
Abstract
Aim: We previously reported changes of DNA methylation and transcription patterns in mammalian cells that carry integrated foreign DNA. Experiments were now designed to assess the epigenetic consequences of inserting a 5.6 kbp plasmid into the human genome. Methods: Differential transcription and CpG methylation patterns were compared between transgenomic and nontransgenomic cell clones by using gene chip microarray systems. Results: In 4.7% of the 28.869 gene segments analyzed, transcriptional activities were up- or downregulated in the transgenomic cell clones. Genome-wide profiling revealed differential methylation in 3791 of > 480,000 CpG’s examined in transgenomic versus nontransgenomic clones. Conclusion: The data document genome-wide effects of foreign DNA insertions on the epigenetic stability of human cells. Many fields in experimental biology and medicine employ transgenomic or otherwise genome-manipulated cells or organisms without considering the epigenetic consequences for the recipient genomes.
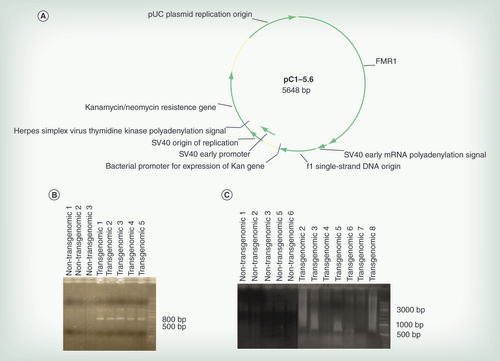
(A) Genetic map of the plasmid. For details see text. (B) Results of PCR analyses on the DNA from HCT116 cell clones as indicated for the presence of the transgenome. Primers placed inside the plasmid were described under Methods. Lanes 01–03 – DNA from nontransgenomic control clones; lanes 04–08 – DNA from plasmid pC1–5.6-transgenomic cell clones. (C) PCR results on DNA from cell clones as indicated using one primer inside the plasmid, the second one in an Alu sequence. The positioning of one primer in Alu sequences of the neighboring cellular DNA and of the second primer inside the plasmid sequence facilitated detection of the integrated plasmid genomes. All primers used were listed under section Methods.
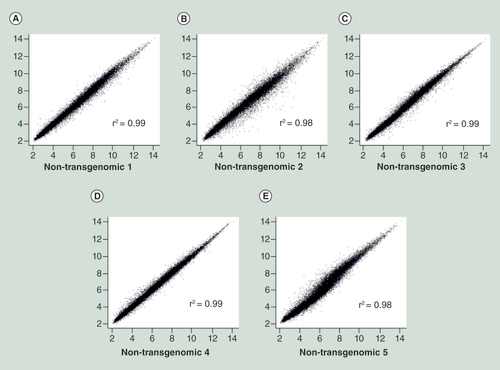
Scatter plots of double determinations from five different single HCT116 cell clones (A–E). X and Y axes display the same clones analyzed in double determinations. See text for details.
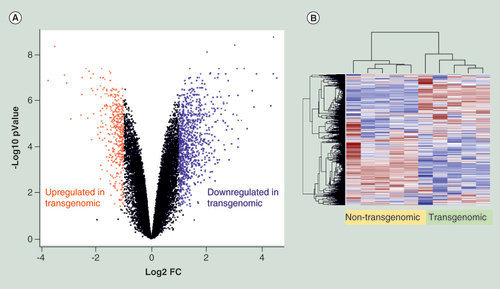
(A) Volcano plot displays nonstandardized signal (log2 fold-change) on the X-axis against standardized signal (-log10 false discovery rate adjusted p-value) on the Y-axis for the comparison of five nontransgenomic against seven transgenomic cell clones of all 28,869 genes analyzed. Upregulated genes in transgenomic cell clones were displayed in red and downregulated genes in blue (fold change ± 2, adjusted p-values < 0.05; n = 1343 genes). (B) Hierarchical cluster analysis of differentially expressed genes. Parameters for cluster calculation: distance = ‘correlation’, linkage = ‘average’. Expression signals were z-transformed. Upregulated genes are indicated in carmine, downregulated genes in blue.
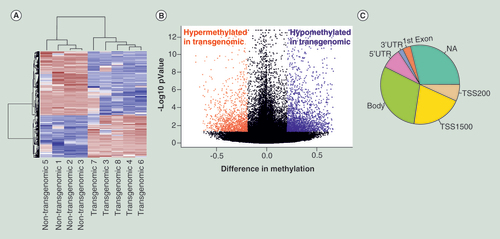
(A) Hierarchical cluster analysis of differentially methylated CpG’s. Parameters for cluster calculation: distance = ‘correlation’, linkage = ‘average’. Upregulated genes are indicated in carmine, downregulated genes in blue. (B) Volcano plot displays differences in methylation on the X-axis against standardized methylation (-log10 false discovery rate adjusted p-value) on the Y-axis for the comparison of four nontransgenomic against five pC1–5.6 transgenomic cell clones of all 361,983 CpG’s interrogated. Hypermethylated CpG’s in transgenomic cell clones are displayed in red and hypomethylated CpG’s in blue (Δβ value ≥0.2, adjusted p-value < 0.05; n = 3,791 CpG’s). (C) Pie chart indicates structural characteristics of differentially methylated CpG sites. Methylation changes found in different genes have been assigned to specific parts of these genes:
Body of gene (body); 5′UTR, 3′UTR, first exon are self- explanatory; NA not assigned; TSS transcription start site.
Studies on mammalian cells with integrated viral (adenovirus type 12, Ad12) DNA [Citation1–4] revealed that cellular genomes as foreign DNA recipients exhibited genome-wide alterations of DNA methylation patterns [Citation5]. Similar changes were documented in cells carrying bacterial plasmid or bacteriophage λ DNA as foreign genome inserts [Citation5,Citation6]. As a corollary, transcriptional profiles in Ad12 DNA or λ DNA transgenomic hamster cell genomes were significantly altered [Citation7]. It has been suggested earlier that these epigenomic destabilizations of transgenomic cells or organisms play a role in (viral) oncogenesis and possibly in evolution, since foreign DNA insertions in evolutionary times might have generated cells with novel functional profiles [Citation8–10].
Additional evidence from several experimental systems also supports the notion of epigenetic destabilization due to foreign DNA integration. In Epstein–Barr virus (EBV)-transformed human lymphoblastoid cell lines, in EBV-infected cells [Citation11,Citation12] or in human macrophages carrying integrated HIV-1 proviral genomes [Citation13], cellular DNA methylation and transcription patterns have been significantly altered. In induced pluripotent human stem cells with multiple integrated foreign genes, genome-wide alterations of CpG methylation profiles have been described [Citation14]. A detailed analysis of the methylation boundary [Citation15] upstream of the human FMR1 (fragile X mental retardation 1) gene in EBV- or telomerase gene-transformed human cells has revealed stability of the boundary itself, but extensive loss of CpG methylation far upstream of the boundary [Citation16]. The same upstream FMR1 region is CpG hypermethylated in nontransgenomic human cells.
In the present report, we have documented widespread alterations in cellular transcription and methylation patterns in human cells that carry a 5.6 kbp bacterial plasmid as integrated transgenome. SNP array karyotyping failed to detect major chromosomal alterations in transgenomic as compared with nontransgenomic cells.
Methods
Cells & cell culture
The human colon tumor cell line HCT116 [Citation17] was cultivated under standard conditions at 37°C using Dulbecco modified medium supplemented with 10% fetal bovine serum in a 5% CO2, 95% air atmosphere. Individual cell clones of nontransgenomic or of plasmid-transgenomic cells were isolated by the serial dilution method and recloned twice in 96-well plastic dishes.
Bacterial plasmid & transfection of HCT116 cells
The structure of the plasmid pC1–5.6 is shown in . Plasmid pC1–5.6 was constructed in the following way: From plasmid pEGFP-C1 (Clontech Laboratories Inc. Mountain View, CA, USA), the EGFP gene and the CMV promoter were removed and a 2298 bp FMR1 segment devoid of coding sequences was inserted. The pC1–5.6 plasmid was purified from bacterial strain DH5α by standard procedures [Citation18]. HCT116 cells (ATCC Cat. No. CCL-247) were transfected with plasmid pC1–5.6 by using the nucleofection protocol Cell Line Nucleofector® Kit V which had been optimized by Amaxa Biosystems Inc. (Cologne, Germany), specifically for this cell line. By following the manufacturer’s instructions, transfection efficiency was reported to be about 80%. Upon transfection, kanamycin resistant cells were selected and continuously maintained in the presence of 150 µg of Geneticin (G418) per ml culture medium. Successfully transfected HCT116 clones were identified by analyzing the total intracellular DNA from the transgenomic clones by the polymerase chain reaction (PCR) using the following primer pairs ( and B): Kanamycin: Kan.pC1–5.6 forward 5′-ATGATTGAACAAGATGGATT-3′; Kan.pC1–5.6 reverse 5′-TCAGAAGAACTCGTCAACAA-3′. Plasmid genome integration was ascertained by PCR using the primer pair: Alu – pC1–5.6: Alu A3: 5′-GACTGCACTCCAGCCTGGGC-3′; and pC1–5.6: 5′-GAATAGACCGAGATAGGGTTGAGTGTTG-3′ [Citation19].
Extraction of DNA & RNA
DNA from cultured cells was purified by phenol/chloroform extraction and ethanol precipitation [Citation1]. Total RNA from cultured cells was isolated by the RNeasy® Mini Kit from Qiagen and was treated with DNase by using the ‘RNase-Free DNase Set’ again from Qiagen and the manufacturer’s protocol.
Molecular karyotyping by SNP microarrays
Illumina’s CoreExome BeadChip was used for molecular karyotyping. Genotyping was performed according to the manufacturer’s manual on a fully automated iScan system. Briefly, the genomic DNA was amplified overnight, then enzymatically fragmented and subsequently hybridized to the array in another overnight step. After hybridization allele specificity was achieved by a single base extension using chemically modified deoxynucleoside triphosphates (dNTPs). After staining, the arrays were read out by using an Illumina iScan system. Analyses were performed by using Genome Studio (version 2011.1) and copy number variation (CNV) partition (version 3.1.6).
Affymetrix gene chip microarray assays
Sample preparations for microarray hybridization were carried out as described in the Ambion WT Expression Kit Protocol (Thermo Scientific) and the Affymetrix WT Terminal Labeling and Hybridization User Manual (Affymetrix, Inc., CA, USA). In brief, 300 ng of total RNA were used to generate double-stranded cDNA, followed by an in vitro transcription reaction. Twelve micrograms of RNA were purified and reverse transcribed into deoxyuridine triphosphate-(d-UTP)-containing sense-strand-(ss)-cDNA. Purified ss-cDNA was fragmented by using a combination of uracil DNA glycosylase and apurinic/apyrimidinic endonuclease 1 (APE 1), followed by terminal labeling with biotin. An amount of 3.8 µg of fragmented and biotin-labeled ss-cDNA was hybridized to Affymetrix Human Gene 1.0 ST arrays for 16 h at 45°C in a rotating chamber. Hybridized arrays were washed and stained in an Affymetrix Fluidics Station FS450, and the fluorescent signals were measured with an Affymetrix GeneChip Scanner 3000–7G. According to the Affymetrix manual, the GeneChip® Human Gene 1.0 ST Array for Gene Expression Analysis interrogates 28,869 annotated human genes with 764,885 distinct probes, in other words, an average of 26 probes per gene with 25-mer oligonucleotides. Sample processing was performed at an Affymetrix Service Provider and Core Facility, KFB – Center of Excellence for Fluorescent Bioanalytics, Regensburg, Germany. The laboratory that performed the activity determinations (KFB, Regensburg) had not been informed about the nature of these analyses or the derivation of the RNA samples.
Methylation assays
DNA was sodium-bisulfite converted using the Eazy DNA Methylation kit (Zymo Research, Freiburg, Germany). Samples were processed on the Human Methylation 450 K Bead Chips from Illumina (San Diego, CA, USA) according to the manufacturer’s manual. The laboratory process was already described under molecular karyotyping.
Expression & methylation data analyses
Expression assay quality control was monitored through hybridization controls. Summarized expression probe set signals were calculated by the RMA algorithm [Citation20] using the Bioconductor in R (statistical programming language). Multiple probes on different exons were summarized into accumulative expression values representing all transcripts from the same gene. Genes with a fold change above twofold and a Student’s t test false discovery rate (FDR) adjusted p-value < 0.05 were considered as significantly regulated.
For the analysis of the methylation data, a subset quantile normalization approach was applied [Citation21]. This processing pipeline included signal corrections for the adjustment of color balance and background level corrections as well as the Infinium I/Infinium II shift correction between sample normalization. By using the ‚Genome Studio’ software, technical quality parameters such as hybridization, extension, bisulfite conversion and specificity were evaluated. Signal distributions of β-values, representing a continuous measurement for methylation levels were inspected by density plots. Sample variation was visualized by unsupervised hierarchical cluster and principal component analysis. The data were analyzed by using Bioconductor in R program. Cross-reactive probes and polymorphic CpG’s as well as CpG’s with SNP’s in the CpG 50-mer probe defined by [Citation22] were removed leaving 361,983 CpG’s for analysis. Differentially methylated CpG loci were identified using a t-test; p-values were corrected for multiple testing using Benjamin-Hochberg corrections [Citation23]. A difference in mean ß-values of 0.2 and FDR-adjusted p-values of < 0.05 were chosen as thresholds to call differential methylation. Pathway analysis of differentially expressed genes and differentially methylated CpG’s was performed using Ingenuity Pathway Analysis.
Results & discussion
Design of experiments & structure of pC1–5.6 plasmid transgenome
Experiments with a human cell line were designed to assess the consequences of the integration of a plasmid transgenome on transcription and methylation patterns in the recipient genomes. Five independently isolated and propagated nontransgenomic single cell clones of the human colon tumor cell line HCT116 [Citation17] were analyzed as controls. These cells showed nearly identical expression (mean r2 = 0.99, ) and very comparable methylation profiles (mean r2 = 0.97, see below). This finding documented the stability and similarity of cellular methylation and transcription profiles in individual cell clones. These results established the basis for comparisons of these profiles to those of seven plasmid-transgenomic cell clones.
The 5648 bp bacterial plasmid pC1–5.6 () served as transgenome that contained the kanamycin/neomycin resistance gene under early SV40 promoter control as the sole functional eukaryotic promoter in the construct. The plasmid also carried the bacterial kanamycin promoter and the nontranslated first exon with parts of the 5′-UTR and the CGG repeat of the human fragile X mental retardation gene 1 (FMR1) [Citation24]. Total length of the FMR1 fragment was 2298 bps. Persistence and integrated state of this plasmid in transgenomic cell clones were ascertained by kanamycin resistance of the cell clones and by PCR ( and C). Primers selected for PCR were listed under the section Methods. The positioning of one primer in Alu sequences of the neighboring cellular DNA sequences and of the second primer inside the plasmid sequence facilitated documentation of the integrated state of the plasmid genomes (). The positive PCR products in which were derived from the DNA of transgenomic clones 2, 3, 5, 6 and 8 were all different in size and in part heterogeneous in lengths. They span the plasmid sequence and the Alu primer-targeted sequences of the abutting integration sites (). The profiles indicate that the insertion sites of plasmid pC1–5.6 are different in each of these clones. Moreover, there might be more than one integration site per clone, because some of the PCR products indicate size heterogeneity.
Molecular karyotyping
To exclude major chromosomal abnormalities introduced through transfection of the pC1–5.6 plasmid into human cells, we analyzed all DNA samples from transgenomic and nontransgenomic cells by using SNP microarrays. Hybridization to the arrays allows measurements of chromosomal abnormalities through changes in the signal intensity of the probes (log-R ratio) and the distribution of heterozygous genotypes (b-allele frequency). By molecular karyotyping, shared copy number variations (CNV’s) including duplications, deletions and copy-neutral loss of heterozygosity were detected across all samples (Supplementary Figure 1). However, comparisons between nontransgenomic and transgenomic cell clones revealed no significant clonal aberrations and high congruency across all samples.
Analysis of transcription activities in nontransgenomic versus transgenomic human cell clones
The results of comparisons of transcriptional activities between nontransgenomic and transgenomic cell clones can be critically interpreted only when independently isolated clones of nontransgenomic cells have identical or very similar transcription patterns across the genome. We have isolated five clones of nontransgenomic HCT116 cells and expanded and propagated them under identical conditions. From each of these cell populations total RNA was isolated by the Qiagen RNeasy® Mini Kit. By using an Affymetrix Expression Array system with fluorescent hybridization probes, transcriptional activities were measured in 28,869 genes covered by 764,885 distinct probes (see section Methods). Expression profiles of all five independently isolated and propagated nontransgenomic cell clones analyzed in double determinations had a pair-wise Pearson’s correlation coefficient above 0.98 for all transcripts measured on the microarrays (). Moreover, the mean correlation coefficient was 0.99 across all samples with a minimum correlation of 0.97. We conclude that individual HCT116 cell clones exhibit identical, or very similar, transcription activities across the analyzed parts of the genome. This result renders meaningful the comparison of transcriptional activities between nontransgenomic and plasmid-transgenomic cell clones.
Differential gene expression was next assessed between mean values of two biological replicates from five nontransgenomic control clones against seven plasmid-transgenomic cell clones by a fold-change (FC)/p-value filter. Genes were called differentially expressed when the FC was above ±2 and the FDR-adjusted p-value < 0.05. Among the 28,869 human genes, 1343 genes were differentially expressed (), 907 were upregulated and 436 were downregulated in transgenomic cell clones (). Genes with the highest changes are summarized in . Gene set enrichment analysis was performed using Ingenuity Pathway Analysis for all identified differentially expressed genes (n = 1343 genes). A significant enrichment was found in 43 canonical pathways which included EIF2 signalling, Regulation of eIF4 and p70S6K signalling, Glutathione-mediated Detoxification, FAK signalling, Insulin Receptor signalling and ErbB4 signalling (). The predominant upregulation effect was found in snoRNA (small nucleolar RNA) genes raising important questions about far-reaching effects on the biochemical modifications of many classes of RNA (ribosomal, transfer and small nuclear) and their consequences for cell function [Citation25]. It is also worth noting that several signalling genes show differential expression. The data indicate that the insertion of foreign DNA, in other words, of bacterial plasmid pC1–5.6, into the recipient human cell genome led to alterations of transcriptional activities in 4.7% of the 28,869 analyzed genes.
Analysis of DNA methylation patterns in plasmid-transgenomic versus nontransgenomic human cell clones
Genome-wide methylation profiling was performed by assessing over 480,000 CpG sites in four nontransgenomic clones and five pC1–5.6 transgenomic clones using Illumina 450K Infinium arrays. Similarly to expression profiling, the mean correlation across all nontransgenomic human cell clones was calculated as 0.97 ± 0.02 SD; thus rendering meaningful the comparison between nontransgenomic and transgenomic cell clones. Using both the stringent criteria of Δβ values ≥0.2 between nontransgenomic and transgenomic cell clones as well as a FDR-adjusted p < 0.05 in the comparison between both groups, 3791 CpG’s were found differentially methylated. Of them, 1504 were hyper- and 2287 were hypomethylated in transgenomic clones ( and B). Most CpG’s were located in gene bodies (29.91%), followed by transcription start sites (27.49%) as summarized in .
IPA pathway analysis using 2622 genes corresponding to the 3791 probes identified as differentially methylated revealed a significant enrichment in 109 canonical pathways including several important functional classes of genes associated with neuron signalling (). We conclude that in the pC1–5.6 plasmid transgenomic cell lines, levels of CpG methylation are significantly altered as compared with methylation profiles in nontransgenomic cells.
Conclusion
This study extends earlier work on changes of cellular transcription and methylation patterns in mammalian cells upon the chromosomal insertion of viral or plasmid DNAs [Citation5–7,Citation16]. We have now chosen a targeted approach and used human cells in culture with a small integrated bacterial plasmid (pC1–5.6), which carried the kanamycin gene under SV40 promoter control. SNP karyotyping documented that, when compared with nontransgenomic clones, the transgenomic cells had not been subject to larger insertions, deletions or copy number variations in their exome.
Differential transcription and methylation was assessed by comparing transgenomic cell clones to nontransgenomic control cell clones. In 4.7% of the 28.869 gene segments analyzed, the transcriptional activities were upregulated (907 genes) or downregulated (436 genes) in the cell clones that carried the bacterial plasmid pC1–5.6. A significant gene set enrichment was found in 43 canonical pathways. Genome-wide methylation profiling was performed for > 480,000 CpG sites in four nontransgenomic and five pC1–5.6 transgenomic clones. Stringent statistical analyses documented that in a comparison of methylation levels in transgenomic versus nontransgenomic cell clones 3791 CpG’s were differentially methylated, 1504 CpG’s were hyper- and 2287 were hypomethylated in the transgenomic clones. The different transgenomic clones differ in their profiles.
Viewed under the perspective of previously published investigations from our [Citation5–7,Citation16] and from other [Citation11–14] laboratories, it is apparent that the insertion of foreign DNA into an established mammalian genome can lead to genome-wide destabilizations of cellular transcription and methylation patterns. We have not yet conducted a systematic investigation into the epigenetic trans effects of transgenome size, its gene or CpG content or copy number of the integrated foreign DNA. From the sum total of all our previous investigations on this topic, we conclude that the integrations of Ad12, bacteriophage λ or plasmid DNA as well as the currently used pC1–5.6 plasmid have all led to alterations of methylation and transcriptional profiles in the recipient cells [Citation5–7]. There is also evidence that cells carrying EBV genomes or the integrated gene for telomerase are subject to DNA methylation changes [Citation16]. Hence, the integration of a number of quite different foreign genomes has elicited epigenetic destabilization which then does not appear to depend on one type of DNA sequence. These genome-wide alterations in DNA methylation patterns go far beyond local CpG methylation changes at the site of foreign DNA integration which were reported earlier [Citation26,Citation27].
The mechanism(s) underlying the observed epigenetic alterations are presently not understood. It is conceivable that extent and location of alterations in genome activities and CpG methylation depend on the site(s) of foreign DNA insertion. In the transgenomic cells analyzed here, the highest upregulations have been found in snoRNA (small nucleolar RNA) genes. A number of genes involved in signalling pathways have also been markedly affected. More general functional discussions on the affected genes will have to await analyses on the interdependency of foreign DNA integration site locations and affected genes. In this context it is of interest to note that massive reporter gene integration has been shown to lead to extensive variation in cellular gene expression levels [Citation28]. Foreign DNA insertions into noncoding parts of the genome would possibly affect a multitude of genetic elements as identified by the ENCODE or FANTOM project [Citation29,Citation30]. In this way, a large number of cellular functions might be fundamentally altered.
Future perspective
In published investigations, work on the epigenetic consequences of genome manipulations in mammalian (human) cells, including the insertion of foreign DNA, has so far received almost no attention. There are, however, numerous unresolved problems in this context, and one might expect increased research activities along these lines. By the same token, genome manipulations, the addition of foreign DNA to mammalian cells by various methods of transfection, and the selection of transgenomic or knocked cells and organisms, have been everyday practice in a large number of laboratories. With further refinement of epigenetic technologies, it will be but a question of (possibly a very short) time that hitherto unsuspected complications in the evaluation of experiments with genome manipulated cells and organisms will come to the fore. It is the intent of this article, not only to document further these complexities of epigenetic mechanisms, but to alert researchers in all fields of biomedicine to possibly far-reaching problems with their interpretations.
Table 1. Differentially expressed genes.
Table 2. Top canonical pathways for differentially expressed genes.
Table 3. IPA top canonical pathways for differentially methylated CpGs.
In studies on the integrated state of adenovirus type 12 (Ad12) DNA in Ad12-transformed hamster cells, we discovered that the CpG methylation profiles in their endogenous retrotransposon sequences and in several cellular genes were increased. This augmented methylation persisted in revertants of the transformed cells that had lost all Ad12 genomes. Moreover, alterations of DNA methylation and transcription profiles were documented in Ad12 DNA- and in bacteriophage λ DNA-transgenomic cells.
We previously hypothesized that epigenetic effects in mammalian genomes due to the insertion of foreign DNA are a general phenomenon. These alterations might play a role in (viral) oncogenesis and are possibly instrumental during evolution as a consequence of multiple retroviral DNA insertions into ancient genomes. Over evolutionary times, these alterations of transcription profiles might have led to novel phenotypes that were then selected for or against depending on environmental conditions during evolution.
To examine the general significance of these observations, we designed a model system for proof of principle assessment. Human cells from cell line HCT116 were rendered transgenomic by transfecting a 5.6 kbp bacterial plasmid and selecting cell clones with foreign plasmids stably integrated, most likely at different genomic sites.
In five nontransgenomic HCT116 control clones without the plasmid, transcription and methylation patterns proved similar, if not identical, among individual cell clones. This finding opened the possibility for comparisons of these patterns between nontransgenomic and transgenomic clones.
Molecular karyotyping demonstrated shared copy number variations including duplications, deletions and copy-neutral loss of heterozygosity across all DNA samples. A comparison of nontransgenomic and transgenomic clones, however, revealed no significant clonal aberrations but high congruency.
In 4.7% of the 28,869 gene segments analyzed, the transcriptional activities were upregulated (907 genes) or downregulated (436 genes) in plasmid-transgenomic cell clones in comparison to control clones. A significant gene set enrichment was found in 43 canonical pathways. Frequent upregulations were noted in small nucleolar RNA genes that regulate RNA metabolism and in genes involved in signalling pathways.
Genome-wide methylation profiling was performed for >480,000 CpG sites. In comparisons of methylation levels in five transgenomic versus four nontransgenomic cell clones, 3791 CpG’s were differentially methylated, 1504 CpG’s were hyper- and 2287 were hypo-methylated.
Thus, the epigenetic effects in the wake of foreign DNA integration events can be considered a general effect also in human cells. We still lack insights into the role of transgenome size, gene or CG content or copy number. The mechanism(s) underlying the observed epigenetic alterations are unknown. Extent and location of alterations in genome activities and CpG methylation might depend on the site(s) of foreign DNA insertion.
We note that genome manipulations in general – work with transgenomic or knocked cells and organisms – have assumed a major role in molecular biology and medicine. The consequences of cellular genome manipulations for epigenetic stability have so far received unwarrantedly limited attention. Before drawing far-reaching conclusions from work with cells or organisms with manipulated genomes, critical considerations for and careful analyses of their epigenomic stability will prove prudent.
Supplemental image
Download TIFF Image (11.4 MB)Acknowledgements
The transcriptional profiles (primary data) were determined by KFB in Regensburg, Germany. We are indebted to the Institute of Clinical and Molecular Virology, University of Erlangen-Nürnberg Medical School for their continued support of W Doerfler’s senior research group. S Weber performed most of the laboratory experiments and was involved in the planning and interpretation of the project. A Hofmann performed all statistical analyses, was involved in the interpretation of data and wrote part of the manuscript. S Weber and A Hofmann have contributed equally to this work. S Herms performed molecular karyotyping. P Hoffmann carried out methylation profiling, interpreted data and wrote part of the manuscript. W Doerfler initiated and planned the project, was involved in the interpretation of data and wrote the manuscript. KFB in Regensburg did the transcription analyses.
Financial & competing interests disclosure
This research was made possible by grants to W Doerfler from the Thyssen Foundation, Köln (Az. 10.07.2.138.) and from the Deutsche Forschungsgemeinschaft, Bonn (DO 165/28-1). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Sutter D , WestphalM , DoerflerW . Patterns of integration of viral DNA sequences in the genomes of adenovirus type 12-transformed hamster cells . Cell14 , 569 – 585 ( 1978 ).
- Doerfler W , GahlmannR , StabelSet al. On the mechanism of recombination between adenoviral and cellular DNAs: the structure of junction sites . Curr. Top. Microbiol. Immunol.109 , 193 – 228 ( 1983 ).
- Hochstein N , MuiznieksI , MangelL , BrondkeH , DoerflerW . The epigenetic status of an adenovirus transgenome upon long-term cultivation in hamster cells . J. Virol.81 , 5349 – 5361 ( 2007 ).
- Doerfler W . DNA methylation and gene activity . Ann. Rev. Biochem.52 , 93 – 124 ( 1983 ).
- Heller H , KämmerC , WilgenbusP , DoerflerW . Chromosomal insertion of foreign (adenovirus type 12, plasmid, or bacteriophage lambda) DNA is associated with enhanced methylation of cellular DNA segments . Proc. Natl Acad. Sci. USA92 , 5515 – 5519 ( 1995 ).
- Remus R , KämmerC , HellerH , SchmitzB , SchellG , DoerflerW . Insertion of foreign DNA into an established mammalian genome can alter the methylation of cellular DNA sequences . J. Virol.73 , 1010 – 1022 ( 1999 ).
- Müller K , HellerH , DoerflerW . Foreign DNA integration. Genome-wide perturbations of methylation and transcription in the recipient genomes . J. Biol. Chem.276 , 14271 – 14278 ( 2001 ).
- Doerfler W . A new concept in (adenoviral) oncogenesis: integration of foreign DNA and its consequences . BBA Rev. Cancer1288 , F79 – F99 ( 1996 ).
- Doerfler W . Epigenetic consequences of foreign DNA integration: Global alterations of methylation and transcription patterns in recipient genomes . Rev. Med. Virol.21 , 336 – 346 ( 2011 ).
- Doerfler W . The impact of foreign DNA integration on tumor biology and evolution via epigenetic alterations . Epigenomics4 , 41 – 49 ( 2012 ).
- Grafodatskaya D , ChoufaniS , FerreiraJCet al. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines . Genomics95 , 73 – 83 ( 2010 ).
- Birdwell CE , QueenKI , KilgorePCet al. Genome-wide DNA methylation as an epigenetic consequence of Epstein–Barr virus infection of immortalized keratinocytes . J. Virol.88 , 11442 – 11458 ( 2014 ).
- Soto-Girón MJ , Garcia-VallejoF . Changes in the topology of gene expression networks by human immunodeficiency virus type 1 (HIV-1) integration in macrophages . Virus Res.163 , 91 – 97 ( 2012 ).
- Doi A , ParkIH , WenBet al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts . Nat. Genet.41 , 1350 – 1353 ( 2009 ).
- Naumann A , HochsteinN , WeberS , FanningE , DoerflerW . A distinct DNA methylation boundary in the 5′-upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome . Am. J. Hum. Genet.85 , 606 – 616 ( 2009 ).
- Naumann A , KrausC , HoogeveenA , RamirezCM , DoerflerW . Stable DNA methylation boundaries and expanded trinucleotide repeats: Role of DNA insertions . J. Mol. Biol.426 , 2554 – 2566 ( 2014 ).
- Brattain MG , FineWD , KhaledFM , ThompsonJ , BrattainDE . Heterogeneity of malignant cells from a human colonic carcinoma . Cancer Res.41 , 1751 – 1756 ( 1981 ).
- Sambrook J , RussellDW . Molecular Cloning, 3rd Edition . Cold Spring Harbor Laboratory Press , Cold Spring Harbor, New York, NY, USA ( 2001 ).
- Minami M , PoussinK , Bre’ChotC , PaterliniP . A novel PCR technique using Alu-specific primers to identify unknown flanking sequences from the human genome . Genomics29 , 403 – 408 ( 1995 ).
- Irizarry RA , HobbsB , CollinFet al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data . Biostatistics4 , 249 – 264 ( 2003 ).
- Touleimat N , TostJ . Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation . Epigenomics4 , 325 – 341 ( 2012 ).
- Chen YA , LemireM , Choufaniet al. Discovery of cross-reactive probes and polymorphic CpG’s in the Illumina Infinium Human Methylation450 microarray . Epigenetics8 , 203 – 209 ( 2013 ).
- Benjamin Y , HochbergY . Controlling of false discovery rate: a practical and powerful approach to multiple testing . J. R. Stat. Soc.289 , 289 – 300 ( 1995 ).
- Verkerk AJMH , PierettiM , SutcliffeSSet al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome . Cell65 , 905 – 914 ( 1991 ).
- McMahon M , ContrerasA , RuggeroD . Small RNAs with big implications: new insights into H/ACA snoRNA function and their role in human disease . Wiley Interdiscip. Rev. RNA6 , 173 – 189 ( 2015 ).
- Jähner D , JaenischR . Retrovirus-induced de novo methylation of flanking host sequences correlaes with gene inactivation . Nature315 , 594 – 597 ( 1985 ).
- Lichtenberg U , ZockC , DoerflerW . Integration of foreign DNA into mammalian genome can be associated with hypo-methylation at site of insertion . Virus Res.11 , 335 – 342 ( 1988 ).
- Akhtar W , de JongJ , PindyurinAVet al. Chromatin position effects assayed by thousands of reporters integrated in parallel . Cell154 , 914 – 927 ( 2013 ).
- The ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome . Nature489 , 57 – 74 ( 2012 ).
- FANTOM Consortium and the RIKEN PMI and CLST (DGT) . A promoter-level mammalian expression atlas . Nature507 , 462 – 470 ( 2014 ).