
Abstract
Chromatin is a macromolecular complex composed of DNA and histones that regulate gene expression and nuclear architecture. The concerted action of DNA methylation, histone post-translational modifications and chromatin-associated proteins control the epigenetic regulation of the genome, ultimately determining cell fate and the transcriptional outputs of differentiated cells. Deregulation of this complex machinery leads to disease states, and exploiting epigenetic drugs is becoming increasingly attractive for therapeutic intervention. Mass spectrometry (MS)-based proteomics emerged as a powerful tool complementary to genomic approaches for epigenetic research, allowing the unbiased and comprehensive analysis of histone post-translational modifications and the characterization of chromatin constituents and chromatin-associated proteins. Furthermore, MS holds great promise for epigenetic biomarker discovery and represents a useful tool for deconvolution of epigenetic drug targets. Here, we will provide an overview of the applications of MS-based proteomics in various areas of chromatin biology.
hPTM: Histone post-translational modification; MS: Mass spectrometry; PTM: Post-translational modification.
Epigenetic features, which include DNA methylation, nucleosome positioning and histone post-translational modifications (hPTMs), play a critical role in the regulation of gene activity and nuclear architecture, ultimately defining the fate of the different cell types of an organism starting from the same DNA sequence by determining the way genetic information is used in differentiated cells. In eukaryotes, DNA compaction depends on chromatin, a macromolecular complex composed of nucleosomes, which consist of 147 bp of DNA wrapped around a protein core formed by one histone H3–H4 tetramer and two histone H2A–H2B dimers. Histones contain a variety of dynamic and reversible PTMs. The type, location and combination of such modifications contribute to regulate the underlying DNA by affecting chromatin configuration and accessibility and by generating binding sites for the recruitment of protein complexes that mediate downstream events [Citation1,Citation2]. Such effector proteins, which translate the information encoded by the modification patterns into specific biological outcomes, are known as chromatin ‘readers’, while the enzymes responsible for placing and removing the hPTMs are known as ‘writers’ and ‘erasers’, respectively. In addition, histone variants – namely, histones encoded by separate genes and characterized by distinct sequences and functionality – are locally enriched at distinct chromatin regions and also influence chromatin-related processes [Citation3,Citation4].
Current DNA sequencing technologies have enabled the comprehensive genomic mapping of various features of the epigenome, including DNA and histone modifications, nucleosome positioning and higher order chromatin structures. However, it is essential not only to determine where in the genome epigenetic modifications are occurring, but also to understand how they exert their biological functions. Mass spectrometry (MS) provides data complementary to high-throughput genomics, and as such has gained an increasingly important role in epigenetic research. The recent advent of high-resolution MS has further increased the impact of MS-based hPTM analysis by allowing the discrimination of near-isobaric modifications, either singly or in combinations, while the introduction of different chemical and metabolic labeling strategies has enabled the accurate quantitation of modifications as well as protein levels, which have been exploited for both basic and clinical research.
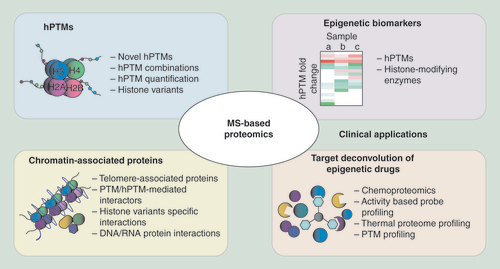
In this review, we will provide an overview of the contributions of MS-based proteomics to the epigenetics field, focusing on three main aspects: identification and quantitation of hPTMs, identification of chromatin constituents and chromatin-associated proteins, and clinical applications, including epigenetic biomarker discovery and target deconvolution of epigenetic drugs ().
Analysis of hPTMs
In recent years, MS has become an invaluable tool for the identification and quantitation of hPTMs. In MS, PTMs are first identified by detecting a ‘delta-mass’ (Δm) between the theoretical and experimentally measured masses of a peptide and then their exact position is assigned by MS/MS fragmentation. This allows the characterization of single modifications as well as their combinations without requiring a priori knowledge of the modification site or type. In addition to its unbiased nature, MS offers several other advantages over traditional antibody-based methods, such as immunoblotting (WB), immunofluorescence (IF) and immunohistochemistry (IHC): it offers a comprehensive view of histone modification patterns, it can detect combinations of modifications and does not suffer from the cross-reactivity and epitope masking (poor efficiency to detect a modification when another one is present nearby) effects typically associated with antibody-based assays.
While referring to a more specialized review [Citation5] for a detailed description of MS-based hPTMs analysis, we will offer here an conceptual overview of MS-based approaches to identify and quantitate hPTMs, together with some examples on how they contributed to the epigenetics field.
Identification of hPTMs & their combinations
Three main MS-based analytical strategies can be used to identify hPTMs in biological samples: bottom–up, middle–down and top–down approaches. In bottom–up approaches, MS histones are enzymatically digested into peptides prior to MS analysis and separated in an LC/MS set up with reversed-phase HPLC, using gradients adapted to the highly hydrophilic nature of histones [Citation6]. This chromatographic step allows the separation of very complex peptide mixtures prior to MS analysis and is particularly important when analyzing hypermodified proteins such as histones, where the identification and quantitation of isobaric peptides often relies on the ability to chromatographically separate them. Due to the elevated presence of basic residues in the histone sequences, trypsin (which cuts at positively charged Lys and Arg side chains) produces peptides that are too small to be easily detected by LC-MS. Therefore Arg-C, which specifically cleaves at the C-terminal of arginine residues, is usually the protease of choice for bottom–up studies [Citation7]. Although Arg-C does not perform well when used in in-gel digestions, Arg-C-like digestions can be obtained starting from gel bands by chemically alkylating unmodified and mono-methylated lysines with agents that impair trypsin digestion at lysine sites, such as propionic or deuterated acetic anhydride [Citation8–11]. Other advantages of chemical alkylation include changes in the molecular weight and hydrophobicity of the alkylated peptides – and, as a consequence, in their retention time during HPLC separation – which can be exploited to better discriminate isobaric species. For instance, the methylated forms of histone H3 peptide 27–40 can be easily distinguished after deuterated acetic anhydride derivatization [Citation12]. In addition, chemical derivatization can improve HPLC retention and detectability for very short peptides, as recently shown for histone H3 peptide 3–8 [Citation13].
A peptide-centric strategy such as bottom–up, however, is not ideal when analyzing co-occurring histone modifications that may be distant from each other in the protein sequence. As an alternative, intact histones or large protein fragments can be analyzed by top–down or middle–down MS approaches [Citation14], respectively, which provide a more complete view on the proteoforms present in a sample. In top–down, intact histones are chromatographically separated and directly analyzed, while in middle–down long histone peptides (>5 kDa) are obtained through enzymatic digestion with endoproteases that cleave at less frequently occurring residues, such as Glu-C and Asp-N, and are usually separated using weak-cation exchange combined with hydrophilic interaction liquid chromatography (WCX-HILIC) [Citation15]. One of the major challenges in MS-based hPTM analysis is the ability to distinguish isobaric species (peptides carrying the same modification(s), but on different positions), a problem that becomes more relevant for the larger molecules analyzed in middle– and top–down. Using these approaches, isobaric species are often co-isolated and fragmented at the MS/MS level, and as a consequence they cannot be discriminated. Another issue related to top– and middle–down approaches is that long peptides have much wider charge state distributions than bottom–up peptides, which reduces the overall signal intensity of each charged state. This sensitivity issues may be partially solved for middle–down strategies by prefractionation of the digestion products. However, top– and middle–down approaches are fundamental to detect long-range PTM associations and enabled to uncover a long-distance link between methylation and acetylation on histone H3 and H4 N-terminal tails [Citation16,Citation17]. Different MS strategies have also been used in parallel to take advantage on the one hand of the unambiguous PTM identification obtainable with bottom–up methods and on the other hand of the more global view of PTM occupancy offered by top– and middle–down. For instance, the effects of the knockdown of the lysine methyltransferase G9a and its interaction partner Glp1 on the most abundant histone H3 PTMs were analyzed by bottom–up and middle–down MS [Citation18], while a combination of top–down and bottom–up approaches were used to dissect the hPTM pattern of linker histone H1 in Drosophila melanogaster [Citation19].
Thanks to these diverse approaches, MS has significantly expanded the list of existing hPTMs and their combinations [Citation20]. For example, histone H4R3me1 was initially discovered by MS and was then linked to transcriptional repression associated with the protein arginine methyltransferase PRMT1 [Citation21]. A gene regulation role for modifications not located on histone tails has also been described thanks to MS-approaches, such as the discovery of H3R42 as a new methylation site that positively affects transcription [Citation22]. Remarkably, MS-based studies helped uncover completely novel types of modifications, including O-GlcN acetylation and lysine propionylation, butyrylation, formylation, succinylation and malonylation [Citation23–27].
hPTM quantitation
An accurate quantitation of the various modified forms of histone peptides is often as essential for the characterization of a biological sample as their identification. Different MS-based strategies have been used to comprehensively and accurately quantify hPTMs (reviewed in [Citation5]). Label-free analysis of histones involves the direct comparison of unlabeled samples through ion intensity-based quantitation, where the raw abundance of each modified peptide is obtained using MS-extracted ion chromatograms (XICs). Isobaric peptides that cannot be chromatographically separated can be quantitated based on the relative ratios of fragment ions at the MS/MS level [Citation28,Citation29]. Label-free strategies have been used in bottom–up [Citation30–33] (see below) as well as middle– and top–down studies. For instance, label-free was used in combination with a middle–down approach to demonstrate that nucleosomes are asymmetrically modified with respect to H3K27 di-/tri-methylation and H4K20 methylation [Citation34].
Histones can also be quantified by in vivo metabolic labeling, for instance by stable isotope labeling by amino acids in cell culture (SILAC), where up to three cell populations are labeled with isotope-encoded amino acids that are distinguishable by MS [Citation35]. SILAC-labeled cell populations can be combined at a very early stage of the MS-proteomics workflow, reducing the experimental variation due to sample preparation and improving quantitation accuracy. SILAC was employed to quantify hPTMs in several studies addressing modification dynamics during the cell cycle [Citation36,Citation37], nutrient-induced cell cycle re-entry from quiescence [Citation38], chromatin maturation [Citation39] and hPTM cross-talk [Citation40]. Modified versions of the SILAC approach employ histones from a reference cell line [Citation41] or a pool of representative cell lines [Citation42,Citation43] (which represents a histone-focused version of the super-SILAC approach [Citation44]) as a spike-in standard to which the different biological samples can be compared. Differently from classical SILAC, these approaches can be used to analyze any number of samples and also allow studying samples that cannot be metabolically labeled. Spike-in approaches have been used to dissect the hPTM patterns in breast cancer cell lines [Citation41], to profile global histone modifications in human cancers [Citation42] and, recently, to detect differences among breast cancer subtypes in primary samples (see also below) [Citation43]. Another variation of SILAC, heavy methyl SILAC (hmSILAC) [Citation45], allows one to discriminate true methylations from other isobaric modifications or amino acid substitutions, thus increasing the confidence in the identification of methylations at lysine and arginine residues. While for quantitative studies addressing hPTM changes among functional states both SILAC and label-free techniques can be used, SILAC is the method of choice for pulse experiments. For instance, hmSILAC was employed to study H3K9me3 turnover in pericentric chromatin [Citation46] and to globally profile the turnover of histone lysine methylation [Citation47]. The progression of H3K79 methylations through the cell cycle was also carried out through a combination of standard and heavy methyl- SILAC in pulse-chase experiments [Citation48], while classical SILAC was used to profile both histone variants and hPTMs [Citation49].
Another labeling strategy that can be exploited for hPTM quantitation relies on chemical derivatization. Samples in different functional states are either left unlabeled or labeled using deuterated acetic or propionic anhydride, which allows to distinguish them by MS when mixed in a 1:1 ratio, similarly to metabolic labeling [Citation50]. An interesting application of a quantitative SILAC approach combined with chemical labeling of histone and nonhistone proteins to quantify acetylation stoichiometry has recently been used to analyze acetylation dynamics in distinct subcellular compartments [Citation51].
Finally, hPTMs can also be quantitated using MS-targeted approaches [Citation52], which are particularly useful to reproducibly and quantitatively analyze a set of peptides known a priori across multiple samples. When synthetic isotope-labeled peptides are employed as internal standards, targeted approaches can be used for the absolute quantitation of histones and their PTMs. This type of approach has been employed to analyze H2B ubiquitination [Citation53] and to determine the absolute abundance of 42 histone-modified peptides in cancer cell lines [Citation54]. Recently, a library of 93 synthetic Protein-Aqua™ peptides was used to analyze hPTMs in histones H3, H4 and H2A. This study highlighted how the presence of PTMs can affect peptide MS-based detection, thus biasing the calculation of their relative intensity, and provided a useful resource to correct for detection biases [Citation55]. MS-targeted approaches were also recently applied on a high-resolution MS spectrometer to analyze and quantify hPTMs in a label-free manner [Citation56].
Histone variants
With the exception of histone H4, histones have genetically encoded variants that often show differences in expression patterns, in the enrichment at specific genomic regions or cell-cycle phases and in their PTM signatures [Citation3], thus mediating different chromatin-related processes. Aberrations in the deposition of histone variants have been linked to various diseases, including cancer [Citation4,Citation57]. Due to their limited sequence differences, identifying some variants is particularly challenging using conventional antibody-based methods. Top–down and middle–down approaches have been used to characterize all canonical histones [Citation14,Citation58], including histone H1 variants [Citation59], 12 unique H2A sequences [Citation60,Citation61] and H3 variants from human, rat and yeast, which were found to have interesting differences in their PTM profiles [Citation14]. A combination of top–down and middle–down was also applied to study histone H2B and H3 N-terminal tail clipping (the proteolytic cleavage of histone N-terminal tails), revealing different hPTMs in intact and clipped histones [Citation62]. Although bottom–up analysis of variants that have very limited sequence differences is challenging, this type of approach has also been used to characterize and even discover new histone variants from different organisms [Citation6]. For instance, the very similar histone variants H3.1 and H3.3 were distinctly characterized thanks to one amino acid difference in peptide 27–40, which was sufficient to distinguish them by MS [Citation63].
Identification of chromatin-associated proteins
Chromatin-mediated functions are exerted through the involvement of chromatin-associated proteins, which comprise enzymes able to modulate the histone-code (like writers and erasers), DNA-binding proteins (such as transcription and replication factors) and chromatin remodelers that bind to the DNA and regulate gene expression and other DNA functions. All together these proteins constitute the chromatin proteome, or ‘chromatome’ [Citation64], whose dissection is fundamental in order to acquire insights into the higher-order structures and functions of chromatin. Characterizing thoroughly the chromatome is not trivial, as many proteins are transiently associated to chromatin or present at low abundance, such as transcription factors. A further level of complexity is represented by the existence of protein variants, isoforms and different post-translational modified forms. In recent years, MS – coupled with strategies to both decrease sample protein complexity and enrich for chromatin components, histone readers or locus-specific regions – has greatly contributed to the dissection of the chromatin-associated proteome () [Citation65].
Chromatin constituents
A first level of characterization of the chromatome consists in determining which proteins constitute chromatin. Different enrichment protocols have been proposed in the last 10 years but a ‘gold standard’ method to map the chromatin proteome still does not exist and the choice of the protocol depends on the downstream application [Citation106]. The first reported chromatin purification strategy took advantage of the of insolubility of chromatin in nonionic detergents, leading to the identification and quantitation of 282 differentially expressed transcription factors and chromatin regulators in c-Myc-expressing and non-expressing human-B lymphocytes [Citation66]. More recently, a number of modified procedures to obtain chromatin pellets have been reported that employ different detergents [Citation67,Citation68], acid, salt or DNaseI extraction to improve purification efficiency [Citation69–71]. For instance, the recently proposed differential chromatin-associated proteins (D-CAP) approach allowed identifying proteins that are differentially associated with chromatin during differentiation, unraveling the role of SMARCD1 in the restriction of pluripotency and activation of differentiation pathways through the regulation of H3K27 methylation [Citation72].
Contrarily from the methods described above, the recently developed chromatin enrichment for proteomics (ChEP) strategy enriches chromatin on the basis of its propensity to be cross-linked rather than solubility, and was used to obtain a comprehensive inventory of interphase chromatin-associated proteins [Citation73,Citation74]. The cross-linking step minimizes the loss of transiently binding chromatin factors and allows for more stringent washes, thus reducing the risk of purification artifacts. Although very promising, ChEP also has limitations, such as the inability to follow local changes in the protein abundance as a consequence of protein re-localization in specific chromatin regions and the fact that not all chromatin components may be fixed by formaldehyde. Chromatin constituents can also be characterized at the PTM level, as recently done by the phosphorylation specific-chromatin affinity purification (PS-ChAP) to monitor phosphorylation changes following activation or inhibition of global transcriptional patterns [Citation75].
Chromatin readers
Modified histones serve as scaffolds for binding of regulatory proteins that translate hPTMs into a physiological response. A typical experimental set-up used to identify histone readers relies on pull-downs using peptides bearing a specific modification, followed by MS analysis. This strategy has evolved from the analysis of single gel bands [Citation76,Citation77] to more sophisticated and quantitative versions employing SILAC labeling [Citation78,Citation79] or the BAC-GFP transfenomics technology [Citation80], which allowed the identification of trimethyl-lysine interactors. H3K4me3 readers were also analyzed using a chemical proteomics approach, cross-linking-assisted SILAC protein identification (CLASPI), which adopts a photo-cross-linking-based peptide probe to covalently stabilize labile interactions [Citation81].
The three-dimensional organization of chromatin, the cross-talk among hPTMs and DNA methylation all contribute to the recruitment of chromatin readers. To take into account this complex environment, recombinant mono-nucleosomes bearing specific combinations of histone methylations or methylation on cytosine at CpG dinucleotides have been used as bait in SILAC nucleosome affinity purification (SNAP) as an alternative to isolated modified histone tail peptides [Citation82]. Using a similar approach, recombinant uniformly modified oligo-nucleosomes in conjunction with SILAC-based quantitative MS led to the identification of specific readers of the prototypic H3K4me3 and H3K9me3 peptides and of H2BK120ub1 [Citation83,Citation84].
DNA readers can also be identified in pull-down experiments using DNA sequences as bait. For instance, a one-step affinity purification from SILAC-labeled extracts was used to identify methyl CpG-binding proteins [Citation85] and readers of 5-methylcytosine (mC) and 5-hydroxymethyl-cytosine (hmC), two oxidative cytosine to which distinct biological roles were ascribed based on the results of this study [Citation86].
Alternative approaches to study chromatin readers are based on the use of antibodies raised against a specific hPTM, histone variant or chromatin-associated protein, which allow investigating chromatin interactors in more physiological conditions. In the last 10 years, the chromatin immunoprecipitation strategy combined with massively parallel DNA sequencing (ChIP-seq) allowed studying the genome-wide localization of a specific chromatin determinant with a high resolution. The availability of well-established protocols and reagents for this genomic approach, together with the technological improvements in MS have paved the way for the development of ChIP strategies coupled to proteomic approaches. Such strategies allow the identification and quantitation of hPTMs, variants and chromatin-binding proteins enriched after immunoprecipitation of a specific chromatin determinant. The first successful example of this type of strategy is represented by the modified ChIP (mChIP) approach, where TAP-tagged versions of the budding yeast Hta2p (the ortholog of mammalian H2A) and Htz1p (H2A.Z in mammals) were expressed in Saccharomyces cerevisae, leading to the reconstruction of the network of their chromatin-associated proteins [Citation87]. The lack of quantitative information obtainable through this approach was overcome in the chromatin proteomics (ChroP) strategy, which combines SILAC-based quantitative proteomics with ChIP to obtain a comprehensive view of histone modification, variants and interacting protein associated with a specific hPTM through a single immunoprecipitation experiment [Citation88]. Variations of this type of approach were recently used to identify proteins associated with the genomic regions marked by different histone variants [Citation89] or by histones modified on specific lysines [Citation90]. A similar strategy was also exploited to study the cofactors and transcription factors associated with hormone receptors in breast cancer using the rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) approach, which consists of an antibody-based affinity purification of chromatin in cross-linking conditions, followed by on-bead digestion and MS analysis (see also below) [Citation91,Citation92].
A number of other approaches were developed for the in vivo tagging of endogenous proteins or DNA. The first example is the chromatin-interacting protein MS (ChIP-MS), where biotinylated forms of two male-specific lethal (MSL) proteins were expressed in D. melanogaster [Citation93]. This approach allowed the proteomic dissection of MSL-bound hPTMs and chromatin-bound proteins after streptavidin affinity purification. Similarly, proteins associated with nascent DNA at replication forks were investigated through the isolation of proteins on nascent DNA (iPOND) strategy, where newly synthesized DNA is labeled with the thymidine analog 5-ethynyl-2′deoxyuridine (EdU) [Citation94]. The nascent chromatin capture (NCC) represents a refined and SILAC-based version of this approach, where the replicating DNA is labeled with biotin-dUTPs. This strategy was used to determine the chromatin dynamics of approximately 4000 proteins during DNA replication [Citation95] and to investigate how histone marks propagate through the cell cycle [Citation96].
Differently from pull-down strategies, antibody-based approaches point toward the proteomic characterization of enriched native chromatin marked by a common determinant; however none of these procedures describe the chromatome at a specific genomic locus.
Locus-specific chromatin-interacting proteins
The major hurdle in the dissection of a locus-specific chromatome is the isolation of a genomic locus and associated proteins in sufficient quantity and purity to allow the identification of bound proteins by MS. The first approach addressing this aspect was the proteomics of isolated chromatin (PICh), which employs a nucleic acid probe to specifically isolate telomeric DNA, thus allowing the characterization of the telomeric chromatome [Citation97,Citation98]. This approach represented a breakthrough in the field and stimulated the development of other proteomics-based strategies for the identification of locus-specific determinants. For instance, proteins associated with the origin of replication were identified by purifying a LacO-tagged mini-chromosome from whole cell extract of S. cerevisiae during specific phases of the cell cycle [Citation99]. The chromatin affinity purification with mass spectrometry (ChAP-MS) strategy allowed a more focused analysis of proteins associated with a single genomic locus. In first generation ChAP-MS a LexA DNA affinity handle was engineered upstream of the GAL1 gene and the locus was enriched under transcriptionally active as well as repressive conditions [Citation100]. The second generation ChAP-MS relied on the engineering of the transcription activator-like (TAL)-protein A fusion protein (TAL-PrA) in order to enrich the GAL1 locus (TAL-ChAP-MS) [Citation101]. Recently, techniques based on molecules able to bind to a specific DNA sequence that are generated by engineering its DNA-binding recognition motif have been developed. Among them, the clustered regularly interspersed short palindromic repeats (CRISPR) system is a flexible and cost-effective strategy based on a catalytically inactive Cas9 (dCas9) and a small guide RNA (gRNA) [Citation102]. This system was recently adopted in the third generation ChAP-MS, which uses a tagged dCas9 and guides the complex to a specific genomic locus by engineering the gRNA (CRISPR-ChAP-MS) [Citation103]. Through a similar approach, the engineered DNA-binding molecule-mediated chromatin immunoprecipitation coupled with MS (enChIP-MS) used the CRISPR system to efficiently isolate specific genomic regions retaining molecular interactions [Citation104]. EnChIP-MS was also employed to isolate of telomere-binding proteins by using a TAL protein recognizing telomere repeats [Citation105].
RNA-associated proteins
Increasing evidence supports the notion that gene expression is regulated by a complex interplay among mRNAs, ncRNAs and RNA-binding proteins (RBPs), which all together constitute a multitude of ribonucleoprotein complexes (RNPs) [Citation107,Citation108]. RNA–protein interactions may be unraveled through protein-centric or RNA-centric approaches, where RNPs are enriched by using either a specific RBP or RNA, respectively [Citation109]. Protein-centric strategies like RNA immunoprecipitation (RIP) and UV cross-linking and immunoprecipitation (CLIP) based methods usually associate immunoprecipitations with deep sequencing to identify RNAs belonging to a specific RNP, but have also been used in combination with MS analysis. One example is the photoactivatable-ribonucloside enhanced CLIP (PAR-CLIP) approach, through which a large number of proteins without a predicted RNA-binding domain were identified as RBPs [Citation110,Citation111].
RNA-centric approaches aim at identifying the protein complexes associated to an RNA of interest. Quantitative MS was used in combination with RNase treatment of affinity-purified RNA–protein complexes to identify proteins that bind to RNA concurrently with an RBP of interest, like Pab1, Nab2 and Puf3 [Citation112], while a workflow combining UV-based stabilization of RNA–protein interactions, high-resolution MS and automated MS spectra analysis was employed to identify both the cross-linked amino acid and RNA moiety, leading to the discovery of specific RNA interaction sites on proteins [Citation113]. Different RNA-centric biochemical approaches (reviewed in [Citation114]) have also been developed to study the function of long noncoding RNAs (lncRNAs), which can regulate gene expression by guiding protein complexes to target genes and by serving as scaffold for RNP assembly or as decoy to sequester away regulatory proteins [Citation115]. For instance, the Chromatin Isolation by RNA Purification (ChIRP) protocol was adapted to MS analysis and led to the identification of 81 proteins that bind the lncRNA Xist, which is essential for X chromosome inactivation [Citation116]. Using a similar approach, numerous proteins interacting with NEAT1 and MALAT1 ncRNAs were isolated by combining the capture hybridization analysis of RNA targets (CHART) approach with MS [Citation117]. Xist was also investigated in a recent study that combined RNA antisense purification and high-resolution MS to identify direct Xist interactors during the initiation of X chromosome inactivation [Citation118].
Applications in clinical epigenetics
Altered gene expression caused by aberrant regulation of chromatin structure and function can lead to the development of various pathologies. In particular, alterations in histone modification patterns have been linked to many diseases [Citation119]. For instance, histone hypoacetylation has been observed in neurological disorders, including ALS, Parkinson’s and Huntington’s diseases and Friedreich’s ataxia, while increased levels of H3K9me2 have been found in lymphocytes from Type-1 diabetes patients. Various aberrations in hPTM levels have also been observed in cancer. For instance, decreased acetylation of histone H4 and of H4K20me3 is generally found in different types of cancer [Citation30], while changes in the levels of other histone marks, including H3K4me3, H3K9me1 and H3K27me3 have been linked with specific tumors [Citation119]. Acetylation and methylation patterns in histone H3 and H4 correlate with disease progression and clinical outcome in different tumors [Citation120,Citation121], highlighting the potential of hPTM signatures as biomarkers useful for diagnostic, prognostic and therapeutic patient stratification.
Alteration of histone acetylation and methylation levels is mainly caused by aberration of the activity or expression of histone-modifying enzymes, such as histone deacetylases, methyltransferases and demethylases [Citation122]. Targeting these enzymes for therapeutic purposes is gaining increasing attention and many epigenetic drugs are already approved or are currently being evaluated in clinical trials for the treatment of different forms of cancer [Citation123]. Below, we discuss the contribution that MS-based approaches can provide to two important areas of clinical epigenetics: epigenetic biomarker discovery and target deconvolution of epigenetic drugs.
Identification of epigenetic biomarkers
Genome-wide techniques are providing an increasingly detailed knowledge regarding aberrant DNA methylation in disease, which could be exploited for biomarker discovery [Citation124]. Recent advances in MS-based hPTM analysis, together with the growing evidence regarding an epigenetic involvement in various diseases, has prompted the application of MS strategies – in addition to genomic approaches – to epigenetic biomarkers discovery. In a seminal study, bottom–up MS in combination with other techniques led to the discovery of H4K16 acetylation and H4K20 trimethylation as histone marks decreased in cancer cells [Citation30]. Bottom–up MS approaches were also used to identify H3 and H4 histone marks associated with cigarette smoking [Citation32] and with the grade of invasiveness in esophageal squamous cell carcinoma [Citation33].
Histone H3 and H4 hypoacetylation was investigated through a bottom–up approach in splenocytes from a mouse model of lupus compared with control, identifying differentially expressed modifications such as H3K18 and H4K31 methylation and H4K31 acetylation [Citation125]. In vivo administration of the histone deacetylase inhibitor trichostatin A (TSA) corrected these differences and improved the disease phenotype, thus establishing an association between altered histone modification patterns and the pathogenesis of lupus. A label-free approach was used to profile histone modification patterns in 24 normal and cancer cell lines, providing a proteomic atlas that could be potentially used to classify cancer cells based on their histone modification landscape [Citation31]. In particular, an enrichment of H3K27me1 was observed in breast cancer cell lines and, accordingly, knockdown of the main H3K27 methyltransferase (EZH2) significantly reduced tumor growth in a mouse mammary xenograft model. These studies demonstrated the predictive power of MS-based techniques and support the possibility to exploit MS-derived epigenetic biomarkers to design epigenetic therapies. Another potential epigenetic biomarker found in breast cancer that was discovered by MS is phosphorylation of T146 of histone H1, which correlates with tumor grade [Citation126]. The role of H1 proteoforms in breast cancer was further investigated in a top–down study, demonstrating differences in the phosphorylation of H1 variants H1.2 and H1.4 through the cell cycle [Citation59]. Top–down MS also helped in characterizing combination of methylations and acetylations on full-length histone H3 from human cell lines derived from multiple myeloma patients overexpressing of the histone methyltransferase MMSET [Citation127].
A super-SILAC-based, high-information-content MS approach was employed to profile global histone modifications in 115 cancer cell lines, identifying a chromatin signature containing increased H3K36me2 in cell lines harboring mutations in the methyltransferase NSD2, which hence emerged as a potential therapeutic target for pediatric acute lymphoblastic leukemia [Citation42]. We also used a spike-in SILAC approach [Citation41] to compare modifications occurring in breast cancer relative to normal cells. We identified hPTM changes already known to occur in cancer, as well as previously unknown PTM variations, such as the increase of H3K4me1 and H3K27me2/3 and the decrease of H3K9me2. More recently, we generated a super-SILAC histone mixture to be used as an internal standard in a histone-focused version of the super-SILAC approach to study breast cancer primary samples [Citation43]. We combined this super-SILAC strategy with the PAT-H-MS (pathology tissue analysis of histones by mass spectrometry) approach, which allowed for the first time to dissect hPTM patterns from formalin-fixed paraffin-embedded (FFPE) tissues. By analyzing breast cancer samples, we revealed significant changes in histone H3K9me3, K36me1/me2 and K27me3 marks in tumors belonging to two distinct disease subtypes with different prognosis, the Luminal A-like and the more aggressive Triple Negative breast cancer [Citation43]. These data may suggest candidate epigenetic targets for the treatment of Triple Negative breast cancers. The development of this method represents an important step for the epigenetic analysis of clinical samples and paves the way for large retrospective epigenetic studies that combine the power of MS-based hPTM analysis with the extensive clinical information associated with FFPE archives, which has been previously hindered by technical limitations. By using the same super-SILAC approach, we analyzed a panel of breast cancer cell lines and primary breast cancer cells that are either sensitive or resistant to four HDAC inhibitors with different specificities, revealing hPTM signatures associated with the cellular response to the drugs and pinpointing the marks affected by HDAC inhibitors targeting different members of the HDAC family (unpublished observations). Interestingly, in a recent study, the pan-HDAC inhibitor SAHA was found to increase the levels not only of acetylated, but also butyrylated histones, in neuroblastoma, possibly identifying candidate biomarkers for the use of SAHA for new clinical indications [Citation128].
MS-based strategies to dissect chromatin-associated proteins can also contribute to the discovery of nonhistone biomarkers and the understanding of their mechanisms. One example is represented by GREB1, a potential clinical biomarker linked with good clinical outcome in breast cancer with previously unknown functions, whose key role as an estrogen-specific estrogen receptor (ER)-associated protein was uncovered by analyzing the ER interactome [Citation91]. The same approach was also used to investigate the proteins associated with the progesterone receptor (PR), revealing that activated PR functions as a proliferative brake that controls the binding of ER to chromatin and its transcriptional activity, with important implications for therapy and prognosis [Citation92].
As an alternative to LC-MS-MS analysis, MALDI imaging has gained much attention for biomarker discovery [Citation129]. MALDI imaging MS combines MS ability to analyze complex mixtures with the capability of obtaining a spatial distribution of proteins within biological samples through the analysis of thin sections of tissue that have been spray-coated or micro-spotted with MALDI matrix. Recently, whole cell MALDI MS biotyping, which analyzes whole cells homogenized in a solvent/MALDI matrix mixture, and MALDI imaging were used to follow histone acetylation changes during treatment with HDAC inhibitors in gastrointestinal cancer cells and mouse tissue, respectively [Citation130]. In addition, protein patterns obtained by MALDI imaging were combined with LC-MS/MS information to compare hepatocellular carcinoma samples with and without microvascular invasion, which represents a major risk factor in postoperative mortality and tumor recurrence in hepatocellular carcinoma [Citation131].
Target deconvolution of epigenetic drugs
Knowledge of the full spectrum of proteins interacting with small molecule inhibitors is crucial to identify in the early phases of drug development off-targets responsible for side effects and to possibly lead to the identification of additional targets that could suggest alternative applications of the inhibitor. This level of compound characterization has been technically unfeasible until the advent of MS-based target deconvolution methods. Chemoproteomics (or chemical proteomics) consists of an affinity chromatography purification followed by MS analysis. A bioactive molecule of interest, which is immobilized on a resin or is chemically conjugated to an affinity moiety, such as biotin, is used as an affinity probe to isolate from cell extracts and then identify by MS all the proteins that are bound to the probe [Citation132,Citation133]. False-positive targets can be identified using different strategies, the most common of which consists in competition-binding experiments, where the affinity probe is incubated with the cell extract in the presence or absence of the unmodified compound. In this experimental set-up genuine targets show significantly reduced binding to the resin in the presence of the competitor compared with vehicle control, while nonspecific binders are not affected by the presence of the soluble compound. When coupled with SILAC or isobaric labeling tags, IC50 and KD values can be precisely calculated for all the identified targets of a given drug. This approach has been successfully applied to a number of small molecule inhibitors, including epigenetic drugs. In particular, a combination of affinity capture and quantitative MS was used to study the selectivity of 16 HDAC inhibitors, providing information not only regarding individual target proteins, but also native Megadalton protein complexes [Citation134], a type of characterization that is uniquely attainable through MS techniques. Interestingly, results obtained with native drug target complexes deviated from values obtained using recombinant enzymes and indicated a certain degree of selectivity for some inhibitors. HDACs were also investigated through active site-directed chemical probe profiling (ABPP), which employs a reactive probe that binds covalently to a target protein active site, allowing to profile the activity state of HDACs and of their neighboring interactors [Citation135].
The nuclear complexes associated with the ubiquitously expressed BET (bromodomain and extra terminal) family of proteins were investigated through a combined proteomics approach that included the target deconvolution of a BET inhibitor by chemical proteomics, immunoprecipitation with selective antibodies against the BRD2/3/4 family members and pull downs with bead-immobilized histone H4 acetylated peptides. This multi-tier proteomic strategy led to the discovery as BET-associated complexes of the super elongation complex (SEC) and the polymerase-associated factor complex, which are crucial for malignant transformation by MLL fusions. These results set the basis for therapeutic intervention with a BET inhibitor in MLL-fusion leukemia, a disease with currently limited treatment options [Citation136].
While target deconvolution by chemoproteomics represents a valuable tool in classical target-based drug discovery, it is a truly essential step in phenotypic drug discovery, where compound libraries are screened using phenotypic/mechanistic readouts, leading to the discovery of hits for which the molecular targets are unknown. Proteomics target deconvolution of compounds identified through phenotypic screens based on their ability to upregulate apolipoprotein A-I led to the discovery of BET bromodomain targets, identifying for the first time this class of proteins as tractable targets [Citation137,Citation138]. Similar strategies were used to find the targets of compound hits identified in phenotypical screens based on the inhibition of a cell-based β-catenin-dependent transcriptional reporter [Citation139] or β-catenin/TCF-dependent transcription [Citation140].
The major drawback of chemical proteomics methods is the requirement for the synthesis of functionalized analogs suitable for binding to beads, which is often the rate-limiting step of these procedures. Recently, a proteomic-based method for target deconvolution that overcomes this limitation has been proposed [Citation141]. This approach, termed thermal proteome profiling (TPP), is based on the biophysical principle of ligand-induced thermal stabilization of target proteins and consists in monitoring changes in protein thermal stability across the proteome through multiplexed quantitative MS, allowing to follow compound binding to its targets without requiring any derivatization. In addition, by comparing results obtained using living cells (where both compound binding and downstream signaling occur) and cell extracts (where only binding occurs) it is possible to discriminate direct from indirect targets. The effects of the HDAC nonselective inhibitors panobinostat were recently analyzed by TPP, identifying not only known HDAC targets and HDAC complex members, but also an unexpected effects on other targets [Citation142].
Histone PTM profiling can also assess the specificity of histone modifiers-inhibitors. One example is a recent work where the acetylation signatures for 19 different KDAC inhibitors were determined and their effect monitored on both histone and non-histone proteins [Citation143]. This study revealed that most KDAC inhibitors increased acetylation of a small, specific subset of the acetylome and provided a useful reference of the acetylated proteins directly or indirectly affected by each drug.
Future perspective
MS-based proteomics has significantly contributed to the epigenetics field, potentiating and complementing other commonly used genomics techniques, such as ChIP-Seq. The development of bioinformatics tools to integrate data obtained through genomics and proteomics approaches and to efficiently visualize them will be fundamental to fully exploit the information obtained using these powerful analytical methods. In addition, the contribution of MS-based approaches in different areas of epigenetic research, from hPTM analysis to drug discovery, will benefit from further technical advances.
For instance, hPTM analysis will benefit from improvements in the ability to comprehensively define modification patterns in a single analysis, including low stoichiometry and less frequent modifications. Most current data acquisition routines are based on data-dependent acquisition (DDA) methods, which are designed to stochastically select the ion precursors with the highest relative abundance in the full MS scan for subsequent fragmentation. Due to this intensity-bias, the less abundant PTMs are less efficiently and reliably detected compared with the canonical and more abundant histone marks. Alternative methods, such as data-independent acquisition (DIA), where MS/MS fragmentation spectra are collected for all the detectable peptide precursors present in a biological sample, independently of precursor abundance/information, might represent a solution to increase the detection of low abundance histone modifications. Supporting this hypothesis, a recent report employing for the first time a data-independent acquisition method, SWATH™ analysis, to study hPTMs in mouse trophoblast stem cells before and after differentiation detected significant differences for very low abundance marks [Citation144]. In addition, DIA methods allow discriminating isobaric forms at the MS/MS level, with no need to selectively target them. Another critical aspect of hPTM analysis is the ability to detect the combinatorial aspect of the histone code. Recent advances in middle– and top–down approaches greatly improved the detection of combinatorial marks, but the implementation of straightforward analytical workflows and bioinformatics tools compatible with large-scale analyses are still lagging behind the current needs. Improvements in middle– and top–down workflows could also facilitate discriminating among histone variants [Citation59,Citation62], a challenging task in bottom–up studies because of their small sequence differences. These improvements in MS-based hPTM analysis would be important also for clinical applications, to evaluate the most complete panel of modifications during the biomarker discovery phase, to reproducibly measure less abundant but relevant marks in patient samples and to define modification signatures of pathological states, which may provide improved predictive power as compared with single biomarkers.
Despite having revolutionized hPTM analysis, MS still cannot replace antibody-based strategies for various epigenetic applications. For instance, very low abundance hPTMs may only be detected by western blotting, and IHC allows detecting marks at the level of single cell and has a higher throughput for the analysis of single/few marks. In addition, expensive instruments allowing high-resolution MS and MS/MS and alternative fragmentation methods are required when analyzing larger biomolecules, thus limiting the number of centers that can perform such analyses. Therefore, antibody-based strategies and MS-proteomics must be considered as complementary tools that can be used in combination (for instance, exploiting MS to discover potential new biomarkers that can be then validated by IHC on a larger panel of samples).
Large archives of paraffin-embedded tissues are a valuable source of clinical samples and retrospective information, which has not been exploited so far for epigenetic biomarker discovery due to technical limitations. The development of the PAT-H-MS approach for the analysis of hPTMs in FFPE tissues represents an important step for the identification of epigenetic biomarkers or epigenetic signatures that could be developed into diagnostic and prognostic tools. However, a limit of this approach is related to the intrinsic tissue heterogeneity, for instance the presence of normal cells in a cancer specimen, which impairs the possibility of analyzing specific cell populations. An interesting implementation of the technique would involve procedures able to increase the purity and homogeneity of cell populations, such as core needle biopsies and laser microdissection. These techniques have been successfully used to implement the pathology tissue chromatin immunoprecipitation (PAT-ChIP) [Citation145,Citation146], a method for the extraction and high-throughput genomic analysis of chromatin from FFPE tissues that represents the genomic counterpart of PAT-H-MS.
Although few successful examples of MS-based target deconvolution of epigenetic drugs exist, this area is still relatively unexploited. As phenotypic drug discovery screening approaches are gaining popularity, we envision that MS-based target deconvolution, including the novel and derivatization-free thermal proteome profiling approach, will become increasingly important in drug discovery. One attractive application would be screening compound libraries directly by chemoproteomic approaches. This strategy would not only address compound potency and selectivity toward all its targets early in the discovery process, but would also allow to perform the screening in a more physiological situation, with reduced requirements for the expression and purification of active proteins.
Table 1. Mass spectrometry-based strategies to dissect chromatin-associated proteins.
Mass spectrometry (MS)-based proteomics has emerged as a powerful analytical method that significantly contributes to the epigenetic field, potentiating and complementing other commonly used genomics techniques.
MS has become an invaluable tool to analyze histone post-translational modifications, thanks to its unbiased nature, accuracy and its ability to quantify modifications and detect their combinations.
Approaches coupling MS with strategies to enrich for chromatin components, histone readers, locus-specific regions or RNA-associated proteins have greatly contributed in recent years to the dissection of the chromatin-associated proteome.
Aberrations in the epigenetic machinery have been linked with many diseases and proteomics has increasingly been exploited for epigenetic biomarker discovery.
Target deconvolution through chemical proteomics and the recently proposed thermal proteome profiling approach represent fundamental tools in epigenetic drug discovery, allowing to identify off-targets responsible for side effects early during the drug development process and to identify additional targets and alternative applications of small molecule inhibitors.
Acknowledgements
The authors thank G Natoli and M Soldi for critical reading of the manuscript.
Financial & competing interests disclosure
T Bonaldi’s research group is supported by grants from the Giovanni Armenise-Harvard Foundation Career Development Program, the Italian Association for Cancer Research (AIRC), the Italian Ministry of Health and CNR-EPIGEN flagship project. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Jenuwein T , AllisCD . Translating the histone code . Science293 ( 5532 ), 1074 – 1080 ( 2001 ).
- Bannister AJ , KouzaridesT . Regulation of chromatin by histone modifications . Cell Res.21 ( 3 ), 381 – 395 ( 2011 ).
- Yuan G , ZhuB . Histone variants and epigenetic inheritance . Biochim. Biophys. Acta1819 ( 3–4 ), 222 – 229 ( 2012 ).
- Skene PJ , HenikoffS . Histone variants in pluripotency and disease . Development140 ( 12 ), 2513 – 2524 ( 2013 ).
- Huang H , LinS , GarciaBA , ZhaoY . Quantitative proteomic analysis of histone modifications . Chem. Rev.115 ( 6 ), 2376 – 2418 ( 2015 ).
- Soldi M , CuomoA , BremangM , BonaldiT . Mass spectrometry-based proteomics for the analysis of chromatin structure and dynamics . Int. J. Mol. Sci.14 ( 3 ), 5402 – 5431 ( 2013 ).
- Bonaldi T , ImhofA , RegulaJT . A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications . Proteomics4 ( 5 ), 1382 – 1396 ( 2004 ).
- Smith CM , HaimbergerZW , JohnsonCOet al. Heritable chromatin structure: mapping “memory” in histones H3 and H4 . Proc. Natl Acad. Sci. USA99 ( Suppl. 4 ), 16454 – 16461 ( 2002 ).
- Garcia BA , MollahS , UeberheideBMet al. Chemical derivatization of histones for facilitated analysis by mass spectrometry . Nat. Protoc.2 ( 4 ), 933 – 938 ( 2007 ).
- Bonaldi T , RegulaJT , ImhofA . The use of mass spectrometry for the analysis of histone modifications . Methods Enzymol.377 , 111 – 130 ( 2004 ).
- Sidoli S , YuanZF , LinSet al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom–up proteomics PTM analysis . Proteomics15 ( 9 ), 1459 – 1469 ( 2015 ).
- Soldi M , CuomoA , BonaldiT . Improved bottom–up strategy to efficiently separate hypermodified histone peptides through ultra-HPLC separation on a bench top Orbitrap instrument . Proteomics14 ( 19 ), 2212 – 2125 ( 2014 ).
- Maile TM , Izrael-TomasevicA , CheungTet al. Mass spectrometric quantification of histone post-translational modifications by a hybrid chemical labeling method . Mol. Cell. Proteomics14 ( 4 ), 1148 – 1158 ( 2015 ).
- Thomas CE , KelleherNL , MizzenCA . Mass spectrometric characterization of human histone H3: a bird’s eye view . J. Proteome Res.5 ( 2 ), 240 – 247 ( 2006 ).
- Young NL , DimaggioPA , Plazas-MayorcaMD , BalibanRC , FloudasCA , GarciaBA . High throughput characterization of combinatorial histone codes . Mol. Cell. Proteomics8 ( 10 ), 2266 – 2284 ( 2009 ).
- Taverna SD , UeberheideBM , LiuYet al. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini . Proc. Natl Acad. Sci. USA104 ( 7 ), 2086 – 2091 ( 2007 ).
- Nicklay JJ , ShechterD , ChittaRKet al. Analysis of histones in Xenopus laevis. II. mass spectrometry reveals an index of cell type-specific modifications on H3 and H4 . J. Biol. Chem.284 ( 2 ), 1075 – 1085 ( 2009 ).
- Plazas-Mayorca MD , BloomJS , ZeisslerUet al. Quantitative proteomics reveals direct and indirect alterations in the histone code following methyltransferase knockdown . Mol. Biosyst.6 ( 9 ), 1719 – 1729 ( 2010 ).
- Bonet-Costa C , VilasecaM , DiemaCet al. Combined bottom–up and top–down mass spectrometry analyses of the pattern of post-translational modifications of Drosophila melanogaster linker histone H1 . J. Proteomics75 ( 13 ), 4124 – 4138 ( 2012 ).
- Zhao Y , GarciaBA . Comprehensive catalog of currently documented histone modifications . Cold Spring Harb. Perspect. Biol.7 ( 9 ), a025064 ( 2015 ).
- Strahl BD , BriggsSD , BrameCJet al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1 . Curr. Biol.11 ( 12 ), 996 – 1000 ( 2001 ).
- Casadio F , LuX , PollockSBet al. H3R42me2a is a histone modification with positive transcriptional effects . Proc. Natl. Acad. Sci. USA110 ( 37 ), 14894 – 14899 ( 2013 ).
- Wisniewski JR , ZougmanA , MannM . Nepsilon-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function . Nucleic Acids Res.36 ( 2 ), 570 – 577 ( 2008 ).
- Sakabe K , WangZ , HartGW . Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code . Proc. Natl. Acad. Sci. USA107 ( 46 ), 19915 – 19920 ( 2010 ).
- Zhang K , ChenY , ZhangZ , ZhaoY . Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software . J. Proteome Res.8 ( 2 ), 900 – 906 ( 2009 ).
- Chen Y , SprungR , TangYet al. Lysine propionylation and butyrylation are novel post-translational modifications in histones . Mol. Cell. Proteomics6 ( 5 ), 812 – 819 ( 2007 .
- Xie Z , DaiJ , DaiLet al. Lysine succinylation and lysine malonylation in histones . Mol. Cell. Proteomics11 ( 5 ), 100 – 107 ( 2012 ).
- Pesavento JJ , MizzenCA , KelleherNL . Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4 . Anal. Chem.78 ( 13 ), 4271 – 4280 ( 2006 ).
- Garcia BA , PesaventoJJ , MizzenCA , KelleherNL . Pervasive combinatorial modification of histone H3 in human cells . Nat. Methods4 ( 6 ), 487 – 489 ( 2007 ).
- Fraga MF , BallestarE , Villar-GareaAet al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer . Nat. Genet.37 ( 4 ), 391 – 400 ( 2005 ).
- Leroy G , DimaggioPA , ChanEYet al. A quantitative atlas of histone modification signatures from human cancer cells . Epigenetics Chromatin6 ( 1 ), 20 ( 2013 ).
- Sundar IK , NevidMZ , FriedmanAE , RahmanI . Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer . J. Proteome Res.13 ( 2 ), 982 – 996 ( 2014 ).
- Zhang K , LiL , ZhuMet al. Comparative analysis of histone H3 and H4 post-translational modifications of esophageal squamous cell carcinoma with different invasive capabilities . J. Proteomics112 , 180 – 189 ( 2014 ).
- Voigt P , LeroyG , DruryWJ3rdet al. Asymmetrically modified nucleosomes . Cell151 ( 1 ), 181 – 193 ( 2012 ).
- Ong SE , BlagoevB , KratchmarovaIet al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics . Mol. Cell. Proteomics1 ( 5 ), 376 – 386 ( 2002 ).
- Pesavento JJ , YangH , KelleherNL , MizzenCA . Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle . Mol. Cell. Biol.28 ( 1 ), 468 – 486 ( 2008 ).
- Bonenfant D , TowbinH , CoulotM , SchindlerP , MuellerDR , Van OostrumJ . Analysis of dynamic changes in post-translational modifications of human histones during cell cycle by mass spectrometry . Mol. Cell. Proteomics6 ( 11 ), 1917 – 1932 ( 2007 ).
- Mews P , ZeeBM , LiuS , DonahueG , GarciaBA , BergerSL . Histone methylation has dynamics distinct from those of histone acetylation in cell cycle reentry from quiescence . Mol. Cell. Biol.34 ( 21 ), 3968 – 3980 ( 2014 ).
- Scharf AN , MeierK , SeitzV , KremmerE , BrehmA , ImhofA . Monomethylation of lysine 20 on histone H4 facilitates chromatin maturation . Mol. Cell. Biol.29 ( 1 ), 57 – 67 ( 2009 ).
- Guan X , RastogiN , ParthunMR , FreitasMA . Discovery of histone modification crosstalk networks by stable isotope labeling of amino acids in cell culture mass spectrometry (SILAC MS) . Mol. Cell. Proteomics12 ( 8 ), 2048 – 2059 ( 2013 ).
- Cuomo A , MorettiS , MinucciS , BonaldiT . SILAC-based proteomic analysis to dissect the “histone modification signature” of human breast cancer cells . Amino Acids41 ( 2 ), 387 – 399 ( 2011 ).
- Jaffe JD , WangY , ChanHMet al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia . Nat. Genet.45 ( 11 ), 1386 – 1391 ( 2013 ).
- Noberini R , UggettiA , PruneriG , MinucciS , BonaldiT . Pathology tissue-quantitative mass spectrometry analysis to profile histone post-translational modification patterns in patient samples . Mol. Cell Proteomics doi:10.1074/mcp.M115.054510 ( 2015 ) ( Epub ahead of print ).
- Geiger T , CoxJ , OstasiewiczP , WisniewskiJR , MannM . Super-SILAC mix for quantitative proteomics of human tumor tissue . Nat. Methods7 ( 5 ), 383 – 385 ( 2010 ).
- Ong SE , MittlerG , MannM . Identifying and quantifying in vivo methylation sites by heavy methyl SILAC . Nat. Methods1 ( 2 ), 119 – 126 ( 2004 ).
- Fodor BD , KubicekS , YonezawaMet al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells . Genes Dev.20 ( 12 ), 1557 – 1562 ( 2006 ).
- Zee BM , LevinRS , XuB , LeroyG , WingreenNS , GarciaBA . In vivo residue-specific histone methylation dynamics . J. Biol. Chem.285 ( 5 ), 3341 – 3350 ( 2010 ).
- Sweet SM , LiM , ThomasPM , DurbinKR , KelleherNL . Kinetics of re-establishing H3K79 methylation marks in global human chromatin . J. Biol. Chem.285 ( 43 ), 32778 – 32786 ( 2010 ).
- Zee BM , LevinRS , DimaggioPA , GarciaBA . Global turnover of histone post-translational modifications and variants in human cells . Epigenetics Chromatin3 ( 1 ), 22 ( 2010 ).
- Plazas-Mayorca MD , ZeeBM , YoungNLet al. One-pot shotgun quantitative mass spectrometry characterization of histones . J. Proteome Res.8 ( 11 ), 5367 – 5374 ( 2009 ).
- Weinert BT , LesmantaviciusV , MoustafaTet al. Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae . Mol. Syst. Biol.10 , 716 ( 2014 ).
- Picotti P , AebersoldR . Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions . Nat. Methods9 ( 6 ), 555 – 566 ( 2012 ).
- Darwanto A , CurtisMP , SchragMet al. A modified “cross-talk” between histone H2B Lys-120 ubiquitination and H3 Lys-79 methylation . J. Biol. Chem.285 ( 28 ), 21868 – 21876 ( 2010 ).
- Gao J , LiaoR , YuYet al. Absolute quantification of histone PTM marks by MRM-based LC-MS/MS . Anal. Chem.86 ( 19 ), 9679 – 9686 ( 2014 ).
- Lin S , WeinS , Gonzales-CopeMet al. Stable-isotope-labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry . Mol. Cell. Proteomics13 ( 9 ), 2450 – 2466 ( 2014 ).
- Tang H , FangH , YinE , BrasierAR , SowersLC , ZhangK . Multiplexed parallel reaction monitoring targeting histone modifications on the QExactive mass spectrometer . Anal. Chem.86 ( 11 ), 5526 – 5534 ( 2014 ).
- Schwartzentruber J , KorshunovA , LiuXYet al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma . Nature482 ( 7384 ), 226 – 231 ( 2012 ).
- Jiang L , SmithJN , AndersonSL , MaP , MizzenCA , KelleherNL . Global assessment of combinatorial post-translational modification of core histones in yeast using contemporary mass spectrometry. LYS4 trimethylation correlates with degree of acetylation on the same H3 tail . J. Biol. Chem.282 ( 38 ), 27923 – 27934 ( 2007 ).
- Chen Y , HooverME , DangXet al. Quantitative mass spectrometry reveals that intact histone H1 phosphorylations are variant specific and exhibit single molecule hierarchical dependence . Mol. Cell. Proteomics doi:10.1074/mcp.M114.046441 ( 2015 ) ( Epub ahead of print ).
- Siuti N , RothMJ , MizzenCA , KelleherNL , PesaventoJJ . Gene-specific characterization of human histone H2B by electron capture dissociation . J. Proteome Res.5 ( 2 ), 233 – 239 ( 2006 ).
- Boyne MT 2nd , PesaventoJJ , MizzenCA , KelleherNL . Precise characterization of human histones in the H2A gene family by top down mass spectrometry . J. Proteome Res.5 ( 2 ), 248 – 253 ( 2006 ).
- Tvardovskiy A , WrzesinskiK , SidoliS , FeySJ , Rogowska-WrzesinskaA , JensenON . Top–down and middle–down protein analysis reveals that intact and clipped human histones differ in post-translational modification patterns . Mol. Cell. Proteomics doi:10.1074/mcp.M115.048975 ( 2015 ) ( Epub ahead of print ).
- Loyola A , BonaldiT , RocheD , ImhofA , AlmouzniG . PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state . Mol. Cell24 ( 2 ), 309 – 316 ( 2006 ).
- Imhof A , BonaldiT . “Chromatomics” the analysis of the chromatome . Mol. Biosyst.1 ( 2 ), 112 – 116 ( 2005 ).
- Soldi M , BremangM , BonaldiT . Biochemical systems approaches for the analysis of histone modification readout . Biochim. Biophys. Acta1839 ( 8 ), 657 – 668 ( 2014 ).
- Shiio Y , EisenmanRN , YiEC , DonohoeS , GoodlettDR , AebersoldR . Quantitative proteomic analysis of chromatin-associated factors . J. Am. Soc. Mass Spectrom.14 ( 7 ), 696 – 703 ( 2003 ).
- Kubota T , HiragaS , YamadaK , LamondAI , DonaldsonAD . Quantitative proteomic analysis of chromatin reveals that Ctf18 acts in the DNA replication checkpoint . Mol. Cell. Proteomics10 ( 7 ), M110 005561 ( 2011 ).
- Kim DR , GidvaniRD , IngallsBP , DunckerBP , McconkeyBJ . Differential chromatin proteomics of the MMS-induced DNA damage response in yeast . Proteome Sci.9 , 62 ( 2011 ).
- Khoudoli GA , GillespiePJ , StewartG , AndersenJS , SwedlowJR , BlowJJ . Temporal profiling of the chromatin proteome reveals system-wide responses to replication inhibition . Curr. Biol.18 ( 11 ), 838 – 843 ( 2008 ).
- Chou DM , AdamsonB , DephoureNEet al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage . Proc. Natl. Acad. Sci. USA107 ( 43 ), 18475 – 18480 ( 2010 ).
- Franklin S , ChenH , Mitchell-JordanS , RenS , WangY , VondriskaTM . Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth . Mol. Cell. Proteomics11 ( 6 ), M111 014258 ( 2012 ).
- Alajem A , BiranA , HarikumarAet al. Differential association of chromatin proteins identifies BAF60a/SMARCD1 as a regulator of embryonic stem cell differentiation . Cell Rep.10 ( 12 ), 2019 – 2031 ( 2015 ).
- Kustatscher G , HegaratN , WillsKLet al. Proteomics of a fuzzy organelle: interphase chromatin . EMBO J.33 ( 6 ), 648 – 664 ( 2014 ).
- Kustatscher G , WillsKL , FurlanC , RappsilberJ . Chromatin enrichment for proteomics . Nat. Protoc.9 ( 9 ), 2090 – 2099 ( 2014 ).
- Han Y , YuanZF , MoldenRC , GarciaBA . Monitoring cellular phosphorylation signaling pathways into chromatin and down to the gene level . Mol. Cell. Proteomics doi:10.1074/mcp.M115.053421 ( 2015 ) ( Epub ahead of print ).
- Wysocka J , SwigutT , MilneTAet al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development . Cell121 ( 6 ), 859 – 872 ( 2005 ).
- Wysocka J , SwigutT , XiaoHet al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling . Nature442 ( 7098 ), 86 – 90 ( 2006 ).
- Vermeulen M , EberlHC , MatareseFet al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers . Cell142 ( 6 ), 967 – 980 ( 2010 ).
- Vermeulen M , MulderKW , DenissovSet al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4 . Cell131 ( 1 ), 58 – 69 ( 2007 ).
- Poser I , SarovM , HutchinsJRet al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals . Nat. Methods5 ( 5 ), 409 – 415 ( 2008 ).
- Li X , FoleyEA , MolloyKR , LiY , ChaitBT , KapoorTM . Quantitative chemical proteomics approach to identify post-translational modification-mediated protein-protein interactions . J. Am. Chem. Soc.134 ( 4 ), 1982 – 1985 ( 2012 ).
- Bartke T , VermeulenM , XhemalceB , RobsonSC , MannM , KouzaridesT . Nucleosome-interacting proteins regulated by DNA and histone methylation . Cell143 ( 3 ), 470 – 484 ( 2010 ).
- Nikolov M , StutzerA , MoschKet al. Chromatin affinity purification and quantitative mass spectrometry defining the interactome of histone modification patterns . Mol. Cell. Proteomics10 ( 11 ), M110 005371 ( 2011 ).
- Shema-Yaacoby E , NikolovM , Haj-YahyaMet al. Systematic identification of proteins binding to chromatin-embedded ubiquitylated H2B reveals recruitment of SWI/SNF to regulate transcription . Cell Rep.4 ( 3 ), 601 – 608 ( 2013 ).
- Mittler G , ButterF , MannM . A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements . Genome Res.19 ( 2 ), 284 – 293 ( 2009 ).
- Spruijt CG , GnerlichF , SmitsAHet al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives . Cell152 ( 5 ), 1146 – 1159 ( 2013 ).
- Lambert JP , MitchellL , RudnerA , BaetzK , FigeysD . A novel proteomics approach for the discovery of chromatin-associated protein networks . Mol. Cell. Proteomics8 ( 4 ), 870 – 882 ( 2009 ).
- Soldi M , BonaldiT . The proteomic investigation of chromatin functional domains reveals novel synergisms among distinct heterochromatin components . Mol. Cell. Proteomics12 ( 3 ), 764 – 780 ( 2013 ).
- Won KJ , ChoiI , LeroyGet al. Proteogenomics analysis reveals specific genomic orientations of distal regulatory regions composed by non-canonical histone variants . Epigenetics Chromatin8 , 13 ( 2015 ).
- Ji X , DadonDB , AbrahamBJet al. Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions . Proc. Natl Acad. Sci. USA112 ( 12 ), 3841 – 3846 ( 2015 ).
- Mohammed H , D’SantosC , SerandourAAet al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor . Cell Rep.3 ( 2 ), 342 – 349 ( 2013 ).
- Mohammed H , RussellIA , StarkRet al. Progesterone receptor modulates ERalpha action in breast cancer . Nature523 ( 7560 ), 313 – 317 ( 2015 ).
- Wang CI , AlekseyenkoAA , LeroyGet al. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila . Nat. Struct. Mol. Biol.20 ( 2 ), 202 – 209 ( 2013 ).
- Sirbu BM , CouchFB , CortezD . Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA . Nat. Protoc.7 ( 3 ), 594 – 605 ( 2012 ).
- Alabert C , Bukowski-WillsJC , LeeSBet al. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components . Nat. Cell Biol.16 ( 3 ), 281 – 293 ( 2014 ).
- Alabert C , BarthTK , Reveron-GomezNet al. Two distinct modes for propagation of histone PTMs across the cell cycle . Genes Dev.29 ( 6 ), 585 – 590 ( 2015 ).
- Antao JM , MasonJM , DejardinJ , KingstonRE . Protein landscape at Drosophila melanogaster telomere-associated sequence repeats . Mol. Cell. Biol.32 ( 12 ), 2170 – 2182 ( 2012 ).
- Dejardin J , KingstonRE . Purification of proteins associated with specific genomic loci . Cell136 ( 1 ), 175 – 186 ( 2009 ).
- Unnikrishnan A , GafkenPR , TsukiyamaT . Dynamic changes in histone acetylation regulate origins of DNA replication . Nat. Struct. Mol. Biol.17 ( 4 ), 430 – 437 ( 2010 ).
- Byrum SD , RamanA , TavernaSD , TackettAJ . ChAP-MS: a method for identification of proteins and histone posttranslational modifications at a single genomic locus . Cell Rep.2 ( 1 ), 198 – 205 ( 2012 ).
- Byrum SD , TavernaSD , TackettAJ . Purification of a specific native genomic locus for proteomic analysis . Nucleic Acids Res.41 ( 20 ), e195 ( 2013 ).
- Qi LS , LarsonMH , GilbertLAet al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression . Cell152 ( 5 ), 1173 – 1183 ( 2013 ).
- Waldrip ZJ , ByrumSD , StoreyAJet al. A CRISPR-based approach for proteomic analysis of a single genomic locus . Epigenetics9 ( 9 ), 1207 – 1211 ( 2014 ).
- Fujita T , FujiiH . Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR . Biochem. Biophys. Res. Commun.439 ( 1 ), 132 – 136 ( 2013 ).
- Fujita T , AsanoY , OhtsukaJet al. Identification of telomere-associated molecules by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) . Sci. Rep.3 , 3171 ( 2013 ).
- Torrente MP , ZeeBM , YoungNLet al. Proteomic interrogation of human chromatin . PLoS ONE6 ( 9 ), e24747 ( 2011 ).
- Chen M , ManleyJL . Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches . Nat. Rev. Mol. Cell Biol.10 ( 11 ), 741 – 754 ( 2009 ).
- Licatalosi DD , DarnellRB . RNA processing and its regulation: global insights into biological networks . Nat. Rev. Genet.11 ( 1 ), 75 – 87 ( 2010 ).
- McHugh CA , RussellP , GuttmanM . Methods for comprehensive experimental identification of RNA-protein interactions . Genome Biol.15 ( 1 ), 203 ( 2014 ).
- Baltz AG , MunschauerM , SchwanhausserBet al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts . Mol. Cell46 ( 5 ), 674 – 690 ( 2012 ).
- Castello A , FischerB , EichelbaumKet al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins . Cell149 ( 6 ), 1393 – 1406 ( 2012 ).
- Klass DM , ScheibeM , ButterF , HoganGJ , MannM , BrownPO . Quantitative proteomic analysis reveals concurrent RNA-protein interactions and identifies new RNA-binding proteins in Saccharomyces cerevisiae . Genome Res.23 ( 6 ), 1028 – 1038 ( 2013 ).
- Kramer K , SachsenbergT , BeckmannBMet al. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins . Nat. Methods11 ( 10 ), 1064 – 1070 ( 2014 ).
- Yang Y , WenL , ZhuH . Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein . Cell Biosci.5 , 59 ( 2015 ).
- Rinn JL , ChangHY . Genome regulation by long noncoding RNAs . Annu. Rev. Biochem.81 , 145 – 166 ( 2012 ).
- Chu C , ZhangQC , Da RochaSTet al. Systematic discovery of Xist RNA binding proteins . Cell161 ( 2 ), 404 – 416 ( 2015 ).
- West JA , DavisCP , SunwooHet al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites . Mol. Cell55 ( 5 ), 791 – 802 ( 2014 ).
- Mchugh CA , ChenCK , ChowAet al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3 . Nature521 ( 7551 ), 232 – 236 ( 2015 ).
- Portela A , EstellerM . Epigenetic modifications and human disease . Nat. Biotechnol.28 ( 10 ), 1057 – 1068 ( 2010 ).
- Seligson DB , HorvathS , McBrianMAet al. Global levels of histone modifications predict prognosis in different cancers . Am. J. Pathol.174 ( 5 ), 1619 – 1628 ( 2009 ).
- Seligson DB , HorvathS , ShiTet al. Global histone modification patterns predict risk of prostate cancer recurrence . Nature435 ( 7046 ), 1262 – 1266 ( 2005 ).
- Chi P , AllisCD , WangGG . Covalent histone modifications – miswritten, misinterpreted and mis-erased in human cancers . Nat. Rev. Cancer10 ( 7 ), 457 – 469 ( 2010 ).
- Nebbioso A , CarafaV , BenedettiR , AltucciL . Trials with ‘epigenetic’ drugs: an update . Mol. Oncol.6 ( 6 ), 657 – 682 ( 2012 ).
- Heyn H , EstellerM . DNA methylation profiling in the clinic: applications and challenges . Nat. Rev. Genet.13 ( 10 ), 679 – 692 ( 2012 ).
- Garcia BA , BusbySA , ShabanowitzJ , HuntDF , MishraN . Resetting the epigenetic histone code in the MRL-lpr/lpr mouse model of lupus by histone deacetylase inhibition . J. Proteome Res.4 ( 6 ), 2032 – 2042 ( 2005 ).
- Harshman SW , HooverME , HuangCet al. Histone H1 phosphorylation in breast cancer . J. Proteome Res.13 ( 5 ), 2453 – 2467 ( 2014 ).
- Zheng Y , FornelliL , ComptonPDet al. Unabridged analysis of human histone H3 by differential top–down mass spectrometry reveals hypermethylated proteoforms from MMSET/NSD2 overexpression . Mol. Cell. Proteomics doi:10.1074/mcp.M115.053819 ( 2015 ) ( Epub ahead of print ).
- Xu G , WangJ , WuZet al. SAHA regulates histone acetylation, butyrylation, and protein expression in neuroblastoma . J. Proteome Res.13 ( 10 ), 4211 – 4219 ( 2014 ).
- Cazares LH , TroyerDA , WangB , DrakeRR , SemmesOJ . MALDI tissue imaging: from biomarker discovery to clinical applications . Anal. Bioanal. Chem.401 ( 1 ), 17 – 27 ( 2011 ).
- Munteanu B , MeyerB , Von ReitzensteinCet al. Label-free in situ monitoring of histone deacetylase drug target engagement by matrix-assisted laser desorption ionization-mass spectrometry biotyping and imaging . Anal. Chem.86 ( 10 ), 4642 – 4647 ( 2014 ).
- Pote N , AlexandrovT , Le FaouderJet al. Imaging mass spectrometry reveals modified forms of histone H4 as new biomarkers of microvascular invasion in hepatocellular carcinomas . Hepatology58 ( 3 ), 983 – 994 ( 2013 ).
- Bantscheff M , ScholtenA , HeckAJ . Revealing promiscuous drug-target interactions by chemical proteomics . Drug Discov. Today14 ( 21–22 ), 1021 – 1029 ( 2009 ).
- Rix U , Superti-FurgaG . Target profiling of small molecules by chemical proteomics . Nat. Chem. Biol.5 ( 9 ), 616 – 624 ( 2009 ).
- Bantscheff M , HopfC , SavitskiMMet al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes . Nat. Biotechnol.29 ( 3 ), 255 – 265 ( 2011 ).
- Salisbury CM , CravattBF . Activity-based probes for proteomic profiling of histone deacetylase complexes . Proc. Natl Acad. Sci. USA104 ( 4 ), 1171 – 1176 ( 2007 ).
- Dawson MA , PrinjhaRK , DittmannAet al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia . Nature478 ( 7370 ), 529 – 533 ( 2011 ).
- Chung CW , CosteH , WhiteJHet al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains . J. Med. Chem.54 ( 11 ), 3827 – 3838 ( 2011 ).
- Nicodeme E , JeffreyKL , SchaeferUet al. Suppression of inflammation by a synthetic histone mimic . Nature468 ( 7327 ), 1119 – 1123 ( 2010 ).
- Huang SM , MishinaYM , LiuSet al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling . Nature461 ( 7264 ), 614 – 620 ( 2009 ).
- Emami KH , NguyenC , MaHet al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] . Proc. Natl Acad. Sci. USA101 ( 34 ), 12682 – 12687 ( 2004 ).
- Savitski MM , ReinhardFB , FrankenHet al. Tracking cancer drugs in living cells by thermal profiling of the proteome . Science346 ( 6205 ), 1255784 ( 2014 ).
- Franken H , MathiesonT , ChildsDet al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry . Nat. Protoc.10 ( 10 ), 1567 – 1593 ( 2015 ).
- Scholz C , WeinertBT , WagnerSAet al. Acetylation site specificities of lysine deacetylase inhibitors in human cells . Nat. Biotechnol.33 ( 4 ), 415 – 423 ( 2015 ).
- Sidoli S , LinS , XiongLet al. Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH) Analysis for characterization and quantification of histone post-translational modifications . Mol. Cell. Proteomics14 ( 9 ), 2420 – 2428 ( 2015 ).
- Fanelli M , AmatoriS , BarozziIet al. Pathology tissue-chromatin immunoprecipitation, coupled with high-throughput sequencing, allows the epigenetic profiling of patient samples . Proc. Natl Acad. Sci. USA107 ( 50 ), 21535 – 21540 ( 2010 ).
- Amatori S , BallariniM , FaversaniAet al. PAT-ChIP coupled with laser microdissection allows the study of chromatin in selected cell populations from paraffin-embedded patient samples . Epigenet. Chromatin7 , 18 ( 2014 ).