
Abstract
Background: Targeted recruitment of chromatin-modifying enzymes to clusters of CpG dinucleotides contributes toward the formation of accessible chromatin. By interprimate comparison we previously identified the set of nonpolymorphic human-specific CpGs (CpG ‘beacons’) and revealed that these loci were enriched for human disease traits. Due to their human-specific CpG density change, extreme CpG ‘beacon’ clusters (≥20 CpG beacons/kb) were predicted to identify permissive chromatin peaks within the human genome. Aim: We set out to explore these sequence-defined regions for evidence of an active chromatin signature. Results: Using available comparative primate epigenomic data from neurons of the prefrontal cortex, we show that these CpG ‘beacon’ clusters are indeed enriched for being human-specific H3K4me3 peaks (χ2: p < 2.2 × 10−16) and thus predictive of permissive chromatin states. These sequence regions had a higher predictive value than previous selective analyses. We also show that both human-specific H3K4me3 and CpG ‘beacon’ clusters are increased within current and ancestral telomeric regions, supporting an association with recombination, which is higher towards the distal ends of chromosomes. Conclusion: Therefore, CpG-focused comparative sequence analysis can precisely pinpoint chromatin structures that contribute to the human-specific phenotype and further supports an integrated approach in genomic and epigenomic studies.
All figures are viewable in color online at www.tandfonline.com/doi/full/10.2217/epi.13.74
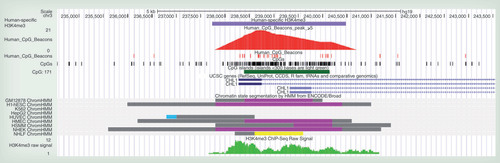
Outer ring: Chromosomes with banding patterns. Dense edge ring: Enredo-Pecan-Ortheus [Citation10,Citation11] h1c1o1 coverage of these chromosomes. Blue peaks: extreme CpG ‘beacon’ peak locations. Human-specific H3K4me3 location with false discovery rate log p values compared to chimpanzee (orange), macaque (green) and overlapping chimpanzee and macaque (olive). Location of ANKRD11, CHL1, DPP10, HAR1A and PSD4 indicated with colocating CpG ‘beacon’ clusters and human-specific H3K4me3. The highlighted yellow wedge is the location of the chromosome 2qFus (2q13–2q14.1) with enrichment of both extreme CpG ‘beacon’ clusters and of Human-specific H3K4me3 peaks (χ2: p = 1.045 × 10-5).
![Figure 2. Human chromosomes 2, 3, 16 and 20.Outer ring: Chromosomes with banding patterns. Dense edge ring: Enredo-Pecan-Ortheus [Citation10,Citation11] h1c1o1 coverage of these chromosomes. Blue peaks: extreme CpG ‘beacon’ peak locations. Human-specific H3K4me3 location with false discovery rate log p values compared to chimpanzee (orange), macaque (green) and overlapping chimpanzee and macaque (olive). Location of ANKRD11, CHL1, DPP10, HAR1A and PSD4 indicated with colocating CpG ‘beacon’ clusters and human-specific H3K4me3. The highlighted yellow wedge is the location of the chromosome 2qFus (2q13–2q14.1) with enrichment of both extreme CpG ‘beacon’ clusters and of Human-specific H3K4me3 peaks (χ2: p = 1.045 × 10-5).](/cms/asset/eef5f4c8-5d84-4f4e-90a0-a07f438f0f03/iepi_a_12324878_f0002.jpg)
This analysis is within the primate-alignable (EPO [Citation10,Citation11]) portion of the human genome. EPO: Enredo-Pecan-Ortheus; FDR: False discovery rate.
![Figure 3. Stronger human-specific H3K4m3 peaks identified (FDR p value) with CpG ‘beacon’ density in both human versus chimpanzee (A), and versus rhesus macaque (B) (Wilcoxon test CpG ‘beacon’ 0 vs > = 20, p = 3.57 × 10-3 chimpanzee and p = 2.50 × 10-2 macaque (p = 0 values capped at 10-200).This analysis is within the primate-alignable (EPO [Citation10,Citation11]) portion of the human genome. EPO: Enredo-Pecan-Ortheus; FDR: False discovery rate.](/cms/asset/1d78347f-4911-4893-a8a6-8b0bfc8e6b19/iepi_a_12324878_f0003.jpg)
CpG ‘beacon’ clusters identify hs-H3K4me3 peaks more successfully than extreme human nucleotide change or studies of selection. Scatter plot of permutated data for random selection of sets of one to 1000 loci from the total of 18,665 human peaks (1000× randomization each) that then overlap the 410 hs-H3K3me4 loci. Upper line: Loess regression line of maximum simulation value; golden points: average for each random selection with average Loess regression line overlaid in grey; red squares: CpG ‘beacon’ clusters; green squares: CpG ‘beacon’ clusters adjusted for the fact that the primate EPO alignment only contains 257 hs-H3K4me3 peaks; blue squares: selective studies – HACNS (HACNS – 0/992) [Citation43] and ANC (ANC – 6/1356) [Citation44], Select_2x, _3x and _4+x are overlaps with studies combined by Akey [Citation42] with loci colocating in 2, 3 or 4 or more studies. HARs are the 49 top HARs [Citation21]. HAR_202 is an expanded set of 202 HARs, with this set the only nonbeacon analysis to perform well, as this larger grouping has a strong overlap with CpG ‘beacons’ (∼85.7% of the hs-H3K4me3 overlapping CpG ‘beacons’ ≥10/kb). ANC: Accelerated noncoding; CB_10: CpG ‘beacon’ clusters ≥10/kb; CB_15: CpG ‘beacon’ clusters ≥15/kb; CB_20: CpG ‘beacon’ clusters ≥20/kb; HAR: Human accelerated regions; HACNS: Human accelerated conserved noncoding; hs-H3K4me3: Human-specific H3K4me3.
![Figure 4. Comparison of studies: proportion of identified loci with overlapping human-specific H3K4me3.CpG ‘beacon’ clusters identify hs-H3K4me3 peaks more successfully than extreme human nucleotide change or studies of selection. Scatter plot of permutated data for random selection of sets of one to 1000 loci from the total of 18,665 human peaks (1000× randomization each) that then overlap the 410 hs-H3K3me4 loci. Upper line: Loess regression line of maximum simulation value; golden points: average for each random selection with average Loess regression line overlaid in grey; red squares: CpG ‘beacon’ clusters; green squares: CpG ‘beacon’ clusters adjusted for the fact that the primate EPO alignment only contains 257 hs-H3K4me3 peaks; blue squares: selective studies – HACNS (HACNS – 0/992) [Citation43] and ANC (ANC – 6/1356) [Citation44], Select_2x, _3x and _4+x are overlaps with studies combined by Akey [Citation42] with loci colocating in 2, 3 or 4 or more studies. HARs are the 49 top HARs [Citation21]. HAR_202 is an expanded set of 202 HARs, with this set the only nonbeacon analysis to perform well, as this larger grouping has a strong overlap with CpG ‘beacons’ (∼85.7% of the hs-H3K4me3 overlapping CpG ‘beacons’ ≥10/kb). ANC: Accelerated noncoding; CB_10: CpG ‘beacon’ clusters ≥10/kb; CB_15: CpG ‘beacon’ clusters ≥15/kb; CB_20: CpG ‘beacon’ clusters ≥20/kb; HAR: Human accelerated regions; HACNS: Human accelerated conserved noncoding; hs-H3K4me3: Human-specific H3K4me3.](/cms/asset/7d2f8eaa-986b-4e3f-aedc-0e604356af53/iepi_a_12324878_f0004.jpg)
The histone methylase PRDM9 directs recombination via motif-specificity, with resultant biased gene conversion within these regions increasing GC content and therefore CpG numbers. Dense CpG regions are able to recruit chromatin-modifying factors such as Cpf1, which lead to the formation of permissive H3K4me3 chromatin domains. Due to the strong association of PRDM9 with recombination [Citation51], ancestral alleles of this rapidly evolving DNA-binding factor [Citation52] are hypothesized to be implicated in this recombination-associated biased gene conversion [Citation39].
![Figure 5. The hypothesis associating potential recombination-driven permissive chromatin formation with CPGi formation via CpG ’beacons’.The histone methylase PRDM9 directs recombination via motif-specificity, with resultant biased gene conversion within these regions increasing GC content and therefore CpG numbers. Dense CpG regions are able to recruit chromatin-modifying factors such as Cpf1, which lead to the formation of permissive H3K4me3 chromatin domains. Due to the strong association of PRDM9 with recombination [Citation51], ancestral alleles of this rapidly evolving DNA-binding factor [Citation52] are hypothesized to be implicated in this recombination-associated biased gene conversion [Citation39].](/cms/asset/ed30dd98-b171-447d-9010-1ec3b7319ae7/iepi_a_12324878_f0005.jpg)
Background
Clusters of CpG dinucleotides, termed CpG Islands (CpGis), act as a genomic platform for gene expression by providing an accessible environment for the binding of the basal transcription machinery [Citation1,Citation2]. They facilitate this setting, due to both their distinctive density and predominately unmethylated state, by recruiting specific factors that modify the three dimensional structure of the local chromatin [Citation3,Citation4]. Hence these islands reside within an expanse of repressed genome, forming active or transcriptionally competent promoters [Citation5] and possess the characteristic permissive H3K4me3 histone signature [Citation6]. This chromatin configuration is itself associated with expression, but also may further enable the recruitment of components required for this activity [Citation7,Citation8].
Due to this direct effect on 3D chromatin structure by CpG dinucleotides [Citation5], and the potential for regulatory modulation it creates, we previously examined the primate-alignable portion of human genome for nucleotide sequence change that has led to the formation or maintenance of human-specific CpGs [Citation9]. This was performed by sequence comparison of six primates (Enredo-Pecan-Ortheus [EPO] alignment including: human, chimpanzee, gorilla, orangutan, rhesus macaque and marmoset [Citation10,Citation11]). The resultant nonpolymorphic human-specific CpGs were termed human CpG ‘beacons’ and identify prospective novel human-specific regulatory regions. These CpG ‘beacons’ are now shown to be markedly enriched in the dynamic portion of the human DNA methylome [Citation12]. We then identified 21 regions of extreme CpG ‘beacon’ clusters (empirical p < 1 × 10-3) within the human genome. These were shown to colocate with four causative genes in monogenic mental/developmental disorders (ANKRD11 [Citation13], CHL1 [Citation14], EHMT1 [Citation15] and VLDLR [Citation16,Citation17]), and genes implicated with complex traits such as autism and psychiatric disease (ANKRD11 [Citation18], DLGAP2 [Citation19] and DPP10 [Citation18,Citation20]), as well as reidentifying the noncoding RNA, HAR1A [Citation21], implicated in cortex development, by precisely hitting its CpGi. These loci were significantly statistically enriched for neurological trait-related genes. As methylation is inversely correlated with CpGi density [Citation22,Citation23], we were also able to indicate the functional consequences of species-specific CpG density change, through comparative DNA methylome analysis in human, chimpanzee and macaque, with expected changes in methylation in orthologous CpGi.
A recently published comparative chromatin analysis for human-specific H3K4me3 within the prefrontal cortex (PFC) between human, chimpanzee and macaque identified 410 loci with a human-specific peak gain [Citation24]. This dataset provided the ideal opportunity to test whether the hypothesized structural effect of CpG ‘beacon’ clusters did in fact lead to human-specific chromatin modification. Furthermore, these data are derived from a cortical region fundamental in human-derived higher functioning [Citation25,Citation26].
Therefore, we tested this hypothesis and identified that CpG ‘beacon’ clusters are indeed enriched for these human-specific accessible chromatin regions. Furthermore they are increasingly likely to precisely locate the most significant fraction of these peaks. This supports the idea that human species-specific CpG ‘beacons’ have played a critical role in human evolution, as they locate significant regions of human-specific functional epigenomic change in the genome, and that CpG-focused comparative sequence analysis between the closely related primate family can be highly informative.
Methods
Data
Human-specific H3K4me3 PFC peak locations were taken from the supplementary data (Supplementary Table S3) of Shulha et al. [Citation24].
Location of human Chip-seq H3K4me3 PFC peaks were accessed from Maunakea et al. [Citation27], via the UCSC Genome Browser Table Browser: Regulation; UCSF Brain DNA Methylation; ucsfChipSeqH3K4me3BrainCoverage. The sequencing reads are available through the NCBI SRA (study accession number SRP002318) [Citation28]. The CpG ‘beacon’ data is browser viewable within UCSC [Citation29].
Analysis
All statistical calculations and simulations were performed in R [Citation30]. Wilcoxon Test was used for hs-H3K4me3 and CpG ‘beacon’ comparison between loci with 0 CpG ‘beacons’ and those with ≥20, using the FDR p-values calculated by Shulha et al. for human versus chimpanzee and macaque regions [Citation24]. For this FDR comparison analysis, those loci with an FDR p = 0 were capped at a p = 1 × 10-200. Genome intersect comparisons were via the BEDTools package command intersectBed [Citation31]. Genomic representation plot was constructed with Circos [Citation32]. Genomic enrichment calculations performed with GREAT, by the region-based binominal method, with regional default for associated genomic regions, but with extension reduced to 100 kb [Citation33]. A locus was considered to reside in a distal, or telomeric region, if it overlapped the first or last chromosome band (coordinates via Ensembl Biomart). All human genomic coordinates given are in the GRCh37/hg19 build.
Results
Primate comparative sequence analysis predicts human-specific chromatin state
A set of 410 human-specific peaks of the activating chromatin modification, H3K4me3, were identified from a total of 34,639 human peaks, by a comparative analysis of the PFC of human, chimpanzee and macaque by Shulha et al. [Citation24]. Within the 21 extreme CpG ‘beacon’ peak regions we previously identified [Citation9], available data from Maunakea et al. had shown that 8 possessed H3K4me3 PFC peaks [Citation9,Citation27]. Comparison of these 8 peaks with the Shulha et al. dataset revealed 5 to be identified as human-specific H3K4me3 (hs-H3K4me3) peaks. Therefore the H3K4me3 signal in these CpG ‘beacon’ clusters is strongly enriched for being present only in human and not in chimpanzee or macaque (χ2: p < 2.2 × 10-16). These five hs-H3K4me3 peaks were precisely located by extreme CpG ‘beacon’ clusters (≥20 CpG ‘beacons’/kb) within or near the genes; ANKRD11, CHL1, DPP10, HAR1A and PSD4 (see & Supplementary Figure 1; accessible online at www.tandfonline.com/doi/suppl/10.2217/epi.13.74).
Randomization via ShuffleBed [Citation31] and genome structure correction (GSC) enrichment [Citation34,Citation35] were highly significant (both p < 1 × 10-5). However, both the extreme CpG ‘beacon’ clusters and H3K4me3 peaks are not likely to be found with equal chance throughout the entire genome, but are highly likely to reside within CpG-dense regions. To account for this, we permutated the 21 loci, representing random extreme CpG clusters, only from the total of 18,665 human PFC H3K4me3 peaks, out of the total of 37,902 (identified from Maunakea et al. [Citation27], as the total set genome co-ordinates are not available from Shulha et al. [Citation24]) that overlap CpG-dense regions (22,374 Ensembl CpGis). With 10,000 permutations this did not exceed the observed five peaks (average: 0.466 ± 0.677 overlapping peaks per genome simulation, empirical p < 1 × 10-5). Therefore while as much as ∼83.4% of brain PFC CpGi’s possess the H3K4me3 signature, only ∼1.8% have human-specific H3K4me3, whereas by contrast, the CpG ‘beacon’ clusters are statistically strongly enriched with ∼23.8% having human-specific H3K4me3 (five out of 21 clusters).
Further statistical overlap at lower CpG cluster thresholds
CpG ‘beacon’ H3K4me3 overlaps were not only restricted to the most extreme clusters (≥20 CpG ‘beacons’/kb) but also found at lower levels. At a CpG ‘beacons’ density level of 15/kb, 11 out of a total of 63 cluster regions overlap with hs-H3K4me3 (simulation average: 1.387 ± 1.164) and 20 out of a total of 254 cluster regions overlaps at CpG ‘beacons’ density level of 10/kb (simulation average: 5.604 ± 2.347). Permutation analysis therefore for both of these results are highly statistically unlikely (all empirical p < 1 × 10-5).
When we initially calculated the set of the extreme CpG ‘beacon’ clusters we took a conservative genome-wide empirical cut-off of p < 1 × 10-3, which was reached at a density level of 20/kb CpG ‘beacons’. The permutation however does not become nonsignificant (p > 0.05) until a density level of <17/kb (p = 0.210) and furthermore even at the 17/kb level the binominal genomic enrichment results via GREAT [Citation33] for cognition is significant (p = 7.6 × 10-6, FDR Q = 3.36 × 10-2). The H3K4me3 peak overlap at 17/kb is 7, out of a possible 36, (empirical p < 1 × 10−5, see ). While the lower CpG ‘beacon’ density levels of 15/kb and 10/kb are not statistical outliers in their own right, this still does not negate their potential functional importance, and the enrichment for hs-H3K4me3 in these loci strongly supports this.
Additionally, the defined CpG ‘beacons’ were identified only within the ∼80% of the human genome with rigorous primate homology from the EPO sequence block alignments [Citation10,Citation11] that included, at least, human, chimpanzee and one other primate sequence (abbreviated as the ‘h1c1o1’ genome). Of the total 410 hs-H3K4me3 peak regions, only 257 lie within this majority of the genome, therefore, an under-representation (∼20% loss genome but 34.15% reduction in peaks; χ2: p < 2.2 × 10-16). Thus the primate nonhomologous regions contain more identified hs-H3K4me3 peaks. This may be due to stronger genetic divergence within these regions having a consequential stronger influence on the epigenome, but also could be contributed to by a higher potential analytical bias component. 30 (∼12%) of these 257 peaks overlap with a CpG beacon peak of at least ≥5/kb.
Predicted H3K4me3 peaks have the strongest evidence for a human-specific state
By utilizing the FDR p-value calculated by Shulha et al. for their H3K4me3 peaks [Citation24], we find that the most extreme CpG ‘beacon’ clusters that overlap with hs-H3K4me3 peaks have a significantly higher likelihood of being truly human-specific peaks. These loci have more significant FDR p-values in comparison with both potential chimpanzee and macaque loci (loci with CpG ‘beacons’ = 0, compared with those with ≥20, Wilcoxon p = 3.57 × 10-3 for human vs chimpanzee and p = 2.50 × 10-2 for human versus macaque, see ). Consequently, while the 410 peaks have variable certainty as to being truly human-specific, as represented by these FDR values, the most likely hs-H3K4me3 lie within extreme CpG clusters. Therefore the conservatively calculated enrichment within these hs-H3K4me3 would be even stronger if false-positive hs-H3K4me3 peaks were excluded. Additional datasets from the other closely related primates, including orangutan and gorilla, would aid the reduction in false-positive human-specific peaks, however these are currently not publically available.
Double peaks & telomeric bias
Peaks of hs-H3K4me3 were also identified by Shulha et al. to occur in pairs or groups within 0.5–1 Mb, with an approximately two- to three-fold enrichment. Recent ENCODE data has identified an average peak region of long range interaction of promoters, enhancers and CTCF to be around ∼120 kb upstream of the TSS [Citation36]. This grouping propensity is also seen within the CpG ‘beacon’ overlap peaks (i.e., DPP10 and EXOC2, among others (Supplementary Table 1)). These double peaks had an average genomic distance between them of ∼126 kb. Shulha et al. showed evidence of these close double peak regions having physical interaction by ChIA-PET. ChromHMM segmentation analysis [Citation37] found that colocating with H3K4me1, a high level of H3K4me3 (61–80%) was also in Strong Enhancer Type 4, thus potentially implicating some of these regions in this role.
We previously identified a distal chromosome or telomeric bias in CpG ‘beacon’ clusters (CpG ‘beacon’ cluster 20/kb = 52.3% in terminal chromosome bands [Citation9]), due to the role recombination-associated biased gene conversion (BGC) plays in their formation. This being the biochemical bias in the DNA repair mechanism that occurs in heteroduplexed DNA, formed during crossing over, leading to clustered weak-to-strong ’biased’ substitutions (AT to GC) [Citation38,Citation39]. Therefore, we assessed whether hs-H3K4me3 also have a telomeric bias which could be contributing to these peak groupings. We calculated there is in fact a >twofold enrichment within these telomeric regions compared to all human H3K4me3 peaks (χ2: p < 2.2 × 10-16).
We also previously identified a strong clustering bias of CpG ‘beacon’ clusters, within the historic chromosome 2qFus region, due to past recombination in these archaic telomeric regions [Citation40]. Examining the hs-H3K4me3 peaks, these also show an enrichment for this 2qfus region (2q13–2q14.1 – chr2: 110,200,001–118,800,000) with an average 0.699 peaks/Mb compared with ∼0.131 peaks/Mb across the genome and an approximately sixfold enrichment over the expected hs-H3K4me3 compared to all the human H3K4me3 peaks present here (χ2: p = 1.045 × 10-5; see ). Hence this further supports the model of recombination-associated BGC increase in CpGs leading to the formation of H3K4me3 peaks. Cohen et al. also recently proposed BGC islands, as one of the categories of CpG islands, due to their formation by this process [Citation41].
CpG ‘beacons’ predict human-specific H3K4me3 peaks greater than other comparative sequence change
Shulha et al. compared the identified hs-H3K4me3 peaks with known regions of accelerated human-specific sequence change, such as the human accelerated regions (HARs) [Citation21] and found only minimal overlap (1/49 loci = HAR1A), as well as nine further studies of positive selection collated together by Akey [Citation42], which also showed low intersection. Due to this inadequate overlap they concluded that comparative sequence analysis was poorly informative of potential functional epigenomic differences [Citation24] and thus that important changes in chromatin structure and function cannot be identified by comparative genomic analysis alone. Furthermore, we also compared the hs-H3K4me3 regions with the human accelerated conserved noncoding (HACNS – 0/992 loci) [Citation43] and accelerated noncoding (ANC – 6/1356 loci) [Citation44] sets and these also show minimal colocation. However, as shown, the specific CpG dinucleotide change, indicated by CpG ‘beacons’ through six primate comparison, enabled a far better prediction of hs-H3K4me3 than any of these methods. Therefore important changes in chromatin structure and epigenomic function can be identified by specific comparative genomic analysis.
We simulated these data by permutation with random selections sets of one to 1000 loci from the total of 18,665 human peaks (1000 × randomization each) that then overlap the 410 hs-H3K3me3 and plotted these results on a scatter plot also with the observed overlaps for all these mentioned studies (blue squares) and the CpG ‘beacon’ data (red and green Squares) (see ). This clearly shows that while the selection studies perform either the same or worse than the simulation values, the CpG ‘beacon’ clusters overlap is far in excess of the maximum simulation results (all empirical p < 1 × 10-5), with those adjusted for the reduced numbers within the primate EPO alignment performing the best (green Squares).
The only study that approaches the success of the CpG ‘beacons’ is the expanded set of 202 HARs (∼85.6 kB), as while the top 49 HARs perform poorly, when this set is extended further these loci begin to capture more CpG ‘beacon’ clusters, such that ∼85.7% of the hs-H3K4me3-overlapping HAR_202 regions are CpG ‘beacon’ clusters of ≥10/kb (12 out of 14). This overlap, as discussed previously, indicates the strong role of BGC in the formation of both CpG ‘beacon’ clusters and regions of extreme species-specific dinucleotide divergence in the human genome, driven by recombination [Citation9,Citation38]. Thus, further indicating how this process, with a by-product of modulating sequence, can affect genomic 3D structure in these extreme locations.
The simulation also clearly displays why the comparison against the 9 positive selective studies as used by Shulha et al. [Citation24] is inappropriate to identify human-specific change, as these studies use human population variation and linkage disequilibrium data that is too recent to identify significant genetic changes that have led to variation between primates. Furthermore, the other comparative studies lose power in comparison with the ‘beacon’ analysis, as they regard all nucleotide change as equal, are focused wider than an interprimate analysis, and in those that center exclusively on nontranscript overlapping sequence, such as the ANC regions [Citation44], may exclude many promoter hs-H3K4me3 regions that reside 5′ within first exons, or intragenic promoter CpGi loci. The inaccuracy of using human population data can be starkly seen by the fact that the genome space associated with two or more selective studies from Akey is 244.7 Mb, compared with only 29.9 kb for the extreme CpG ‘beacon’ ≥20/kb peaks regions, or ∼0.01% of this, and yet the latter has 23.8% successful overlap with hs-H3K4me3 peaks versus 2.36% of the former.
Discussion
DNA methylation is essential for mammalian development and is almost exclusively restricted to the CpG dinucleotide in differentiated cells [Citation45]. Therefore, this epigenomic mechanism requires the presence of the CpG genetic sequence template in order to facilitate the modification’s dynamic functionality. Although a lower level of non-CpG cytosine methylation has been identified to occur in the human brain, predominately at CACC motifs [Citation46]. We previously identified, by interprimate comparative analysis, human-specific CpG ‘beacon’ clusters and found these to be enriched for human disease and phenotype loci. The facilitated epigenetic role that these clusters of CpGs provide is not restricted to DNA methylation, but may also be involved in enabling other modifications of DNA, including hydroxymethylation [Citation47]. Furthermore, we predicted that these clusters would influence chromatin structure, due to their significant human-specific change in CpG density. Using the PFC hs-H3K4me3 data from Shulha et al. [Citation24] we have shown strong evidence of this. This sequence relationship is only associated with H3K4me3, and presently only tested in PFC, but further analysis, when other comparative epigenomic tissue signatures become available, may be revealing.
Mechanisms have evolved to reduce the local functional impact of transposable elements, such as Alu elements [Citation48], which while possessing high CpG content, are usually methylated [Citation49]. However sequence divergence of homologous loci through highly dissimilar species-specific recombination [Citation50], driven by the rapidly evolving recognition motif of PRDM9 [Citation51,Citation52], can lead to BGC-associated increase in local GC content [Citation53]. This can concurrently increase CpG dinucleotides and form increasingly dense [Citation54] or new CpGi, within these hotspots [Citation55], even without requiring the force of positive selection [Citation41]. These CpGi regions, recruit chromatin-modifying enzymes, such as Cfp1, as has been experimentally shown by CpG island inclusion experiments in a mouse model by Thomson et al. [Citation3]. This, in turn, leads to the targeted formation of accessible H3K4me3 chromatin structure in the appropriate genomic regions [Citation56]. This chromatin signature may not itself be causative in expression, but is indicative of a permissive promoter formation. Furthermore independent of transcription status, both CpG content and CpGi width correlate with local nucleosome depletion [Citation57].
Dreszer et al. has previously identified outlying regions of high BGC with loci of extreme sequence diverge, and these regions also have a telomeric or distal bias, due to the higher level of recombination that occurs there [Citation38,Citation58]. CpG ‘beacon’ clusters also share these properties [Citation9] and thus link recombination with H3K4me3 formation (see Hypothesis in ). Therefore the extreme CpG increase identified in human, within these loci, is not due to severe concurrent loss of CpGs in all of the other primates. Thus we have shown by our primate-specific comparison that these sequence-directed or associated epigenetic changes, identified in murine models, in fact occur in human, and furthermore within the significant brain tissue of the human PFC.
Comparing DNA sequence can identify human-specific epigenomic variation that can increase the understanding of brain function unique to humans. Epigenetics complements genetics, and is not in conflict with it, justifying an integrated approach in genomic and epigenomic analyses for disease studies [Citation59–61]. Structural effects on the genome through chromatin modification can in fact be deduced by direct sequence comparison, especially when the importance of species–specific CpGs ‘beacons’ in the formation of H3K4me3 is acknowledged and the additional power of comparison between multiple closely-related primates is employed.
Conclusion
We have shown that CpG ‘beacon’ clusters can precisely locate hs-H3K4me3 and, furthermore, are more enriched in the strongest human-specific peaks. This supports our work that the most extreme CpG ‘beacon’ peaks map to tissue-invariant peaks of accessible H3K4me3 chromatin, specific only to the human genome, and implicated in the formation of the human phenotype [Citation9].
Future perspective
The integration of genomic, epigenomic and transcriptomic sequence data will enable an increasingly comprehensive understanding of biology. Rapid advances in sequencing technology will speed this powerful combinatorial analysis and facilitate detailed exploration of the evolution of malignant, population and species–specific variation. Increased understanding of how genetic sequence change, such as in CpGs and transcription factor motifs, may be associated with active or passive epigenomic alterations will aid deciphering these data. Furthermore, through these methods, the identification of functional differences, between humans and other closely related primates, will help explain the human-specific phenotype and increase the understanding of human pathophysiology.
Table 1. Human-specific CpG ‘beacon’ clusters with Human-specific H3K4me3.
We observed that genetic human-specific CpG (CpG ‘beacon’) clusters are enriched for human-specific H3K4me3 peaks identified in the prefrontal cortex.
Therefore, these CpG ‘beacon’ clusters are predictive of the location of this permissive chromatin state within the human genome.
The strongest human-specific H3K4me3 peaks (lowest FDR) have the strongest CpG ‘beacon’ association.
There is also an association with recombination, as both human-specific H3K4me3 and CpG ‘beacon’ clusters are increased within distal or telomeric chromosome regions (terminal chromosome bands).
The process of biased gene conversion is implicated in these dramatic CpG dinucleotide increases.
Financial & competing interests disclosure
This work was supported by the Wellcome Trust (99148), EU-FP7 projects EPIGENESYS (257082) and BLUEPRINT (282510), and a Royal Society Wolfson Research Merit Award to S Beck (WM100023). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Blackledge NP , KloseR . CpG island chromatin: a platform for gene regulation . Epigenetics 6 ( 2 ), 147 – 152 ( 2011 ).
- Illingworth RS , BirdAP . CpG islands–’a rough guide’ . FEBS Lett. 583 ( 11 ), 1713 – 1720 ( 2009 ).
- Thomson JP , SkenePJ , SelfridgeJ et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1 . Nature 464 ( 7291 ), 1082 – 1086 ( 2010 ).
- Blackledge NP , ZhouJC , TolstorukovMY , FarcasAM , ParkPJ , KloseRJ . CpG islands recruit a histone H3 lysine 36 demethylase . Mol. Cell 38 ( 2 ), 179 – 190 ( 2010 ).
- Bird A . The dinucleotide CG as a genomic signalling module . J. Mol. Biol. 409 ( 1 ), 47 – 53 ( 2011 ).
- Kouzarides T . Chromatin modifications and their function . Cell 128 ( 4 ), 693 – 705 ( 2007 ).
- Guenther MG , LevineSS , BoyerLA , JaenischR , YoungRA . A chromatin landmark and transcription initiation at most promoters in human cells . Cell 130 ( 1 ), 77 – 88 ( 2007 ).
- Vermeulen M , MulderKW , DenissovS et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4 . Cell 131 ( 1 ), 58 – 69 ( 2007 ).
- Bell CG , WilsonGA , ButcherLM , RoosC , WalterL , BeckS . Human-specific CpG “beacons” identify loci associated with human-specific traits and disease . Epigenetics 7 ( 10 ), 1188 – 1199 ( 2012 ).
- Paten B , HerreroJ , BealK , FitzgeraldS , BirneyE . Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs . Genome Res. 18 ( 11 ), 1814 – 1828 ( 2008 ).
- Paten B , HerreroJ , FitzgeraldS et al. Genome-wide nucleotide-level mammalian ancestor reconstruction . Genome Res. 18 ( 11 ), 1829 – 1843 ( 2008 ).
- Ziller MJ , GuH , MullerFet al. Charting a dynamic DNA methylation landscape of the human genome . Nature 500 , 477 – 481 ( 2013 ).
- Sirmaci A , SpiliopoulosM , BrancatiF et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia . Am. J. Hum. Genet. 89 ( 2 ), 289 – 294 ( 2011 ).
- Frints SG , MarynenP , HartmannD et al. CALL interrupted in a patient with non-specific mental retardation: gene dosage-dependent alteration of murine brain development and behavior . Hum. Mol. Genet. 12 ( 13 ), 1463 – 1474 ( 2003 ).
- Kleefstra T , BrunnerHG , AmielJ et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome . Am. J. Hum. Genet. 79 ( 2 ), 370 – 377 ( 2006 ).
- Boycott KM , FlavelleS , BureauA et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification . Am. J. Hum. Genet. 77 ( 3 ), 477 – 483 ( 2005 ).
- Trommsdorff M , GotthardtM , HiesbergerT et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2 . Cell 97 ( 6 ), 689 – 701 ( 1999 ).
- Marshall CR , NoorA , VincentJB et al. Structural variation of chromosomes in autism spectrum disorder . Am. J. Hum. Genet. 82 ( 2 ), 477 – 488 ( 2008 ).
- Pinto D , PagnamentaAT , KleiL et al. Functional impact of global rare copy number variation in autism spectrum disorders . Nature 466 ( 7304 ), 368 – 372 ( 2010 ).
- Djurovic S , GustafssonO , MattingsdalM et al. A genome-wide association study of bipolar disorder in Norwegian individuals, followed by replication in Icelandic sample . J. Affect. Disord. 126 ( 1-2 ), 312 – 316 ( 2010 ).
- Pollard KS , SalamaSR , LambertN et al. An RNA gene expressed during cortical development evolved rapidly in humans . Nature 443 ( 7108 ), 167 – 172 ( 2006 ).
- Eckhardt F , LewinJ , CorteseR et al. DNA methylation profiling of human chromosomes 6, 20 and 22 . Nat. Genet. 38 ( 12 ), 1378 – 1385 ( 2006 ).
- Weber M , HellmannI , StadlerMB et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome . Nat. Genet. 39 ( 4 ), 457 – 466 ( 2007 ).
- Shulha HP , CrisciJL , ReshetovD et al. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons . PLoS Biol. 10 ( 11 ), e1001427 ( 2012 ).
- Semendeferi K , TefferK , BuxhoevedenDP et al. Spatial organization of neurons in the frontal pole sets humans apart from great apes . Cereb. Cortex 21 ( 7 ), 1485 – 1497 ( 2011 ).
- Varki A , GeschwindDH , EichlerEE . Explaining human uniqueness: genome interactions with environment, behaviour and culture . Nat. Rev. Genet. 9 ( 10 ), 749 – 763 ( 2008 ).
- Maunakea AK , NagarajanRP , BilenkyM et al. Conserved role of intragenic DNA methylation in regulating alternative promoters . Nature 466 ( 7303 ), 253 – 257 ( 2010 ).
- NCBI SRA. Evolutionarily conserved role of intragenic DNA methylation in regulating alternative promoters. =www.ncbi.nlm.nih.gov/Traces/sra/?study=SRP002318
- UCL. Medical Genomics. www2.cancer.ucl.ac.uk/medicalgenomics/humanCpGBeacons/trackList.php
- Ihaka R , GentlemanR . R: A Language for Data Analysis and Graphics . J. Comp. Graph. Stat. 5 ( 3 ), 299 – 314 ( 1996 ).
- Quinlan AR , HallIM . BEDTools: a flexible suite of utilities for comparing genomic features . Bioinformatics 26 ( 6 ), 841 – 842 ( 2010 ).
- Krzywinski M , ScheinJ , BirolI et al. Circos: an information aesthetic for comparative genomics . Genome Res. 19 ( 9 ), 1639 – 1645 ( 2009 ).
- Mclean CY , BristorD , HillerM et al. GREAT improves functional interpretation of cis-regulatory regions . Nat. Biotechnol. 28 ( 5 ), 495 – 501 ( 2010 ).
- Bickel PJ , BoleyN , BrownJB , HuangHY , ZhangNR . Subsampling Methods for Genomic Inference . Ann. Appl. Stat. 4 ( 4 ), 1660 – 1697 ( 2010 ).
- Yip KY , ChengC , BhardwajN et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors . Genome Biol 13 ( 9 ), R48 ( 2012 ).
- Sanyal A , LajoieBR , JainG , DekkerJ . The long-range interaction landscape of gene promoters . Nature 489 ( 7414 ), 109 – 113 ( 2012 ).
- Ernst J , KellisM . ChromHMM: automating chromatin-state discovery and characterization . Nat. Methods 9 ( 3 ), 215 – 216 ( 2012 ).
- Dreszer TR , WallGD , HausslerD , PollardKS . Biased clustered substitutions in the human genome: the footprints of male-driven biased gene conversion . Genome Res. 17 ( 10 ), 1420 – 1430 ( 2007 ).
- Capra JA , HubiszMJ , KostkaD , PollardKS , SiepelA . A model-based analysis of GC-biased gene conversion in the human and chimpanzee genomes . PLoS Genet. 9 ( 8 ), e1003684 ( 2013 ).
- Fan Y , NewmanT , LinardopoulouE , TraskBJ . Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions . Genome Res. 12 ( 11 ), 1663 – 1672 ( 2002 ).
- Cohen NM , KenigsbergE , TanayA . Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection . Cell 145 ( 5 ), 773 – 786 ( 2011 ).
- Akey JM . Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res. 19 ( 5 ), 711 – 722 ( 2009 ).
- Prabhakar S , ViselA , AkiyamaJA et al. Human-specific gain of function in a developmental enhancer . Science 321 ( 5894 ), 1346 – 1350 ( 2008 ).
- Bird CP , StrangerBE , LiuM et al. Fast-evolving noncoding sequences in the human genome . Genome Biol. 8 ( 6 ), R118 ( 2007 ).
- Smith ZD , MeissnerA . DNA methylation: roles in mammalian development . Nat. Rev. Genet. 14 ( 3 ), 204 – 220 ( 2013 ).
- Varley KE , GertzJ , BowlingKM et al. Dynamic DNA methylation across diverse human cell lines and tissues . Genome Res. 23 ( 3 ), 555 – 567 ( 2013 ).
- Branco MR , FiczG , ReikW . Uncovering the role of 5-hydroxymethylcytosine in the epigenome . Nat. Rev. Genet. 13 ( 1 ), 7 – 13 ( 2012 ).
- Zarnack K , KonigJ , TajnikM et al. Direct Competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements . Cell 152 ( 3 ), 453 – 466 ( 2013 ).
- Das R , DimitrovaN , XuanZ et al. Computational prediction of methylation status in human genomic sequences . Proc. Natl Acad. Sci. USA 103 ( 28 ), 10713 – 10716 ( 2006 ).
- Myers S , BowdenR , TumianA et al. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination . Science 327 ( 5967 ), 876 – 879 ( 2010 ).
- Baudat F , BuardJ , GreyC et al. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice . Science 327 ( 5967 ), 836 – 840 ( 2010 ).
- Oliver PL , GoodstadtL , BayesJJ et al. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa . PLoS Genet. 5 ( 12 ), e1000753 ( 2009 ).
- Galtier N , DuretL . Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution . Trends Genet. 23 ( 6 ), 273 – 277 ( 2007 ).
- Long HK , SimsD , HegerA et al. Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates . Elife 2 , e00348 ( 2013 ).
- Polak P , ArndtPF . Long-range bidirectional strand asymmetries originate at CpG islands in the human genome . Genome Biol. Evol. 1 , 189 – 197 ( 2009 ).
- Clouaire T , WebbS , SkeneP et al. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells . Genes Dev. 26 ( 15 ), 1714 – 1728 ( 2012 ).
- Fenouil R , CauchyP , KochF et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters . Genome Res. 22 ( 12 ), 2399 – 2408 ( 2012 ).
- Yu A , ZhaoC , FanY et al. Comparison of human genetic and sequence-based physical maps . Nature 409 ( 6822 ), 951 – 953 ( 2001 ).
- Birney E . Chromatin and heritability: how epigenetic studies can complement genetic approaches . Trends Genet. 27 ( 5 ), 172 – 176 ( 2011 ).
- Bell CG . Integration of genomic and epigenomic DNA methylation data in common complex diseases by haplotype-specific methylation analysis . Personalized Med. 8 ( 3 ), 243 – 251 ( 2011 ).
- Mattick JS . RNA driving the epigenetic bus . EMBO J. 31 ( 3 ), 515 – 516 ( 2012 ).