epigenetic profiles [Citation21], smoking, alcohol and drug consumption, diet, physical activity, stress levels and chemical exposure can also cause significant changes in sperm DNA patterns and contribute to male infertility [Citation3,Citation17]. Both hypomethylation and hypermethylation can have positive and negative impacts on health, depending on the affected loci.
Although metabolic disorders in offspring, such as diabetes mellitus, have been linked to the hypomethylation pattern in sperm from older fathers, imprinted genes controlling growth and development (neuronatin, PEG3 and MEST) were also hypomethylated in blood samples from children with obese fathers [Citation17]. An unhealthy paternal diet also negatively influences the methylation of genes encoding some liver functions connected to cholesterol and lipid metabolism. By contrast, oral supplementation of micronutrients such as folate, vitamin B, zinc and cysteine showed a positive increase in DNA methylation levels [Citation17].
A supervised yoga regimen promoted methylation changes, with positive health impacts in almost 400 genes, whereas tobacco smoking had a strong negative impact on health through methylation changes in many genes, not just those related to male infertility [Citation17]. The effect of smoking on sperm DNA methylation is similar to the effect of aging, as smokers tend to be predicted as older than nonsmokers [Citation1,Citation30]. Together, these findings suggest a global link between a healthy lifestyle, proper methylation patterns and reproductive health [Citation17].
Future perspective
This whole field of study is quite fragmented, with often contradictory results. Inconsistencies in study design limit the possibility for inferring any consensus. Unifying the terminology would definitely help (e.g., a clear definition of DMRs in terms of total length, minimum number of contained CpGs and minimum difference between compared sample sets). The easiest way might be to completely stop using DMRs in studies and instead use an unambiguous individual CpG approach. Array-based studies also do not cover all positions where significant differences and associations can be found by the whole genome approach. The use of whole genome sequencing technology in the discovery phase of studies seems feasible, as it provides more data from less researched areas like noncoding and repetitive regions. Those regions can also be interesting targets for future aging-related epigenetic studies. The biggest limitation of sperm and semen DNA methylation studies is the small sample sets, so it is necessary to validate predictive models on larger sets of well-defined samples covering various characteristics of donors in terms of general and reproductive health, lifestyle factors, like obesity and smoking, and age from puberty to senescence.
The differences between methylation profiles from purified sperm cells and whole semen samples should also be researched further. Defining the magnitude of error that the variable ratio of somatic cells in the semen sample brings to age prediction is important for real-life use of this tool and would minimize inconsistencies caused by different sample preprocessing methods. Single-cell sequencing methods should be implemented more widely, as they hold the potential to overcome the problem of sample purity and epigenetic differences between somatic and germ cells. The development of techniques suitable for working with smaller samples would also be very beneficial, especially for forensic use, where the low quantity and quality of crime scene DNA is the biggest limiting factor. For the same reason, bisulfite conversion, which is considered the gold standard for methylation studies, should be replaced with some less destructive method, such as enzymatic conversion or TET-assisted pyridine borane sequencing. Recently, a new quantitative PCR method capable of distinguishing between methylated and unmethylated cytosine in untreated DNA has been created [Citation48]. With this approach, the use of intercalating pseudonucleotides increases the melting temperature of sequences, including methylated cytosine, allowing their preferential amplification.
Background
Compared with somatic cells, age-related methylation changes in sperm are understudied, although they have both clinical and forensic implications.
The role of DNA methylation
The correct methylation signal is important for proper gene expression control, maintaining genome stability, proper cell type differentiation and cellular adaptation to the changing environment.
Development of sperm DNA methylation signals
Because of continuous cell division and higher epigenetic information reproduction error rate, sperm DNA is more affected by epigenetic changes with increasing age than somatic cells or ovum DNA.
During embryonic development, embryonic cells and future sperm cell precursors undergo almost complete remodeling of the methylation signal.
Sperm cell sampling
Methylation is tissue-specific; thus, the highly variable ratio of individual cell types in semen might significantly affect age prediction results.
Differences between somatic&germ cell methylation
Although aging in sperm cells is characterized by global hypermethylation with specific hypomethylated loci, the situation in somatic cells is exactly the opposite.
Multitissue age prediction models report a high mean absolute error for semen samples compared with somatic tissues.
The magnitude of DNA methylation changes in sperm DNA
Results of studies comparing methylation in semen samples of young and old individuals are inconclusive in terms of the ratio between hypomethylated and hypermethylated loci during aging.
Clinical impacts of age-related epigenetic sperm changes
Clinical studies show the connection between older paternal age and longer time to conception, lower percentage of live births and various disorders (mainly metabolic, neurological and behavioral) in offspring. The same applies to younger men with a higher epigenetic age.
Forensic applications of age-related epigenetic sperm changes
Forensic models allow the prediction of an unknown sperm trace donor’s age, which can help to narrow down police investigations, primarily in cases of sexual assault.
Age prediction models
Clinically relevant age prediction models can use a larger number of markers with a smaller individual impact and are more focused on detecting age acceleration or deceleration compared with chronological age. The mean absolute error of prediction is approximately 2.5 years.
Forensically relevant age prediction models must use a lower number of predictors because of the limited quantity of DNA samples and are focused on chronological age itself. The mean absolute error of prediction is approximately 5.0 years.
Limiting factors
Existing studies are mostly burdened with insufficient sample sizes and significant inconsistencies regarding types of samples, sample preprocessing, typing methods and terminology used to describe results.
Future perspective
The entire field of study is fragmented, with contradictory results, and inconsistencies in study designs and terminology limit the possibilities for reaching consensus.
Larger sample sets with various donor characteristics, use of whole genome sequencing in the discovery phase, development of techniques suitable for less DNA input, and alternatives to bisulfite sequencing could lead to the development of better prediction models.
L Kotková contributed to conceptualization, preparing of the original draft, graphics and writing. J Drábek contributed to conceptualization, reviewing, editing and administration. Both authors have read and agreed to the submitted version of the manuscript.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with an interest in or conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending or royalties.
Writing disclosure
The authors thank B Stenglová for help with graphics and J Parrott for proofreading.
Acknowledgments
Graphics were created with BioRender.com.
Financial disclosure
This work was supported by European Infrastructure for Translational Medicine and funded by LM2023053, CZ.02.1.01/0.0/0.0/16_026/0008448, EF16_013/0001674, LX22NPO5102 and IGA LF UP 2023_006. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- Jenkins TG , AstonKI , PfluegerC , CairnsBR , CarrellDT. Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLOS Genet.10(7), e1004458 (2014).
- Ashapkin V , SuvorovA , PilsnerJR , KrawetzSA , SergeyevO. Age-associated epigenetic changes in mammalian sperm: implications for offspring health and development. Hum. Reprod. Update29(1), 24–44 (2023).
- Oluwayiose OA , WuH , SaddikiHet al. Sperm DNA methylation mediates the association of male age on reproductive outcomes among couples undergoing infertility treatment. Sci. Rep.11(1), 3216 (2021).
- Hannum G , GuinneyJ , ZhaoLet al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell49(2), 359–367 (2013).
- Horvath S . DNA methylation age of human tissues and cell types. Genome Biol.14(10), R115 (2013).
- Bormann F , Rodríguez-ParedesM , HagemannSet al. Reduced DNA methylation patterning and transcriptional connectivity define human skin aging. Aging Cell15(3), 563–571 (2016).
- Jin B , LiY , RobertsonKD. DNA methylation: superior or subordinate in the epigenetic hierarchy?Genes Cancer2(6), 607–617 (2011).
- Ghazimoradi MH , HasegawaK , ZolghadrE , MontazeriS , FarivarS. Reprogramming of fibroblast cells to totipotent state by DNA demethylation. Sci. Rep.13(1), 1154 (2023).
- Aston KI , UrenPJ , JenkinsTGet al. Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertil. Steril.104(6), 1388–1397.e1–5 (2015).
- Bernhardt L , DittrichM , PrellAet al. Age-related methylation changes in the human sperm epigenome. Aging (Albany NY)15(5), 1257–1278 (2023).
- Leitão E , DiPersio S , LaurentinoSet al. The sperm epigenome does not display recurrent epimutations in patients with severely impaired spermatogenesis. Clin. Epigenetics12(1), 61 (2020).
- Zhu P , GuoH , RenYet al. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat. Genet.50(1), 12–19 (2018).
- Galan C , SerraRW , SunF , RinaldiVD , ConineCC , RandoOJ. Stability of the cytosine methylome during post-testicular sperm maturation in mouse. PLoS Genet.17(3), e1009416 (2021).
- Ramakrishna NB , MurisonK , MiskaEA , LeitchHG. Epigenetic regulation during primordial germ cell development and differentiation. Sex. Dev.15(5–6), 411–431 (2021).
- Ben Maamar M , BeckD , NilssonE , McCarreyJR , SkinnerMK. Developmental alterations in DNA methylation during gametogenesis from primordial germ cells to sperm. iScience25(2), 103786 (2022).
- Li L , LiL , LiQet al. Dissecting the epigenomic dynamics of human fetal germ cell development at single-cell resolution. Cell Res.31(4), 463–477 (2021).
- Rotondo JC , LanzillottiC , MazziottaC , TognonM , MartiniF. Epigenetics of male infertility: the role of DNA methylation. Front. Cell Dev. Biol.9, 689624 (2021).
- Laurentino S , CremersJF , HorsthemkeBet al. A germ cell-specific ageing pattern in otherwise healthy men. Aging Cell19(10), e13242 (2020).
- Denomme MM , HaywoodME , ParksJC , SchoolcraftWB , Katz-JaffeMG. The inherited methylome landscape is directly altered with paternal aging and associated with offspring neurodevelopmental disorders. Aging Cell19(8), e13178 (2020).
- Ben Maamar M , KingSE , NilssonE , BeckD , SkinnerMK. Epigenetic transgenerational inheritance of parent-of-origin allelic transmission of outcross pathology and sperm epimutations. Dev. Biol.458(1), 106–119 (2020).
- Jenkins TG , JamesER , AstonKIet al. Age-associated sperm DNA methylation patterns do not directly persist trans-generationally. Epigenetics Chromatin12(1), 74 (2019).
- WHO . WHO Laboratory Manual for the Examination and Processing of Human Semen. WHO, Geneva, Switzerland (2021).
- Anamthathmakula P , WinuthayanonW. Mechanism of semen liquefaction and its potential for a novel non-hormonal contraception. Biol. Reprod.103(2), 411–426 (2020).
- Long S , KenworthyS. Round cells in diagnostic semen analysis: a guide for laboratories and clinicians. Br. J. Biomed. Sci.79, 10129 (2022).
- Fedder J . Nonsperm cells in human semen: with special reference to seminal leukocytes and their possible influence on fertility. Arch. Androl.36(1), 41–65 (1996).
- Di Persio S , LeitãoE , WösteMet al. Whole-genome methylation analysis of testicular germ cells from cryptozoospermic men points to recurrent and functionally relevant DNA methylation changes. Clin. Epigenetics13(1), 160 (2021).
- Luján S , CaroppoE , NiederbergerCet al. Sperm DNA methylation epimutation biomarkers for male infertility and FSH therapeutic responsiveness. Sci. Rep.9(1), 16786 (2019).
- Lee HY , JungSE , OhYN , ChoiA , YangWI , ShinKJ. Epigenetic age signatures in the forensically relevant body fluid of semen: a preliminary study. Forensic Sci. Int. Genet.19, 28–34 (2015).
- Weidner CI , LinQ , KochCMet al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol.15(2), R24 (2014).
- Pilsner JR , SaddikiH , WhitcombBWet al. Sperm epigenetic clock associates with pregnancy outcomes in the general population. Hum. Reprod.37(7), 1581–1593 (2022).
- Orioli D , DellambraE. Epigenetic regulation of skin cells in natural aging and premature aging diseases. Cells7(12), 1–30 (2018).
- Simpson DJ , ChandraT. Epigenetic age prediction. Aging Cell20(9), e13452 (2021).
- Heidegger A , PisarekA , dela Puente Met al. Development and inter-laboratory validation of the VISAGE enhanced tool for age estimation from semen using quantitative DNA methylation analysis. Forensic Sci. Int. Genet.56, 102596 (2022).
- Pisarek A , PośpiechE , HeideggerAet al. Epigenetic age prediction in semen – marker selection and model development. Aging (Albany NY)13(15), 19145–19164 (2021).
- Milekic MH , XinY , O’DonnellAet al. Age-related sperm DNA methylation changes are transmitted to offspring and associated with abnormal behavior and dysregulated gene expression. Mol. Psychiatry20(8), 995–1001 (2015).
- Prell A , SenMO , PotabattulaRet al. Species-specific paternal age effects and sperm methylation levels of developmentally important genes. Cells11(4), 1–12 ( 2022).
- Kayser M , SchneiderPM. DNA-based prediction of human externally visible characteristics in forensics: motivations, scientific challenges, and ethical considerations. Forensic Sci. Int. Genet.3(3), 154–161 (2009).
- Freire-Aradas A , PośpiechE , AliferiAet al. A comparison of forensic age prediction models using data from four DNA methylation technologies. Front. Genet.11, 932 (2020).
- Paparazzo E , LaganiV , GeracitanoSet al. An ELOVL2-based epigenetic clock for forensic age prediction: a systematic review. Int. J. Mol. Sci.24(3), 1–14 (2023).
- Daunay A , BaudrinLG , DeleuzeJF , How-KitA. Evaluation of six blood-based age prediction models using DNA methylation analysis by pyrosequencing. Sci. Rep.9(1), 8862 (2019).
- Lee JW , ChoungCM , JungJY , LeeHY , LimSK. A validation study of DNA methylation-based age prediction using semen in forensic casework samples. Leg. Med. (Tokyo)31, 74–77 (2018).
- Zbiec-Piekarska R , SpolnickaM , KupiecTet al. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet.17, 173–179 (2015).
- Thong Z , ChanXLS , TanJYY , LooES , SynCKC. Evaluation of DNA methylation-based age prediction on blood. Forensic Sci. Int. Genet. Suppl. Ser.6, e249–e251 (2017).
- Cho S , JungSE , HongSRet al. Independent validation of DNA-based approaches for age prediction in blood. Forensic Sci. Int. Genet.29, 250–256 (2017).
- Li L , SongF , LangMet al. Methylation-based age prediction using pyrosequencing platform from seminal stains in Han Chinese males. J. Forensic Sci.65(2), 610–619 (2020).
- Vidaki A , KayserM. From forensic epigenetics to forensic epigenomics: broadening DNA investigative intelligence. Genome Biol.18(1), 238 (2017).
- Aliferi A , BallardD , GallidabinoMD , ThurtleH , BarronL , SyndercombeCourt D. DNA methylation-based age prediction using massively parallel sequencing data and multiple machine learning models. Forensic Sci. Int. Genet.37, 215–226 (2018).
- Bendixen KK , MindegaardM , EpistolioSet al. A qPCR technology for direct quantification of methylation in untreated DNA. Nat. Commun.14(1), 5153 (2023).
- Cao M , ShaoX , ChanPet al. High-resolution analyses of human sperm dynamic methylome reveal thousands of novel age-related epigenetic alterations. Clin. Epigenetics12(1), 192 (2020).
- Jenkins TG , AstonKI , CairnsB , SmithA , CarrellDT. Paternal germ line aging: DNA methylation age prediction from human sperm. BMC Genomics19(1), 763 (2018).