
Abstract
Epidemiological evidence suggests that an adverse in utero environment is associated with an increased risk for developing adult onset diseases. The molecular mechanisms for susceptibility to chronic noncommunicable diseases are not fully understood, although recent research has proposed that epigenetic modifications play an important role in fetal programming. Genetic and environmental factors contribute to interindividual and spatiotemporal tissue-specific methylation patterns. Although the diverse environments and high genetic diversity of African populations provide unparalleled potential to investigate the effects of environmental change on the epigenetic profile in humans, only a small percentage of genomic and epigenetic studies have focused on populations from this continent. This emphasizes the need to build capacity in Africa for research that leads to an understanding of the association between genetic, epigenetic and environmental risk factors for noncommunicable diseases on the continent.
Noncommunicable disease in Africa
The burden of noncommunicable diseases (NCDs) is increasing globally and is currently the leading cause of death and disease burden worldwide. These NCDs not only pose a major challenge for healthcare systems but also to affected individuals [Citation1]. Cardiovascular disease (CVD), cancer, chronic lung disease and diabetes result in more than 30 million deaths annually worldwide [Citation1,Citation2]. Previously regarded as diseases of high-income countries, NCDs are increasing at an alarming rate in low- and middle-income countries [Citation3]. This is mainly due to demographic transitions and changing lifestyles of populations associated with urbanization. In Africa it is estimated that 40% of the population live in urban areas but by 2030, this number will exceed 50% as Africa ceases to be a predominantly rural continent. This transformation is evident by the high urban growth rate of 3.6% in sub-Saharan Africa, double the world average. This rapid urbanization has been accompanied by significant shifts in the health patterns of South Africans, thus increasing the prevalence of NCDs [Citation4]. Although public health in African countries has focused primarily on communicable diseases such as HIV/AIDS, malaria and tuberculosis, NCDs are now major sources of morbidity and mortality and are projected to overtake infectious diseases by 2030 [Citation1]. NCDs are to some extent preventable by modifying risk factors such as physical inactivity, high blood cholesterol, high blood pressure, obesity, unhealthy diet and inappropriate use of tobacco and alcohol [Citation3,Citation4]. These lifestyle related risk factors result in various long-term disease processes that lead to high mortality rates attributable to stroke, heart attacks, cancers, obstructive lung diseases and many others [Citation4]. However, research also highlights the importance of in utero and early life environment to an increased susceptibility to developing NCDs later in life. Given the burden of NCD in many African countries where healthcare resources are scarce, it is important to identify prenatal risk factors in an attempt to curb this growing epidemic [Citation3,Citation5].
The Developmental Origins of Health and Disease paradigm (DOHaD) is a multidisciplinary field that examines how ‘environmental factors acting during the phase of developmental plasticity interact with genotypic variation to change the capacity of the organism to cope with is environment in later life’ [Citation6]. Since Barker proposed this model by showing that a low birth weight is associated with an increased risk of CVD later in life, this concept has been extensively studied and given rise to complementary hypotheses [Citation7]. The thrifty-phenotype hypothesis proposes that an early life exposure to malnutrition regulates genes to operate in a manner suited for a nutritionally suboptimal environment. Such an adaption may improve short-term chances of survival but may be deleterious to long-term health given a more resourced environment [Citation3,Citation8]. Fetal metabolic programming is a concept first suggested by Barker and Hales in the early 1990s. They hypothesized that fetal and perinatal events, such as maternal over- and undernutrition, were central to determining the risk of developing chronic metabolic diseases, such as diabetes, CVD and obesity, in adulthood [Citation9]. The theory of a mismatch between a nutritionally adverse in utero environment and an increased susceptibility of developing adult onset disease is of particular interest in low- and middle-income African countries. In these countries, the standard of living increases substantially within one generation [Citation3]. People are born into nutrient scarce rural environments and move to urban areas where they are exposed to NCD risk factors (smoking, drinking, less active, unhealthy diet). Although malnutrition and starvation remain an unresolved problem in Africa, where approximately 27–51% of women of reproductive age are underweight (WHO), the prevalence of NCDs in urban areas is on the rise. The growing burden of disease in these developing economies may be underpinned by early life exposure to limited nutrition [Citation3,Citation4]. There is substantial evidence that supports the hypothesis that increased susceptibility to NCD is influenced by prenatal exposures [Citation10], and some of the most striking examples of developmental plasticity have come from studies on the effects of famine.
Maternal malnutrition in humans
A study which demonstrates the long-lasting effects of an early adverse nutritional environment on health and disease susceptibility is the Dutch Hunger winter cohort [Citation11]. This cohort comprises both men and women who were exposed in utero to the Dutch famine of 1944–1945 [Citation11,Citation12]. Individuals exposed to under nutrition during early gestation exhibited an increased risk for metabolic, cardiovascular and other complex diseases [Citation13]. In rural Gambia, there is seasonal variation in the availability of micronutrients with an alternation between the dry season (when food is plentiful) and the wet season (when there is less food available and therefore poorer nutrition). This dramatic seasonal fluctuation in nutritional status has an important impact on pregnant women. During the nutritionally poor rainy season, a high incidence of deficiencies in several essential micronutrients has been observed in pregnant women [Citation14]. This seasonal deficiency is associated with an increased incidence of low-birth-weight babies, as well as childhood morbidity and mortality [Citation14,Citation15]. In 1967, a civil war broke out in Nigeria after the Igbo people of the south-eastern provinces declared their independence as the Republic of Biafra. The war was a culmination of ethnic, economic and religious tensions among the various peoples of Nigeria. Disapproving of the separation, Nigerian forces pushed the Biafra people into a small enclave and cut off food supply. This resulted in an extensive famine among the Igbo people [Citation16]. The Biafran famine cohort consists of 1339 Igbo individuals who were born between 1965 and 1973, i.e. before, during and after the Biafran famine. Hult et al. studied the risks for hypertension and glucose intolerance 40 years after fetal exposure to the famine. They found that fetal and infant undernutrition is associated with significantly increased risk in 40-year-old Nigerians. This study highlights that the prevention of undernutrition during pregnancy and in infancy must become a high priority in health, education and economic agendas [Citation3,Citation16].
There have been several mechanisms proposed to explain the associations between in utero environment and adult health in human populations [Citation17], however recent research has highlighted epigenetics as the ‘link between genome and environment’ capable of modulating gene transcription [Citation18]. Reflecting on an evolutionary perspective of DOHaD, Hanson and Gluckman proposed the fetal origin hypothesis which states that fetal reprogramming, induced by in utero exposures, is a short-term adaption to the anticipated environment in order to maximize the survival of the individual. These adaptions are believed to occur partly through epigenetic changes [Citation3,Citation19].
Epigenetic effects
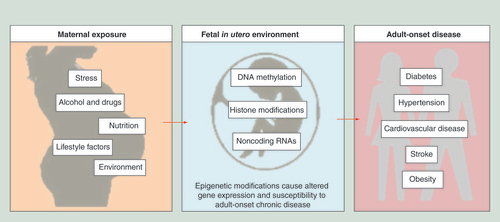
Epigenetic modifications are considered to be relatively stable during development and transmissible from one cell generation to the next (mitotic inheritance). It is believed that epigenetic marks can also be transmitted down organismal generations [Citation20], a mode of inheritance referred to as transgenerational epigenetic inheritance. Some of the earliest evidence for transgenerational epigenetic inheritance comes from studies in plants. One of the oldest examples involves a change in flower symmetry from bilateral to radial in Linaria vulgaris. This change can be explained by a change in DNA methylation rather than DNA sequence. The phenotype of the flower correlates with the degree of DNA methylation at the promoter of the Lcyc gene, and the presence or absence of DNA methylation at the promoter correlates with its silent or active state, respectively [Citation21]. In order for transgenerational epigenetic inheritance to occur, the reprogramming of epigenetic modifications that take place in the primordial germ cells and in the zygote, to ensure the totipotency of cells in the early embryo, must be bypassed [Citation21]. Epigenetic modifications are influenced by several factors including age, the environment/lifestyle and disease state. New and ongoing research is uncovering the role of epigenetics in a variety of NCDs. DNA methylation, the most prominently studied epigenetic modification, is dynamic across an individual’s lifetime and is affected by environmental influences. DNA methylation has been implicated in developmental disorders and cancer [Citation22] and is of great interest to researchers as a potential link between genome, environment and disease [Citation23,Citation24]. This epigenetic modification is crucial for normal mammalian development. Methylation patterns are established during embryogenesis in a spatiotemporal manner and remain mostly unchanged in adult cells. Modifications to the environment during early development can lead to permanent changes in the pattern of epigenetic modifications () [Citation25]. An impressive example for fetal origins of adult disease is gestational diabetes mellitus (GDM). GDM is referred to as a carbohydrate intolerance that develops during pregnancy. In GDM pregnancies the balance between increased insulin resistance and maternal insulin production is disturbed, resulting in maternal hyperglycemia, fetal hyperinsulemia and consequently fetal overnutrition [Citation13]. There have been a number of genome-wide and candidate gene DNA methylation studies designed to highlight the effects that overnutrition in utero (caused from GDM) has on the DNA methylation levels in the fetus. In 2013, Ruchat et al. reported on the impact of GDM exposure on offspring DNA methylation levels across the genome in placental and cord blood samples [Citation26]. Genome wide DNA methylation was measured using the Infinium HumanMethylation 450 Bead Chip and an Ingenuity Pathway Analysis was done to identify metabolic pathways epigenetically affected by GDM. They observed that a large number of genes in the placenta and cord blood are differentially methylated between samples exposed or not exposed to GDM and these genes are predominantly involved in metabolic disease pathways. They also observed a correlation between the level of DNA methylation at 326 genes in the placenta and 117 genes in cord blood with newborn weight. Together, these findings suggest that GDM exposure does have an impact on the newborn’s epigenome. Since DNA methylation is mitotically fairly stable, the DNA methylation adaptions that occur in certain genes in response to GDM may have a profound long-term phenotypic effect. This could also explain why newborns exposed to adverse intrauterine environments such as GDM have an increased risk of developing chronic diseases later in life [Citation26]. These studies have the potential to provide new insights into disease pathogenesis as well as provide disease predictive biomarkers. Despite the success of such epigenome-wide association studies (EWAS) in identifying common disease-associated loci in humans, a large proportion of disease causality remains unexplained [Citation27]. Although EWAS studies have become more feasible, the candidate gene approach is still valuable and relevant, specifically in a resource poor environment and continues to prove successful in identifying genes involved in disease progression [Citation9]. This approach avoids the multiple testing problems and allows one to detect significant changes of small effect size [Citation13]. In two separate studies, Bouchard et al. examined whether a newborn exposed to a GDM intrauterine environment displayed an altered DNA methylation profile in two well-known obesity and diabetes candidate genes, leptin (LEP) and adiponectin (ADIPOQ) [Citation28,Citation29]. The most significant finding from the earlier study was that DNA methylation levels in the leptin gene promoter region, in placental tissue only, was directly correlated with glucose levels in GDM women. In both the placental and cord blood tissue, the DNA methylation level in the leptin gene was associated with a decrease in leptin gene mRNA levels. In the more recent study, they demonstrated that DNA methylation levels were lower in the promoter of ADIPOQ on the fetal side of the placenta when the mother’s blood glucose concentration was high during the second trimester of pregnancy. They also found that the lower DNA methylation levels were associated with higher levels of circulating adiponectin in the mother throughout pregnancy. The results from both studies showed that the DNA methylation profiles of these two genes were deregulated by exposure to maternal hyperglycemia. In a more recent study, the DNA methylation profiles of 14 metabolic programming candidate genes were analyzed in cord blood and placental samples [Citation30]. The maternally imprinted MEST gene and nonimprinted glucocorticoid receptor (NR3C1) gene showed a significant decrease in the DNA methylation levels in GDM samples when compared with controls in both tissue types. The highest DNA methylation difference between groups reached 7.2% in the placenta, which is likely to have a physiological effect [Citation9,Citation26].
Epigenetic effects of the maternal nutrition environment on offspring in animals
DNA methylation has been the topic of research in both animal models and human populations. Animal studies have demonstrated that nutritional factors can modify the epigenome of the developing offspring [Citation11]. In a study done by Plagemann et al., epigenetic changes in the hypothalamic proopiomelanocortin (Pomc) and insulin receptor (Insr) genes were associated with neonatal over feeding of rats [Citation31]. The level of DNA methylation was directly dependent on the amount of glucose given to the rats. The DNA methylation pattern and expression levels of the glucocorticoid receptor (Nr3cl) and peroxisome proliferator-activated receptor α (Ppara) genes, was altered in the liver and brain tissue of pregnant rats that were fed a protein strict diet [Citation32,Citation33]. One of the most impressive animal model examples of the epigenetic effects of maternal nutrition on the fetus is the viable yellow agouti (Avy) mouse model, in which coat color variation is correlated to epigenetic marks established early in development [Citation34]. This mouse model has been used to investigate the impacts of nutritional and environmental influences on the fetal epigenome [Citation34]. The agouti gene is present in all mammals and functions in the determination of coat color. In order to create a metastable epiallele in the mouse that can be switched on or off during early development, a transposable intracisternal A particle (IAP) element was inserted upstream of the Agouti gene transcription start site. The degree of methylation that occurs at this IAP element correlates inversely with Agouti gene expression and hence the phenotype of the mouse. When the degree of methylation at the epiallele was increased by adding methyl donors to the mother’s diet [Citation21], the majority of the offspring appeared healthy and displayed a brown coat. This brown coat color or wild-type phenotype is the result of hypermethylation at the IAP element which suppresses Agouti gene expression. Hypomethylation of the IAP element increases Agouti expression and results in a yellow coat phenotype which is correlated with the susceptibility to metabolic diseases, cancers and obesity [Citation11].
Epigenetic effects of the maternal nutrition environment on offspring in humans
Individuals who were exposed in utero to the Dutch Hunger Winter famine had, almost 60 years later, less DNA methylation at the imprinted IGF2 gene when compared with their same-sex, unexposed siblings [Citation12]. In 2009, Tobi et al. investigated the methylation levels of 15 genes implicated in growth and metabolic disease in exposed and unexposed individuals from the Dutch Hunger Winter Cohort. They observed that the methylation of INSIGF was lower and methylation of IL10, LEP, ABCA1, GNASAS and MEG3 was higher among exposed individuals when compared with their unexposed same-sex siblings. They also observed a significant interaction between the sex of the individuals and level of methylation in INSIGF, LEP and GNASAS. Not only did exposed individuals have significantly altered levels of DNA methylation in a number of genes, they also had a higher incidence of chronic diseases such as obesity, CVD and diabetes when compared with unexposed siblings. The findings from these studies support the hypothesis that DNA methylation changes that occur in certain genes may be the result of prenatal famine exposure and that the level of methylation change depends on the sex of the exposed individual [Citation35]. In 2010, Waterland et al. showed that the seasonal variation in periconceptional nutrition induces significant increases in DNA methylation at certain metastable epialleles (ME) of individuals conceived during Gambia’s rainy season [Citation14]. Some of the observed DNA methylation changes were shown to persist into early infancy [Citation15], highlighting the permanent effect of periconceptional environment on a human epigenome [Citation14]. These annual oscillations in nutrient availability and substrate utilization have long been known to affect fetal growth and development. To test this hypothesis in the Gambian population, Dominguez-Salas et al. observed the influence a mother’s periconceptional dietary intake and plasma concentrations of key methyl-donor pathway substrates (methionin [MET], choline [CHOL], betain [BET]), cofactors (folate [FOL], vitamins B2, B6, B12, active B12 [ACTB12]) and intermediatery metabolites (dimethyl glycine [DMG], S-adenosylmethionine [SAM], S-adenosylhomocysteine [SAH], homocysteine [HCY], cysteine [CYS]) has on their infant’s DNA CpG methylation at six previously described MEs. After measuring the concentration of these 13 plasma biomarkers at the time of conception, they observed significant seasonal differences in the concentrations of eight of these biomarkers. The maternal periconceptional concentrations of FOL, B2, MET, BET and SAM were higher during the nutritionally poor rainy season and ACTB12, DMG, HCY and SAH were lower. They then tested whether these nutritionally driven seasonal differences in maternal periconceptional metabolism has an effect on the methylation profile of six known MEs. They found that methylation at these six MEs in the peripheral blood lymphocytes of infants conceived in the rainy season was consistently higher. This illustrates that maternal nutrition at the time of conception can influence the methylation patterns of MEs in offspring [Citation36]. In 2012, Khulan et al. aimed to demonstrate that periconceptional maternal micronutrient supplementation might affect fetal genome-wide methylation within gene promoters. They obtained cord blood samples from offspring of Gambian mothers taking micronutrient supplementations or placebos during the pre- and periconceptional period. The global methylation patterns showed a significant association between micronutrient supplementation and changes in DNA methylation of CpG loci. These significant changes in the epigenome in cord blood DNA were also present in infant blood DNA samples taken at 9 months, proving that a majority of these changes are persistent. These results not only highlight the importance of micronutrient supplementation during the rainy seasons and around the time of conception in Gambia but also support the idea that the nutritional environment in which a fetus develops can influence the epigenetic programming of gene activity later in life [Citation15]. It is unknown whether any epigenetic analysis has been done on the Biafra famine cohort.
Limitations to epigenetic studies
Germline genetic variation is present and unaltered in virtually every cell in an individual whereas the epigenetic profiles (such as DNA methylation) are subject to temporal and developmental dynamics and are also influenced by environmental factors [Citation27]. Because DNA methylation patterns are cell type and tissue specific [Citation11,Citation37], obtaining readily available and informative tissue is challenging. Ideally, researchers should assess epigenetic modification in tissues that contribute to the phenotype they are studying. This is difficult in epidemiological studies conducted in humans as obtaining disease relevant tissue is often invasive and/or impossible [Citation22,Citation38]. For this reason, surrogate tissues, most commonly whole blood, are used [Citation39]. The assumption is that the cell types present in the surrogate tissue will reflect epigenetic modifications found in the target tissue, or at least yield biomarkers that, although not directly causative of the disease, can still be used for predictive and/or diagnostic purposes as well as provide new pathophysiological insights into the disease [Citation27,Citation40]. Such approaches will not explore epigenetic differences that exist between different cells and tissues [Citation41]. Regardless of whether a target or surrogate tissue is used, the cause-and-effect relationship between epigenetic changes and disease phenotype is not a simple one. The causal link between variation in epigenetic modifications observed between an affected (case) and unaffected (control) individual and the disease (or predisposition to developing the disease) is difficult to determine and changes may simply reflect differences in cellular composition between the disease and nondisease state [Citation27,Citation38,Citation42]. The disease itself could induce epigenetic changes in an individual so it is important to distinguish between causal and noncausal associations [Citation38]. It is therefore necessary to take into account cellular heterogeneity in whole blood by using a post hoc bioinformatic solution for confounding cell-type bias in EWASs [Citation22,Citation43]. Another challenge facing EWAS studies is sample size. The optimal size for an epigenomic study is not yet known, however, it is assumed that if the effects are moderate to small, ideally thousands of participants should be included in an EWAS study to avoid type one errors [Citation38]. Moreover, power analysis, which is used to compute the minimum sample size for a study to give statistically significant results, is almost meaningless without realistic estimates of the degree of possible epigenetic difference between cases and controls [Citation38]. Careful phenotyping can improve statistical power especially in studies with small-to-moderate sample sizes [Citation44].
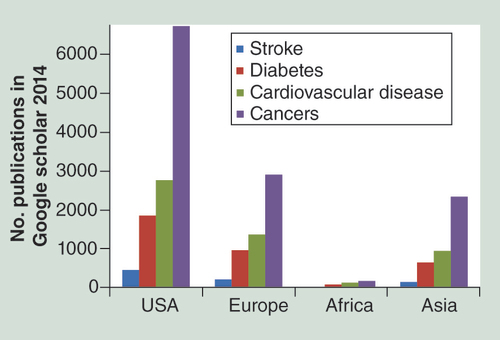
Epigenetic data from African countries is limited, leading to an under-representation of the epigenetic diversity in African populations. To highlight the international disparity in epigenetic NCD studies, the number of papers focusing on DNA methylation in stroke, CVD, diabetes and cancers in specific population groups (African, European, American and Asian) was quantified in the following manner using Google scholar. Search terms included ‘DNA methylation’ in combination with ‘Africa’ and ‘stroke.’ Each condition (stroke, CVD, diabetes and cancers) was limited to being a major subject matter. The search was repeated changing ‘Africa’ to ‘USA’ ‘China’ and ‘Europe.’ The advanced search on Google scholar was used to limit each search to those articles involving humans, having an abstract and written in English (). Limitations of epigenetic studies that are unique to the African continent include lower levels of funding, poor infrastructure and health systems as well as a small number of trained scientists [Citation5,Citation45]. EWAS studies could play a pivotal role in identifying disease-associated biomarkers for screening high-risk populations and in developing strategies for disease control and treatment. The cost involved in conducting such large-scale research projects is currently high, prohibiting this type of research in countries where funding is scarce. One of the biggest challenges to this scale of research in the African continent is poor or unavailable health infrastructure. Hospital records tend to be poor and/or nonexistent and for many individuals, accessibility to a healthcare facility is limited. Many laboratories lack state-of–the art facilities and equipment as well as a critical mass of skilled scientists. Ethics approval and recruitment for large-scale genomic and epigenomic studies requiring broad consenting for the sharing of data and biological material also pose challenges, as do language and cultural barriers [Citation45–47]. On a more positive note, African populations harbor increased genetic diversity and rapidly changing environments (e.g., through urbanization) that provide a unique opportunity to explore genome–environment–epigenome relationships.
The effect of fixed genetic variation on DNA methylation
When interpreting the role of epigenetic variation in a complex disease, it is important to consider both fixed genetic variation and environmental factors. It is well known that environmental factors are a source of epigenetic variation observed between individuals [Citation48], and there is a large amount of epidemiological data that links disease risk directly to the in utero environment which affects the epigenome through stable epigenetic modifications, and these resulting epigenetic marks alter the physiology to influence later disease risk [Citation48,Citation49]. Recent studies have highlighted that a large proportion of DNA methylation variability across individuals and populations is also attributable to underlying genetic variability [Citation48–50]. McRae et al. found that the majority of the similarity in DNA methylation levels between relatives is due to genetic effects which means that approximately 20% of DNA methylation variation between individuals in a population is due to sequence-based DNA variants (SNPs) that are not located within the CpG sites. These identified SNPs, whose genotypes correlate with levels of DNA methylation, are termed methylation quantitative trait loci, or meQTLs [Citation49]. Therefore the interindividual variation in DNA methylation is, in part, a result of nucleotide polymorphisms [Citation48]. To determine whether these meQTLs varied with ancestral population, developmental stage and tissue types, Smith et al. analyzed genome-wide DNA methylation and SNP data from seven cohorts that varied by ancestry (African–American vs Caucasian), developmental stage (neonate vs adults) and tissue type (blood vs brain tissue). They found similarities in genotype-dependent DNA methylation across this diverse range of subject characteristics and tissues [Citation51]. Bell et al. utilized the Illumina Human Methylation 27 Bead Chip to map associations between SNPs and methylation levels at 22,290 CpG dinucleotides in lymphoblastoid cell lines (LCLs). They found 180 CpG sites associated with nearby SNPs [Citation52]. In a similar study using the same DNA methylation platform, Gibbs et al. studied samples from four human brain regions in 150 individuals and reported hundreds of SNP-associated CpG sites in each brain sample, with meQTLs typically located very close to the associated CpG site [Citation53]. Although many DNA methylation studies have examined genome-wide DNA methylation data through array-based or whole-genome bisulphate sequencing technologies, they have not established population-epigenetic principles to guide design, efficient statistics or interpretation [Citation52]. Liu et al. examined the observed correlation in DNA methylation data, generated from an Illumina HumanMethylation450 array, on whole blood from 247 healthy European individuals, focusing particularly on the CpG sites with the largest methylation variation (vCpGs). They observed that the clustering of correlated DNA methylation at these vCpGs was similar to that of linkage disequilibrium correlation in genetic SNP variation but for shorter distances. Furthermore, they aimed to determine the relationship between these contiguous methylation clusters and genotype at a population level by examining whether methylation correlation in clusters is driven by genetic variants. In doing so, they identified that some distinct methylation clusters were associated with the same underlying SNP cluster but were not spatially defined as a single cluster because uncorrelated vCpGs existed between them. These potentially noncontiguous genetically controlled methylation clusters were termed GeMes. Given that SNPs were found to control GeMes and that clusters of methylated CpGs are smaller than LD blocks, it would be of interest to incorporate CpG-methylation data and GWAS-SNP data to better understand the functional effects of genome-wide associations [Citation54].
Although the influences of prenatal environment on future disease risk are intensively studied, it is important to address the degree to which the environmental influences are moderated by genotype. Therefore any study aiming to explore the role of DNA methylation variation in a complex disease needs to carry out a parallel analysis of underlying genetic variation. These findings highlight the importance of understanding the genetic diversity of target populations. Africa is an important region in which to study human genetic diversity because of its complex population history and the dramatic variation in climate, diet and exposure to infectious diseases all of which result in high levels of genetic and phenotypic variation within the continent [Citation55]. African populations are characterized by greater levels of genetic diversity when compared with non-African populations and although Africa is critical for understanding modern human origins and genetic risk factors for disease, only a small percentage of genomic studies have focused on African populations due to a number of challenges (). Genetic studies of human disease are more challenging to perform in heterogeneous populations with higher genetic diversity. In 2009, Tishkoff et al., found that the genetic structure of Africans traces to 14 ancestral population clusters that broadly correlate with ethnicity and culture or language. Given the high levels of substructure and admixture between genetically distinct populations in Africa, even within small geographic regions, the study highlighted the heterogeneity of the African populations. Therefore, there is no single African population that is representative of the diversity present in Africa [Citation56]. Studying patterns of genetic diversity, and how they correlate with epigenetic signatures, among the multitude of ethnically diverse African populations will shed light on the questions regarding the genetic basis of phenotypic variation. However, despite the important contributions that studies of the African populations can make, these populations are vastly understudied. By comparing populations in Africa to populations of African ancestry outside Africa, such as the African–Americans, it may be possible to begin to distinguish the roles of environmental and genetic risk factors that contribute to complex diseases. There have been a number of genome-wide epigenetic studies designed to observe DNA methylation variation in African-American cohorts [Citation57–60].
Population specificity & DNA methylation
Recent advances in high-throughput technologies for measuring quantitative locus specific and genome-wide DNA methylation have provided an opportunity to characterize methylation patterns in the context of human genome variation. DNA methylation can differ in diseases and cell types, or even between monozygotic twins, but while most research has focused on the potential for variation at the cellular level, relatively little research has been done to examine how epigenetic variation affects humans at the population level. DNA methylation patterns are important for establishing cell, tissue and organism phenotypes, but very little is known about their contribution to natural human variation [Citation61]. As the scale of DNA methylation association studies approaches that of genome-wide association studies, issues such as population stratification have to be addressed. In 2012, Fraser et al. measured the DNA methylation near the transcription start sites of over 14,000 genes in 180 cell lines derived from 30 northern European family trios (mother, father and child) as well as from 30 Yoruba (west African) family trios. The data demonstrated a wide range of within population variability in the methylation of individual CpG sites. In addition to the variation within each population, they observed that a third of the genes they studied showed differences in the DNA methylation patterns between the populations. Although the methylation at over a thousand of the analyzed CpG sites is heritable, the heritability also seems to differ between populations, suggesting extensive divergence in the genetic control of DNA methylation [Citation62]. These results suggest that DNA methylation is highly divergent between populations and this is due to a combination of genetic factors and complex gene–environment interactions. In 2013, Heyn et al. generated genome-wide DNA methylation profiles of three distinct human populations (namely Caucasian–American, African–American and Han Chinese–American) using the Human Methylation 450 Bead Chip. They found that 439 CpG sites were significantly differentially methylated. Although many of these identified DNA methylation differences that distinguish the three major human ethnic groups were shown to be associated with underlying genetic changes (highlighting a direct relationship between the genetic and epigenetic modification), about a third stood alone as epigenetic markers and CpG methylation quantitative trait loci associated with natural variation in the human population [Citation61]. These data help to explain phenotype diversity beyond that which is attributable to genomic variation. There is mounting evidence that on a genome-wide scale there is substantial ethnic variation in DNA methylation, only some of which is associated with genetic changes. It is therefore likely that African populations have both genetic and environmental influences on the methylome and it will be a challenge to tease these apart in the context of chronic disease studies.
In 2014, Kwabi-Addo et al. conducted a genome-wide large-scale analysis of DNA methylation changes in prostate tissue from seven unaffected and three prostate cancer patients from African–American participants versus eight unaffected and three prostate cancer patients from European–American participants. Using the Human Methylation 450 Bead Chip they observed significant global DNA methylation differences in prostate tissue when comparing African–Americans to European–Americans. This finding provides a mechanistic explanation for the disease disparity [Citation63]. In 2012, Nieminen et al. found distinct genetic and epigenetic signatures in colorectal cancers that were dependent on the ethnicity (Egyptian vs Finnish) of the patients. They screened 14 tumor suppressor genes for aberrant promoter region methylation in 69 Egyptian and 80 Finnish individuals with colorectal cancer. Of these 14 genes, four (MSS, CDKN2B, p15 and TSGMP) displayed a significant increased level of promoter region methylation when comparing Egyptian patients to Finish patients. They concluded that the possible effects of environmental exposures in carcinogenesis occur through DNA methylation. This observation could have important implications for prevention, diagnosis, prognosis and treatment of the disease [Citation64]. Although these ethnic differences in human DNA methylation in the above mentioned studies have been shown for a number of CpG sites, the genome-wide patterns and extent of these differences are largely unknown [Citation62]. These small but extensive epigenetic differences observed between populations are most likely the result of both genetic [Citation23] and environmental factors [Citation62]. Adkins et al. found that, at birth, there are significantly different DNA methylation levels between African–Americans and Caucasians at a subset of CpG dinucleotides. The DNA methylation levels at 26,485 autosomal CpGs were assayed in 201 newborns (107 African–American and 94 Caucasian) and nonparametric analyses were performed to examine the relationship between these methylation levels and maternal parity and age, newborn gestational age, newborn gender and newborn race. They found that 13.7% (3623) of the autosomal CpGs exhibited significantly different levels of DNA methylation between African–Americans and Caucasians and that 2% of autosomal CpGs had significantly different DNA methylation levels between male and female newborns. Cancer pathway genes, including those involved in four cancers (pancreatic, prostate, bladder and melanoma) with substantial ethnic differences in incidence, were highly represented among the genes containing significant ethnic-divergent CpGs [Citation65].
Environmental effects on DNA methylation variation
With regard to the environment, population specific environmental factors such as socio-economic status, infections and lifestyle may also contribute to differences in DNA methylation. Socio-economic status (SES) is a measure of an individual’s or family’s economic and social position based on education, income and occupation, and has long been a strong predictor of health. In many African countries, there exist inequalities in socio-economic status and the burden of infectious and NCD is greater among individuals of lower SES [Citation66]. In a preliminary study, Borghol et al. revealed an association between exposure to a low SES in childhood and differential DNA methylation in adulthood [Citation67]. Forty adult males from the 1958 British Birth Cohort were selected according to their SES at childhood and in mid adulthood. The SES scores were determined using specific criteria, dividing the men into the most disadvantaged and the least disadvantaged. Genome-wide DNA methylation in the blood of these men was performed using MeDIP (methylated DNA immunoprecipitation). The DNA methylation profiles in adult blood indicated greater association with childhood SES than with adult SES. In a separate study, McGuinness et al., investigated the relationship between SES and DNA methylation in a subset of individuals from the pSoBid cohort. This cohort is characterized by an extreme socio-economic and health gradient. They observed global DNA hypomethylation in the individuals classified as the ‘most socio-economically deprived.’ They also found an association between global DNA methylation and biomarkers of CVD and inflammation, after adjustment for socio-economic factors. Both these studies showed that there is an association between epigenetic modifications and SES. This relationship has direct implications for population health and is reflected in further associations between global DNA methylation content and emerging biomarkers of NCD [Citation68]. Another example of an environmental stress factor that influences epigenetic modifications in humans is that of bacterial infections, a common cause of illness and death in Africa. Studies have shown that bacteria have the ability to change the chromatin structure and transcriptional activity of their host cells. This is achieved through the induction of epigenetic modifications, such as DNA methylation. These bacterial induced epigenetic modifications may affect the host cell function either to promote host defense or to allow pathogen persistence [Citation69].
Conclusion & future perspective
De Vries and Pepper highlighted the importance of the development of genomics research ‘In Africa, for Africa, by Africans.’ We wish to adapt this statement to emphasize both the need for efficient collaboration and the potential global impact of studying African populations in Africa. This highlights the need to build capacity in Africa for research that leads to an understanding of the association between genetic, epigenetic and environmental risk factors for NCD on the continent [Citation70]. Scientific interest in genomics studies in Africa is on the rise [Citation70]. There are a few large research initiatives which specifically aim to understand the genomic and molecular underpinning of health in African populations. One such initiative is the Human Heredity and Health in Africa (H3Africa) Consortium that aims to facilitate a current research approach to study the genomic and environmental determinants of common diseases on the continent [Citation71]. This initiative aims to contribute to the development of the necessary expertise among African scientists, and to establish networks of African investigators. H3Africa also aims to create a network of laboratories that are well-equipped to conduct research focused on the complex interplay between environmental and genetic factors which determine disease susceptibility and drug responses in African populations [Citation70]. It is important that genomic and epigenomic research include populations from the poorer parts of the world, including Africa [Citation70]. Although there are challenges facing this type of research in many African countries, the information obtained on the genomes of African populations would have a positive influence on the health of individuals with African ancestry. The limited research infrastructure, including laboratories, biorespositories and databases, coupled with inadequate funding and other resources have hampered African scientists from carrying out high-quality research that focusses primarily on NCD. Building sustainable, comprehensive and multidisciplinary programs relevant to African research is essential. Although advancing genomic research on the African continent is challenging, time consuming and cost intensive, the potential benefit of this research to improving health in Africa is unlimited. As the world moves toward epigenome-wide association studies to examine the relationship between epigenetic modifications and disease, it is important that African countries keep up with these improved technologies for high-throughput research. The use of epigenetics in a diagnostic setting and for the purpose of informing treatment is in its infancy. Implementing such tests in an African setting would require specific validation and would be challenging in many developing countries. Correct diagnosis is not only critical in a clinical setting, but also for meaningful research and eventually for determining patient outcomes. One way to improve diagnostic testing in African countries may be to introduce twinning with institutions in the more developed regions. Populations are not only genetically diverse but also have unique characteristics at an epigenetic level. Africans are exposed to many adverse environmental influences (such as poor nutrition, infection, low SES) which potentially trigger population-specific epigenetic changes that contribute to a population’s epigenetic diversity. For this reason, epigenetic studies are not readily transferrable from one population to the next and it is necessary to identify disease-specific profiles, obtain knowledge regarding the exposure, the modifiable factors and their effects in order to develop population appropriate interventions.
Table 1. Challenges facing noncommunicable disease studies in Africa.
Noncommunicable disease in Africa
Previously regarded as diseases of high-income countries, noncommunicable diseases (NCDs) are increasing at an alarming rate in low- and middle-income countries. This is mainly due to demographic transitions and changing lifestyles of populations associated with urbanization.
Although public health in African countries has focused primarily on communicable diseases such as HIV/AIDS, malaria and tuberculosis, NCDs are now major sources of morbidity and mortality and are projected to overtake infectious diseases by 2030.
Research highlights the importance of in utero and early life environment to an increased susceptibility to developing NCDs later in life.
Given the burden of NCD in many African countries where healthcare resources are scarce, it is important to identify prenatal risk factors in an attempt to curb this growing epidemic.
Epigenetic effects
Epigenetic modifications are influenced by several factors including age, sex, the environment/lifestyle and disease state.
New and ongoing research is uncovering the role of epigenetics in a variety of NCDs.
Epigenetic data from African countries is limited, leading to an under-representation of the epigenetic diversity in African populations.
The effect of fixed genetic variation on DNA methylation
When interpreting the role of epigenetic variation in a complex disease, it is important to consider both fixed genetic variation and environmental factors.
Recent studies have highlighted that a large proportion of DNA methylation variability across individuals and populations is also attributable to underlying genetic variability.
Most research has focused on the potential for variation at the cellular level, relatively little research has been done to examine how epigenetic variation affects humans at the population level.
Conclusion & future perspective
There is a need to build capacity in Africa for research that leads to an understanding of the association between genetic, epigenetic and environmental risk factors for NCD on the continent.
African populations harbor increased genetic diversity and rapidly changing environments (e.g., through urbanization) that provide a unique opportunity to explore genome–environment–epigenome relationships.
There is a need to identify biomarkers through a systematic comparison of fetal epigenomes using genome-wide methylation arrays concentrating on African populations.
Disclaimer
M Ramsey is a South African Research Chair in Genomics and Bioinformatics of African Populations hosted by the University of the Witwatersrand, funded by the Department of Science and Technology and administered by the National Research Foundation of South Africa (NRF). AH is supported by an NIH Fogarty Training Fellowship. The content of the review is solely the responsibility of the authors and does not necessarily represent the official views of the Fogarty International Centre, the NIH or the NRF.
Financial & competing interests disclosure
The authors wish to acknowledge support from the National Health Laboratory Service and the Sydney Brenner Institute of Molecular Bioscience, as well as a training grant from the Fogarty International Centre (1D43TW008330) (NIH) which supported A Hobbs. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Holmes MD , DalalS , VolminkJet al. Non-communicable diseases in sub-Saharan Africa: the case for cohort studies . PLoS Med.7 ( 5 ), e1000244 ( 2010 ).
- Kim HC , OhSM . Noncommunicable diseases: current status of major modifiable risk factors in Korea . J. Prev. Med. Public Health46 ( 4 ), 165 – 172 ( 2013 ).
- Ueda P . Famine in Nigeria and vitamin D in Sweden: two early life exposures and their relation to cardiovascular risk in adulthood . PhD, Karolinska Institutet , Stockholm, Sweden ( 2013 ).
- Puoane TT L , SandersD , ParkerW . Chronic non-communicable diseases . In : South African Health Review 2008 . BarronP , Roma-ReardonJ ( Eds ). Health Systems Trust , Durban, South Africa ( 2008 ).
- Consortium HA . Research capacity. Enabling the genomic revolution in Africa . Science344 ( 6190 ), 1346 – 1348 ( 2014 ).
- Uauy R , KainJ , CorvalanC . How can the Developmental Origins of Health and Disease (DOHaD) hypothesis contribute to improving health in developing countries?Am. J. Clin. Nutr.94 ( 6 Suppl. ), S1759 – S1764 ( 2011 ).
- Barker DJ , WinterPD , OsmondC , MargettsB , SimmondsSJ . Weight in infancy and death from ischaemic heart disease . Lancet2 ( 8663 ), 577 – 580 ( 1989 ).
- Hales CN , BarkerDJ . Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis . Diabetologia35 ( 7 ), 595 – 601 ( 1992 ).
- Bouchard L . Epigenetics and fetal metabolic programming: a call for integrated research on larger cohorts . Diabetes62 ( 4 ), 1026 – 1028 ( 2013 ).
- Wang G , WalkerSO , HongX , BartellTR , WangX . Epigenetics and early life origins of chronic noncommunicable diseases . J. Adolesc. Health52 ( 2 Suppl. 2 ), S14 – S21 ( 2013 ).
- Lehnen H , ZechnerU , HaafT . Epigenetics of gestational diabetes mellitus and offspring health: the time for action is in early stages of life . Mol. Hum. Reprod.19 ( 7 ), 415 – 422 ( 2013 ).
- Heijmans BT , TobiEW , SteinADet al. Persistent epigenetic differences associated with prenatal exposure to famine in humans . Proc. Natl Acad. Sci. USA105 ( 44 ), 17046 – 17049 ( 2008 ).
- El Hajj N , SchneiderE , LehnenH , HaafT . Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment . Reproduction148 ( 6 ), R111 – R120 ( 2014 ).
- Waterland RA , KellermayerR , LaritskyEet al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles . PLoS Genet.6 ( 12 ), e1001252 ( 2010 ).
- Khulan B , CooperWN , SkinnerBMet al. Periconceptional maternal micronutrient supplementation is associated with widespread gender related changes in the epigenome: a study of a unique resource in the Gambia . Hum. Mol. Genet.21 ( 9 ), 2086 – 2101 ( 2012 ).
- Hult M , TornhammarP , UedaPet al. Hypertension, diabetes and overweight: looming legacies of the Biafran famine . PLoS ONE5 ( 10 ), e13582 ( 2010 ).
- Rinaudo P , WangE . Fetal programming and metabolic syndrome . Annu. Rev. Physiol.74 , 107 – 130 ( 2012 ).
- Koukoura O , SifakisS , SpandidosDA . DNA methylation in the human placenta and fetal growth (review) . Mol. Med. Rep.5 ( 4 ), 883 – 889 ( 2012 ).
- Gluckman PD , HansonMA . The developmental origins of the metabolic syndrome . Trends Endocrinol. Metab.15 ( 4 ), 183 – 187 ( 2004 ).
- Fernandez-Morera JL , Rodriguez-RoderoS , Menendez-TorreE , FragaMF . The possible role of epigenetics in gestational diabetes: cause, consequence, or both . Obstet. Gynecol. Int. 2010 , 605163 ( 2010 ).
- Daxinger L , WhitelawE . Transgenerational epigenetic inheritance: more questions than answers . Genome. Res.20 ( 12 ), 1623 – 1628 ( 2010 ).
- Jaffe AE , IrizarryRA . Accounting for cellular heterogeneity is critical in epigenome-wide association studies . Genome Biol.15 ( 2 ), R31 ( 2014 ).
- Barfield RT , AlmliLM , KilaruVet al. Accounting for population stratification in DNA methylation studies . Genet. Epidemiol.38 ( 3 ), 231 – 241 ( 2014 ).
- Relton CL , GroomA , St PourcainBet al. DNA methylation patterns in cord blood DNA and body size in childhood . PLoS ONE7 ( 3 ), e31821 ( 2012 ).
- Bollati V , BaccarelliA . Environmental epigenetics . Heredity (Edinb)105 ( 1 ), 105 – 112 ( 2010 ).
- Ruchat SM , HoudeAA , VoisinGet al. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases . Epigenetics8 ( 9 ), 935 – 943 ( 2013 ).
- Lowe R , RakyanVK . Correcting for cell-type composition bias in epigenome-wide association studies . Genome Med.6 ( 3 ), 23 ( 2014 ).
- Bouchard L , HivertMF , GuaySP , St-PierreJ , PerronP , BrissonD . Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration . Diabetes61 ( 5 ), 1272 – 1280 ( 2012 ).
- Bouchard L , ThibaultS , GuaySPet al. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy . Diabetes Care33 ( 11 ), 2436 – 2441 ( 2010 ).
- El Hajj N , PliushchG , SchneiderEet al. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus . Diabetes62 ( 4 ), 1320 – 1328 ( 2013 ).
- Plagemann A , HarderT , BrunnMet al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome . J. Physiol.587 ( Pt 20 ), 4963 – 4976 ( 2009 ).
- Lillycrop KA , PhillipsES , JacksonAA , HansonMA , BurdgeGC . Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring . J. Nutr.135 ( 6 ), 1382 – 1386 ( 2005 ).
- Slater-Jefferies JL , LillycropKA , TownsendPAet al. Feeding a protein-restricted diet during pregnancy induces altered epigenetic regulation of peroxisomal proliferator-activated receptor-alpha in the heart of the offspring . J. Dev. Orig. Health Dis.2 ( 4 ), 250 – 255 ( 2011 ).
- Dolinoy DC . The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome . Nutr. Rev.66 ( Suppl. 1 ), S7 – S11 ( 2008 ).
- Tobi EW , LumeyLH , TalensRPet al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific . Hum. Mol. Genet.18 ( 21 ), 4046 – 4053 ( 2009 ).
- Dominguez-Salas P , MooreSE , BakerMS , BergenAW . Maternal nutrition at concepeption modulates DNA methylation of human metastable epialleles . Nat. Commun.5 ( 3746 ), 1 – 7 ( 2014 ).
- Barouki R , GluckmanPD , GrandjeanP , HansonM , HeindelJJ . Developmental origins of non-communicable disease: implications for research and public health . Environ. Health11 , 42 ( 2012 ).
- Petronis A . Epigenetics as a unifying principle in the aetiology of complex traits and diseases . Nature465 ( 7299 ), 721 – 727 ( 2010 ).
- Paul DS , BeckS . Advances in epigenome-wide association studies for common diseases . Trends Mol. Med.20 ( 10 ), 541 – 543 ( 2014 ).
- Rakyan VK , DownTA , BaldingDJ , BeckS . Epigenome-wide association studies for common human diseases . Nat. Rev. Genet.12 ( 8 ), 529 – 541 ( 2011 ).
- Heijmans BT , MillJ . Commentary: the seven plagues of epigenetic epidemiology . Int. J. Epidemiol.41 ( 1 ), 74 – 78 ( 2012 ).
- Verma M . Epigenome-wide association studies (EWAS) in cancer . Curr. Genomics13 ( 4 ), 308 – 313 ( 2012 ).
- Zou J , LippertC , HeckermanD , AryeeM , ListgartenJ . Epigenome-wide association studies without the need for cell-type composition . Nat. Methods11 ( 3 ), 309 – 311 ( 2014 ).
- Sham PC , PurcellSM . Statistical power and significance testing in large-scale genetic studies . Nat. Rev. Genet.15 ( 5 ), 335 – 346 ( 2014 ).
- Ramsay M . Africa: continent of genome contrasts with implications for biomedical research and health . FEBS Lett.586 ( 18 ), 2813 – 2819 ( 2012 ).
- Jallo N , LyonDE , KinserPA , KellyDL , MenziesV , Jackson-CookC . Recruiting for epigenetic research: facilitating the informed consent process . Nurs. Res. Pract.935740 ( 2013 ).
- Ramsay M , De VriesJ , SoodyallH , NorrisSA , SankohO . Ethical issues in genomic research on the African continent: experiences and challenges to ethics review committees . Hum. Genomics8 ( 1 ), 15 ( 2014 ).
- Teh AL , PanH , ChenLet al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes . Genome Res.24 ( 7 ), 1064 – 1074 ( 2014 ).
- McRae AF , PowellJE , HendersAKet al. Contribution of genetic variation to transgenerational inheritance of DNA methylation . Genome Biol.15 ( 5 ), R73 ( 2014 ).
- Wagner JR , BuscheS , GeB , KwanT , PastinenT , BlanchetteM . The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts . Genome Biol.15 ( 2 ), R37 ( 2014 ).
- Smith AK , KilaruV , KocakMet al. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage and tissue type . BMC Genomics15 , 145 ( 2014 ).
- Bell JT , PaiAA , PickrellJKet al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines . Genome Biol.12 ( 1 ), R10 ( 2011 ).
- Gibbs JR , Van Der BrugMP , HernandezDGet al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain . PLoS Genet.6 ( 5 ), e1000952 ( 2010 ).
- Liu Y , LiX , AryeeMJet al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysi of disease . Am. Soc. Hum. Genet.94 , 485 – 495 ( 2014 ).
- Campbell MC , TishkoffSA . African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping . Annu. Rev. Genomics Hum. Genet.9 , 403 – 433 ( 2008 ).
- Tishkoff SA , ReedFA , FriedlaenderFRet al. The genetic structure and history of Africans and African Americans . Science324 ( 5930 ), 1035 – 1044 ( 2009 ).
- Ambrosone CB , YoungAC , SuchestonLEet al. Genome-wide methylation patterns provide insight into differences in breast tumor biology between American women of African and European ancestry . Oncotarget5 ( 1 ), 237 – 248 ( 2013 ).
- Ashktorab H , DaremipouranM , GoelAet al. DNA methylome profiling identifies novel methylated genes in African American patients with colorectal neoplasia . Epigenetics9 ( 4 ), 503 – 512 ( 2014 ).
- Dogan MV , ShieldsB , CutronaCet al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women . BMC Genomics15 , 151 ( 2014 ).
- Wang S , DorseyT , TerunumaA , KittlesR , AmbsS , Kwabi-AddoB . Relationship between tumor DNA methylation status and patient characteristics in African-American and European-American women with breast cancer . PLoS ONE7 ( 5 ), e37928 ( 2012 ).
- Heyn H , MoranS , Hernando-HerraezIet al. DNA methylation contributes to natural human variation . Genome Res.23 ( 9 ), 1363 – 1372 ( 2013 ).
- Fraser HB , LamLL , NeumannSM , KoborMS . Population-specificity of human DNA methylation . Genome Biol.13 ( 2 ), R8 ( 2012 ).
- Kwabi-Addo BW , WangS , DevaneyJ . Epigenome-wide profiling identified significant differences in DNA methylation between African-American and European-American men with prostate cancer . Cancer Res. ( 74 ), 388 ( 2014 ).
- Nieminen TT , ShomanS , EissaS , PeltomakiP , Abdel-RahmanWM . Distinct genetic and epigenetic signatures of colorectal cancers according to ethnic origin . Cancer Epidemiol. Biomarkers Prev.21 ( 1 ), 202 – 211 ( 2012 ).
- Adkins RM , KrushkalJ , TylavskyFAet al. Racial differences in gene specific DNA methylation Levels are present at birth . Birth Defects Res. A. Clin. Mol. Teratol.91 ( 8 ), 728 – 736 ( 2011 ).
- Ataguba JE , AkaziliJ , McintyreD . Socioeconomic-related health inequality in South Africa: evidence from General Household Surveys . Int. J. Equity Health10 ( 1 ), 48 ( 2011 ).
- Borghol N , SudermanM , McardleWet al. Associations with early-life socio-economic position in adult DNA methylation . Int. J. Epidemiol.41 ( 1 ), 62 – 74 ( 2012 ).
- Mcguinness D , McglynnLM , JohnsonPCet al. Socio-economic status is associated with epigenetic differences in the pSoBid cohort . Int. J. Epidemiol.41 ( 1 ), 151 – 160 ( 2012 ).
- Bierne H , HamonM , CossartP . Epigenetics and bacterial infections . Cold Spring Harb. Perspect. Med.2 ( 12 ), a010272 ( 2012 ).
- De Vries J , PepperM . Genomic sovereignty and the African promise: mining the African genome for the benefit of Africa . J. Med. Ethics38 ( 8 ), 474 – 478 ( 2012 ).
- H3Africa – Human hereditary and health in Africa . www.h3africa.org .