SZ: Schizophrenia.
Original figure provided by Tomas Ekström.
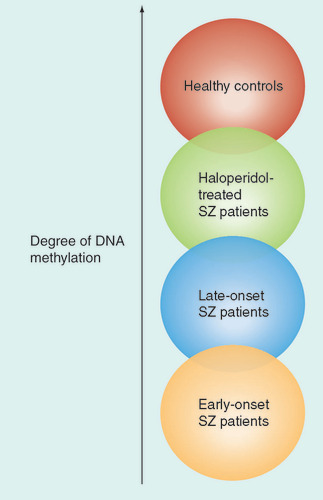
Scientists at the Karolinska Institutet (Stockholm, Sweden) have published new data that demonstrate different DNA methylation patterns in the leukocytes of schizophrenic and healthy subjects. It is well documented that schizophrenia has a strong hereditary component; however, epigenetic research has indicated that DNA methylation could be an explanatory candidate for the disorder‘s nongenetic basis. This particular study adds further evidence to the idea that epigenetics may have an important role in the regulation of mental illness.
To investigate associations between epigenomics and schizophrenia, the researchers examined genome-wide and gene-specific DNA methylation levels in the leukocytes of schizophrenia patients and a control group. These two techniques are indicative of genome stability and gene-expression activity, respectively.
Using genome-wide analysis, the global methylation results demonstrated a highly significant hypomethylation in schizophrenia patients. Linear regression among the patients revealed that disease onset and antipsychotic treatment explained 11% of the global methylation variance. This hypomethylation was most prominent in early-onset schizophrenia patients. In addition, treatment with haloperidol was associated with higher (control-like) methylation. The gene-specific methylation analyses revealed that the analyzed region of S-COMT was hypermethylated in schizophrenia patients.
Tomas Ekström, one of the study authors, spoke to Epigenomics about the importance of their results: “One very significant issue is that the peripheral blood cell epigenome seems to reflect the global epigenetics status of a disease which is characterized to have its main effects in the brain. The fact that the global methylation level in leukocytes could be used as a marker of the severity of schizophrenia opens up completely new opportunities.” Although the biological significance of the methylation is yet to be determined, Ekström explained how their results could be important in the treatment of schizophrenia patients: “Our data suggest that this marker may also be used to follow a patient‘s therapy response, and, as we found, haloperidol partly ‘normalized‘ the global DNA methylation level.”
The authors claim that their results support the notion of a dysregulated epigenome in schizophrenia, which global analysis indicated was more pronounced in early-onset patients, and could be partially reversed by antipsychotic treatment. However, Ekström stresses that the study does have its limiations and further work will need to be performed to further investigate these data: “The current work was primarily descriptive and aimed to assess if the blood epigenome is suited as a diagnostic tool to follow state and treatment response. To understand the deeper relevance and use of this ‘epigenetic tool‘, we need to perform whole-genome methylation profiling to learn whether the actual profile is similar in blood cells as it is in the brain.
“Furthermore, it is important to elucidate if the difference in methylation profile is due to a different profile of leukocyte types, as inflammatory markers have been reported to be upregulated in schizoprehnia. The understanding of the biology of the epigenome in schizophrenia is another, even greater, task that several others are also working on.”
Source: Melas PA, Rogdaki M, Ösby U et al. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. doi:10.1096/fj.11-202069 (2012) (Epub ahead of print).
Researchers at the Cave Western University (OH, USA), have published results reporting the identification of an epigenetic signature for colon cancer. Although previous studies have largely focused on coding sequences and promoters, this study group investigated the histone mark H3K4me1, finding a signature of enhancer histone modifications that indicated the presence of colorectal cancer (CRC).
In their study, the researchers utilized this histone mark to perform a genome-wide comparison of gain and loss of enhancer activity in primary colon cancer lines and normal colon crypts. The investigators identified thousands of variant enhancer loci (VEL) that were differentially lost or gained in CRC cells, thus producing a ‘signature‘ of the disease. This signature appears to be predictive of the in vivo CRC transcriptome.
When analyzing the CRC cell line, it was found that approximately 40% of gained VELs displayed H3K27ac, whereas most of the lost VELs showed no detectable levels of H3K27ac. Speaking to Epigenomics, Peter Scacheri, an author of the study, explained the findings, highlighting the significance of the signature: “The VEL signature demonstrates that a common set of distal regulatory elements are targeted for epigenetic dysregulation in colon cancer. We discovered that approximately 200 VELs are shared across the cohort of nine colon tumors used in our study. To put this in perspective, whole-genome sequencing studies have revealed that less than ten candidate driver genes are frequently targeted for genetic mutations in colon cancer.”
Scacheri spoke further on what these findings mean in the field, commenting: “Our work illustrates an emerging concept in epigenomics, i.e., that identifying disease-specific alterations in chromatin marks at distal enhancer elements can cut through the complexity of heterogenous expression and mutation profiles in cancer to reveal potential ‘driver genes‘ with greater sensitivity and success than approaches that focus solely on tumor transcriptomes, DNA methylation, or epigenetic alterations at promoters.”
Although these findings show great promise in how scientists can study epigenetic activity to identify disease states in the genome, Scacheri emphasized that further work needs to be done: “The next step is to identify the mechanism by which VELs arise in cancer. One possibility is that VELs form as a result of somatic mutations in enhancers that arose over the course of carcinogenesis. Integrating whole-genome sequencing studies and H3K4me1 chromatin immunoprecipitation sequencing analysis on matched normal tumor pairs could help shed light on this hypothesis”.
“Furthermore, the VEL signature has identified a novel set of genes that are potential drivers of carcinogenesis and clearly warrant further investigation. An additional step is to determine if VEL signatures can stratify colon tumors by their clinical phenotypes, including responsiveness to drugs that target chromatin regulators; these are currently in clinical development.”
Source: Akhtar-Zaidi B, Cowper-Sal Lari R, Corradin O et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science 336(6082), 736–739 (2012).
A new study published in Science has, for the first time, detailed the use of a sequencing method for the quantitative mapping of 5-hydroxymethylcytosine (5hmC) in genomic DNA at single-nucleotide resolution. 5hmC is thought to be involved in the stem cell function during development. By analyzing the DNA of embryonic stem cells, the investigators have been able to learn more about the epigenetic role of TET, the enzyme responsible for the conversion of 5-methylcytosine (5mC) to 5hmC.
Previously, sequencing 5hmC with traditional bisulfite sequencing techniques had been problematic due to its similarity to 5mC. However, this study, a collaboration between the Babraham Institute (Cambridge, UK) and the University of Cambridge (Cambridge, UK), successfully developed a bisulfite technique that could differentiate between these two, which they called oxidative bilsulfite sequencing (oxBS-Seq). Michael Booth, one of the inventors of the technique explained: “We developed a chemistry that was specific for this modified DNA base, 5hmC. This allowed us to accurately distinguish between 5mC and 5hmC at single-base resolution in the genome.” This variation of the technique involved treating DNA with potassium perruthenate, which selectively oxidizes 5hmC to 5-formylcytosine; this reads as uracil after bisulfite conversion (like unmethylated cytosine). This produced a high-resolution map of 5hmC that could be analyzed for epigenetic activity.
The researchers implemented oxBS-Seq to map the DNA of mouse embryonic stem cells. In particular, they wanted to look at CpG islands for methylation activity in the mouse genome. The group identified 800 5hmC-containing CpG islands, which showed on average 3.3% hydroxymethylation. It is noted that high levels of 5hmC were found in CpG islands associated with transcriptional regulators such as LINE-1 elements. This indicates that these regions may undergo epigenetic reprogramming in embryonic stem cells. Miguel Branco, joint lead author, explained how this technique has benefited their research: “There was a real need in the field for a technique that would map both 5hmC and 5mC in the genome quantitatively and at high resolution. We applied this new technology to embryonic stem cells and immediately recognized its power in furthering our understanding of the biological functions of these DNA modifications.”
The authors believe that oxBS-Seq could have an important role to play in genome-mapping research, uncovering the dynamics of 5hmC, and the epigenetic role of TET. Wolf Reik, leader of the study at the Babraham Institute, highlights the importance of this work: “It has recently become apparent that in addition to DNA methylation, there are other modifications of DNA, such as hydroxymethylation. This suggests that DNA modifications are more dynamic than we previously thought. With the new method we are now in a position to map these modifications at great precision, and to relate them to stem cell function, aging and perhaps, more generally, to how the environment interacts with the genome.”
Source: Booth MJ, Branco MR, Ficz G et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science doi:10.1126/science.1220671 (2012) (Epub ahead of print); University of Cambridge Research News: www.cam.ac.uk/research/news/scientists-have-demonstrated-a-new-technique-that-will-transform-epigenetics-research.
– All stories written by Jonathan Wilkinson