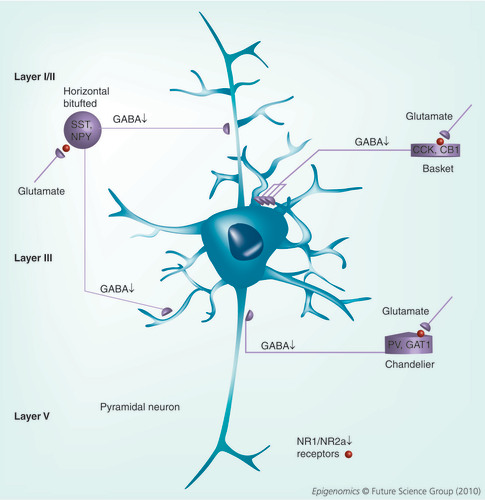
GABAergic interneurons function to modulate the output of pyramidal and other neurons. We propose that reduced expression of glutamate decarboxylase 1 and other GABAergic markers (SST, NPY, CCK, CB1, PV, GAT1 and reelin) along with reduced levels of NR1 and NR2A (shown by a sphere) containing glutamate receptors contributes to reduced GABA release (shown by a down arrow). This GABA hypofunction causes decreased pyramidal neuron synchronization. The model proposes that the reduced signaling at NMDA-selective glutamate receptors present on GABAergic interneurons causes the glutmatergic hypofunction, which then facilitates reduced GABA release onto the main output neurons (pyramidal neurons).
CB: Calbindin; CCK: Cholecystokinin; GAT: GABA transporter; NPY: Neuropeptide Y; NR: NMDA receptor; PV: Parvalbumin; SST: Somatostatin.
The history of our thinking regarding the origins of psychosis has been influenced to a certain degree by the pharmacology of psychotropic drugs as they mimic certain aspects of psychiatric illness. The community of biological psychiatrists were well aware of the effects of lysergic acid diethylamide and other psychedelic drugs, and their capabilities in distorting reality and inducing sensory hallucinations. The same can be said of the dissociative anesthetic phencyclidine, which was commonly mixed and smoked with marijuana (also known as ‘angel dust‘). The main pharmacological action of phencyclidine has been demonstrated to be an inhibitor of neurotransmission at the NMDA subtype of ionotropic glutamate receptor. However, the main psychosis-inducing effects of both lysergic acid diethylamide and phencyclidine are more complicated, and are most likely related to metabolites acting at multiple subtypes of G-protein-coupled receptors, including the family of serotonin and dopamine receptors. During the 1950s, the first neuroleptics (such as chlorpromazine) were being tested and were found to be promising for the treatment of some of the positive symptoms of schizophrenia (SZ). Both the first generation, typical antipsychotics and the newer, atypical antipsychotics exhibit inhibitory activity at dopamine receptors. However, much like the other compounds described, these drugs show a broad range of receptor targets. The neurotransmitter GABA has been implicated in the symptomatology of SZ from studies of postmortem tissue, these studies established that certain mRNAs and proteins decreased in patients with SZ and bipolar disorder with psychosis. Compelling arguments can be made for each of these neurotransmitters (glutamate Citation[1], dopamine Citation[2] and GABA Citation[3]) as being involved in the pathophysiology of SZ. While each of these neurotransmitters plays distinct roles in CNS function, they also overlap in terms of their effects on various cognitive processes and behaviors. Currently lacking is a comprehensive hypothesis that links each neurotransmitter/receptor system that can account for the varied role of each of these in terms of the altered brain circuitry observed in patients diagnosed with SZ. In this article, I attempt to do this by proposing that an epigenetic dysfunction that perturbs cortical GABA neuron transcription impacts both GABA and glutamate neurotransmitter signaling at the level of either presynaptic release (GABA) or postsynaptic hypofunction (glutamate). In many local circuits of the brain, glutamate regulates dopamine release and dopamine is currently thought to be downstream of the primary neurotransmitter deficit Citation[2].
It was first reported some years ago that, compared with nonpsychiatric subjects, reelin is downregulated in the postmortem cortices of SZ patients Citation[4]. We subsequently studied the reelin gene and recognized that the regulation of this promoter was affected by the methylation status of key residues proximal to the RNA start site Citation[5]. Results from additional studies have shown reduced mRNA expression profiles for a large number of mRNAs associated with neuronal function. Many of the mRNAs downregulated in these studies, including glutamic acid decarboxylase I (GAD1 or GAD67), the vesicular GABA transporter (VGAT), GABA transporter (GAT-1), NMDA receptors (NR1 and NR2A), glutamate transporters (VGLUT1), in addition to reelin and others, are expressed in cortical interneurons of the brain Citation[6,7]. These interneurons are inhibitory and use GABA as their neurotransmitter. Interestingly, DNA methyltransferase I (DNMT1) is increased in GABAergic neurons of cortical layers I, II and IV of SZ and bipolar patients. This inverse relationship between reelin, GAD67 and other mRNAs and the increased expression of DNMT1 suggest that promoter hypermethylation may play a role in the reduced expression profiles.
Recent data also support the concept that insufficient stimulation of NMDA-selective glutamate receptors (NR1/NR2A receptor containing assemblies) leads to insufficient GABA release at synapses on cortical pyramidal neurons. This could explain why NMDA receptor antagonists, such as phencyclidine and dizocilpine (MK-801), induce psychotic episodes when these compounds are ingested Citation[1]. It also explains, in part, why metabotropic glutamate receptor agonists that facilitate glutamate release onto GABAergic interneurons may prove beneficial to patients with SZ Citation[8,9]. As most of the observed cortical mRNA changes in SZ have been localized to GABAergic neurons (at least in the cortex), our hypothesis is consistent with a disruption of pyramidal neuron synchronization mediated by an inhibitory hypofunction occurring at the level of presynaptic interneurons. This model was also suggested by recent behavioral findings in mice in which the NR1 subunit (Grin1) of the NMDA-selective glutamate receptor complex was selectively knocked out in cortical and hippocampal GABA interneurons Citation[10]. This hypothesis accounts for both the GABA neuron deficits and the glutamatergic deficits that have thus far been described Citation[1].
The cellular architecture of the cortex consists largely of excitatory pyramidal neurons, the major cortical output, multiple types of smaller interneurons that modulate this output and a host of other cell types (glia) that provide numerous roles in cortical function , cortical layers are formed based on developmental appearance with layer I closest to the pial surface and layer VI the last to migrate. GABAergic neurons modulate the excitability of pyramidal neurons by hyperpolarizing their membranes. Cortical pyramidal neurons are characterized by having a large cell body (located in layers III and V) with apical dendrites and basal dendrites. These are generally excitatory and use glutamate as their neurotransmitter. Layer III pyramidal neurons send outputs to other cortical areas (contralateral) and feed-forward onto GABA interneurons. Layer V pyramidal neurons are the main cortical output and also provide inhibition through GABA interneurons (feed-forward inhibition). These GABAergic neurons are inhibitory to the neurons they synapse with, using GABA as their transmitter. GABAergic neurons are characterized by their morphology, laminar position and the calcium-binding proteins that they express (e.g., parvalbumin, calbindin and calretinin). These neurons form synapses onto the apical dendrites (horizontal, bitufted), cell bodies (basket cell) and axons (chandelier cells) of pyramidal cells. Cortical layer I is comprised almost exclusively of these horizontal cells and these neurons show increased levels of DNMT1 mRNA and protein Citation[11–13]. In addition, GABAergic neurons present in layers II and IV also show increased DNMT1 expression relative to nonpsychiatric subjects Citation[12]. This results in the downregulation of selected mRNAs in these same neurons and a disruption of normal synaptic function at GABA/glutamate synapses. The GABAergic neurons of SZ patients are thought to be compromised and fail to provide sufficient inhibitory tone onto layer III and V pyramidal neuron dendrites (for a review see Citation[14]). The net result is a disruption of the synchronization of pyramidal neuron firing that results in cognitive dysfunction. Collectively, our model predicts that the increased expression of DNMT1 in GABAergic neurons has epigenetic consequences, which results in the downregulation of multiple mRNAs that provide functional support for inhibitory neurotransmission.
The increased expression of DNMTs in GABA neurons of the cortex and striatum is consistent with the increased methylation that has been reported in SZ patients Citation[15]. As noted above, recent studies also support the concept that insufficient stimulation of NMDA-selective glutamate receptors (NR1 [GRIN1] and NMDA2A [GRIN2A] receptor assemblies) on these GABAergic interneurons contributes to the proposed GABA hypofunction at cortical pyramidal neurons. The reduced expression of subunits of the NMDA receptor in GABA interneurons of SZ patients is well documented Citation[16,17]. While it has not been demonstrated as yet, the possibility that many of the corresponding promoters, including those that regulate GABA-related transcripts (GAD67, reelin and GABA transporters) and NMDA receptor subunits may be hypermethylated as a consequence of DNMT overexpression. The difficulties with determining this include the diversity of neuronal subtypes present in various segments of the brain and also knowing where to look for methylation changes in any selected promoter. It is not clear that the studies carried out thus far Citation[15,18] can exclude hypermethylation as a mechanism simply because of these complexities. The tendency to choose regions within CpG islands and upstream of RNA start sites for methylation analyses is still blind as compared with more informed approaches. That is, it would be vastly more informative to look for regulatory elements within promoters and then to examine these regions for alterations in DNA methylation. This would require us to first define the regulatory elements of the genes of interest and to use a laser capture type of approach to avoid the heterogeneity complication. However, until we consistently reach that level of sophistication, we may be looking in the wrong place at the wrong time. A recent review article suggests that the data available thus far make it very unlikely that SZ involves a generalized shift towards increased DNA methylation in the brain Citation[19]. While this may be true, a more plausible explanation is that SZ is associated with changes in the methylation of select promoters at discrete sites relevant to regulation and that these only occur in the GABAergic neurons of the brain. These neurons represent approximately 30% of the total neurons in the brain so the actual percentage change in methylation may not be detectable in the absence of laser capture techniques. Since many drugs (e.g., the histone deacetylase inhibitors Citation[20]) that act at the level of chromatin remodeling also interact with the methylation machinery, it will be increasingly important to analyze the effects of these compounds on defined promoter targets to better understand if these marks can be erased.
The failure to find clear cut differences in DNA methylation in postmortem brain tissue of SZ subjects at specific gene promoters has caused some confusion as to whether DNA methylation plays a key role in the pathogenesis of the disease. However, studying human genomic DNA for subtle differences in methylation is more about knowing where in a promoter to look for these changes. Just as important is utilizing the tools necessary to microdissect and collect individual neuronal subtypes for the analysis.
Acknowledgements
The author would like to thank Dr Alessandro Guidotti for his ongoing dialog regarding this hypothesis. This editorial is dedicated to the memory of my mentor and colleague, Dr Erminio Costa, who passed away 28 November 2009.
Financial & competing interests disclosure
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Bibliography
- Lisman JE , CoyleJT, GreenRWet al.: Circuit based framework for understanding neurotransmitter and risk gene interactions in schizophrenia.Trends Neurosci.31 , 234–242 (2008).
- Howes OD , KapurS: The dopamine hypothesis of schizophrenia: version III – the final common pathway.Schizophrenia Bull.35 , 549–562 (2009).
- Lewis DA , HashimotoT: Deciphering the disease process of schizophrenia: the contribution of cortical GABA neurons.Int. Rev. Neurobiol.78 , 109–131 (2007).
- Guidotti A , AutaJ, DavisJMet al.: Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study.Arch. Gen. Psych.57 , 1061–1069 (2000).
- Chen Y , SharmaRP, CostaRH, CostaE, GraysonDR: On the epigenetic regulation of human reelin promoter expression.Nucleic Acids Res.30 , 2930–2939 (2002).
- Guidotti A , AutaJ, DavisJMet al.: GABAergic dysfunction in schizophrenia: a new treatment on the horizon.Psychopharmacology (Berl.)180 , 191–205 (2005).
- Hashimoto T , ArionD, UngerTet al.: Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia.Mol. Psychiatry13 , 147–161 (2008).
- Conn PJ , LindsleyCW, JonesCK: Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia.Trends Pharmacol. Sci.30 , 25–31 (2008).
- Marek GJ , BehlB, BespalovAY, GrossGet al.: Glutamatergic (N-methyl-D-aspartate receptor hypofrontality in schizophrenia: too little juice or miswired brain?Mol. Pharmacol.77 , 317–326 (2010).
- Belforte JE , ZsirosV, SklarERet al.: Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes.Nat. Neurosci.13 , 76–83 (2010).
- Veldic M , CarunchoHM, LiuWSet al.: DNA-methyltransferase-1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains.Proc. Natl Acad. Sci. USA101 , 348–353 (2004).
- Veldic M , GuidottiA, MalokuE, DavisJM, CostaE: In psychosis, cortical interneurons overexpress DNA-methyltransferase I.Proc. Natl Acad. Sci. USA102 , 2152–2157 (2005).
- Ruzika W , ZhubiA, VeldicM, GraysonDR, CostaE, GuidottiA: Selective epigenetic alteration of Layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection.Mol. Psychiatry12 , 385–397 (2007).
- Lewis DA , HashimotoT, VolkDW: Cortical inhibitory neurons and schizophrenia.Nat. Rev. Neurosci.6 , 312–324 (2005).
- Mill J , TangT, KaminskyZet al.: Epigenomic profiling reveals DNA-methylation changes associated with major psychosis.Am. J. Hum. Genet.82 , 696–711 (2008).
- Woo TUW , WalshJP, BenesFM: Density of glutamic acid decarboxylase 67 messenger RNA containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar diorder.Arch. Gen. Psychiatry61 , 649–657 (2004).
- Woo TUW , KimAM, ViscidiE: Disease-specific alterations in glutamatergic neurotransmission on inhibitory interneurons in the prefrontal cortex in schizophrenia.Brain Res.1218 , 267–277 (2008).
- Huang HS , AkbarianS: GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia.PLoS One29 , E809 (2007).
- Akbarian S : The molecular pathology of schizophrenia – focus on histone and DNA modifications.Brain Res. Bull. DOI:10.1016/j.brainresbull.2009.08.018 (2009) (Epub ahead of print).
- Grayson DR , KundakovicM, SharmaRP: Is there a future for HDAC inhibitors in the pharmacotherapy for psychiatric disorders?Mol. Pharmacol.77 , 126–135 (2010).