
Abstract
Aim: Prenatal alcohol exposure (PAE) can cause fetal alcohol spectrum disorders (FASD). Previously, we assessed PAE in brain tissue from mouse models, however whether these changes are present in humans remains unknown. Materials & methods: In this report, we show some identical changes in DNA methylation in the buccal swabs of six children with FASD using the 450K array. Results: The changes occur in genes related to protocadherins, glutamatergic synapses, and hippo signaling. The results were found to be similar in another heterogeneous replication group of six FASD children. Conclusion: The replicated results suggest that children born with FASD have unique DNA methylation defects that can be influenced by sex and medication exposure. Ultimately, with future clinical development, assessment of DNA methylation from buccal swabs can provide a novel strategy for the diagnosis of FASD.
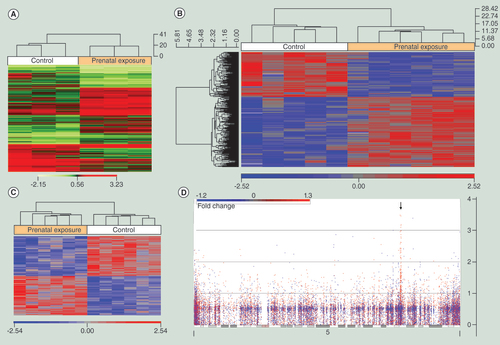
(A) Heatmap of top significant (p < 0.01) contigs from the adult mouse prenatally exposed to alcohol. (B) Heatmap of significantly (p < 0.005) differentially methylated targeted CpG sites generated using hierarchical clustering of buccal epithelial DNA obtained via swab from fetal alcohol spectrum disorders (n = 6) and matched control (n = 5) children from the discovery cohort. (C) Heatmap of significantly (p < 0.005) differentially methylated targeted CpG sites generated using hierarchical clustering of buccal epithelial DNA obtained via swab from fetal alcohol spectrum disorders (n = 6) and matched control (n = 6) children from the replication cohort. (D) Manhattan plot for human chromosome 5 from the discovery sample. Genomic location is plotted on the x-axis and –log10 (p-value) of differential CpG methylation from prenatal alcohol exposure is on the y-axis. Each dot represents a single CpG site. A red dot indicates an increase in methylation and a blue dot indicates a decrease in methylation. The black arrow identifies clustered protocadherin genes on 5q31.
The network highlights preferential and synchronous alteration involving Protocadherin genes in children with fetal alcohol spectrum disorders.

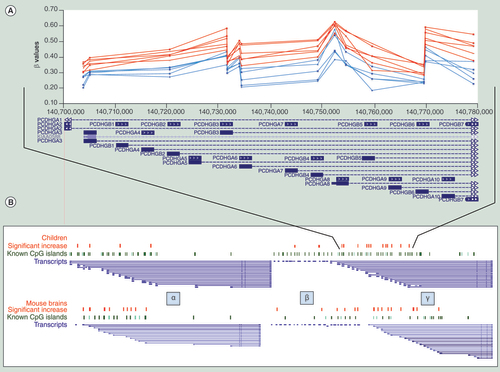
(A) Representative methylation patterns in 14 CpGs in the gamma cluster in six fetal alcohol spectrum disorders children and their five matched controls. (B) Significantly (p < 0.005) increased methylation in the clustered protocadherin region of patients (red dots), known CpG island (green dots) and potential transcripts (blue lines) along with their similarity to the mouse model (p < 0.01). Figure is not to scale as non-informative regions have been removed for clarity.
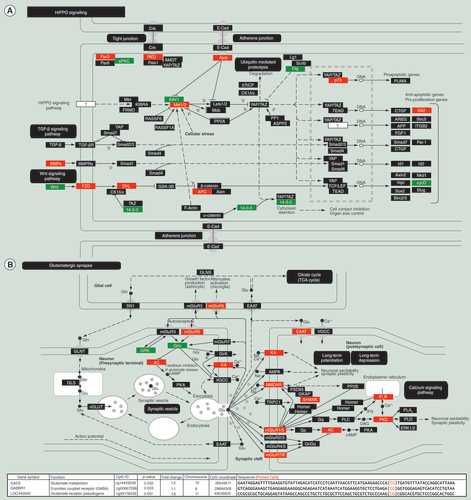
Genes were identified and then assembled as protein complexes. (A) Hippo signaling; (B) glutamatergic synapse pathway. Green marks a decrease in methylation and potentially increased gene expression while red indicates a increase in methylation and potentially decreased gene expression.
Prenatal alcohol exposure (PAE) can cause a continuum of heterogeneous defects termed fetal alcohol spectrum disorders (FASD). Patients affected with FASD show life-long defects, particularly affecting the central nervous system. At the molecular level PAE disturbs critical signaling related to apoptosis and cell-to-cell interactions [Citation1]. Furthermore, PAE causes changes to synaptic signaling and disrupts the neuroendocrine system, where it causes enhanced activity of the hypothalamic–pituitary–adrenal (HPA) axis [Citation2]. Finally, the effects are confounded by genetic variation in pathways related to the immediate toxicity, which include ethanol’s metabolite acetaldehyde and oxidative stress pathways [Citation3].
The question of how fetal alcohol exposure causes long-term molecular and behavioral defects has been difficult to ascertain. It has necessitated the development of mouse models [Citation4] and genome-wide technologies [Citation5]. Results show that exposure to alcohol during neurodevelopment causes behavioral disabilities in the resulting offspring that mimic human FASD patients in a C57BL/6J (B6) mouse model [Citation6]. Furthermore, the resulting mice show life long changes in brain gene expression [Citation7] as well as ‘footprints’ of exposure from critical windows of epigenetic programming, particularly related to DNA methylation and noncoding RNA (ncRNA) expression [Citation8]. This effect of PAE in rodent models has been reported from a number of independent laboratories [Citation9–15] over the last decade. They are now viewed as a milestone in FASD research [Citation16,Citation17]. The genes affected participate in the critical neural processes of synaptogenesis, apoptosis, cellular identity, cell–cell adhesion, and signaling [Citation7,Citation8].
These results are best explained by observations (in alcoholism and FASD research) that alcohol directly affects the dynamics of the epigenome via altering one-carbon metabolic pathways from which methyl groups are derived [Citation18–20]. Developmental alcohol exposure also results in altered expression of the genes that establish, maintain and read DNA methylation, such as Dnmt1, Dnmt3a, Mecp2, and Mtrr [Citation7,Citation21,Citation22]. Furthermore, using genome-wide assays to interrogate the cytosine methylation and ncRNA expression within the brain of adult mice prenatally exposed to moderate amounts of alcohol, we have identified a transcriptomic and epigenomic footprint from exposure during critical windows of development [Citation5,Citation8,Citation23–25]. This ‘footprint’ is characterized by alterations to imprinted regions of the genome that encode multiple developmentally important noncoding RNAs (ncRNAs) and are regulated by CCCTC-binding factor (CTCF), a zinc finger protein that functions as a genomic insulator [Citation26]. The canonical pathways affected include PTEN/PI3K/AKT/mTOR pathway where the alterations affect cell adhesion and proliferation [Citation8,Citation12,Citation27]. These findings suggest that ‘footprints’ in the epigenome may represent past developmental transcriptional aberrations as well as current ones in related cells that are either active or poised for a response. In fact, these processes are developmentally integral to the outcome of larger scale cortical brain structure formation, since the events of both pre- and postnatal neurodevelopment are highly dependent on the (epi)genotype, as well as the molecular environment of the differentiating cells [Citation28]. Taken together, current evidence suggests that ethanol can create long-term changes in the epigenetic landscape that are essential and actively needed for remaining neurodevelopment.
Of special interest to this report are the genes related to the clustered protocadherin gene family and glutamatergic synapses. Protocadherins are expressed in the nervous system where they mediate highly complex cell-to-cell interactions. The protocadherin molecules affect the adhesion of a number of synaptic events, including glutamatergic synapses. Glutamate is the most abundant excitatory neurotransmitter of the nervous system and has a role in synaptic plasticity. As such, it is a key molecular component of learning and memory. Glutamate is known for its involvement in the plasticity process of long-term potentiation and is utilized in the hippocampus and neocortex, among other regions, and the related signaling is particularly sensitive to ethanol. On the other hand, protocadherins are transmembrane proteins with repeats of the cadherin motif or the cadherin extracellular domain. This domain contains a conserved Ca2+ binding sequence and the number of extracellular domains may vary across different cadherin molecules. The protocadherins function as cell adhesion molecules and create individual neuronal identities that allow for large-scale network formation via controlling synaptic interactions. They have been implicated in a number of brain disorders with similar endophenotypes to FASD such as autism spectrum disorders, bipolar disorder, and schizophrenia [Citation29].
In this report we analyzed children (3–6 year olds) with a comprehensive FASD diagnosis and a maternal history of PAE (termed the discovery cohort) for genome-wide DNA methylation changes that are analogous to the results on mice. This effect is also replicated in an independent group of FASD children (termed the replication cohort) collected 6 months apart. The repeated results establish that DNA methylation is affected at specific genomic loci in children with FASD. The analysis allowed for the identification of protocadherins, glutamate, and hippo signaling factors as critical molecules in the etiology of this neurodevelopmental disorder. These results may also explain overlaps involving FASD and other neurodevelopmental disorders of unknown etiology.
Materials & methods
The results included in this report are based on two cohorts (discovery and replication) of young children clinically diagnosed with FASD by J Kapalanga in Ontario, Canada (). The discovery cohort consisted of 6 FASD patients and 5 matched controls, while the replication cohort consisted of a different set of 6 FASD children and 6 matched controls. The diagnosis shows that FASD patients from the discovery cohort are much more homogeneous and better matched to controls with respect to age (3–6 year olds), sex (males only), socioeconomic status, maternal history of drinking, life exposures, and ancestry (A). All subjects had Northern-European ancestry and the matched controls were visiting the clinic for annual check-ups or relatively minor non-neurodevelopmental or behavioral problems like asthma, allergies, musculoskeletal problems, or gastrointestinal problems. Following ethical approval and informed consent from parents or guardians, the study subjects contributed cheek swabs for analysis of buccal epithelial DNA. While four (R2, R3, R5 and R6) of the six patients in the replication cohort were 6–10 year old males and not on medication, two were older 7 and 9 year old females (R1 and R4) and on psychotropic/stimulant medication (B). Thus, while the replication cohort matches with the discovery cohort, the replication cohort is different in that some are females, on medication, and/or have a wider age range.
Human 450K Infinium Methylation BeadChip
Individual cheek swabs were used to isolate genomic DNA using the QIAamp DNA Mini Kit following manufacturer’s protocol. This DNA was processed following sodium bisulfite modification at The Centre for Applied Genomics (Toronto, Canada). The genome-wide CpG methylation for each subject was assessed using the Illumina HumanMethylation450 BeadChip following the manufacturer’s protocol. The array quantifies the methylation status of over 480,000 CpGs in the human genome. Arrays were scanned using Illumina’s GenomeStudio at The Centre for Applied Genomics (Toronto). It generated scan data (.idat files) for each human subject used in this analysis.
The .idat files were analyzed using Partek Genomics Suite® Version 6.6. A site-based DNA methylation workflow for Illumina BeadArray Methylation was performed as the primary analysis. Preprocessing using both control normalization and background subtraction was performed using Illumina’s algorithm to generate β-values. β-values were then normalized, converted to a fold-change, and subjected to a one-way ANOVA analysis to identify regions of differential methylation using annotations from Hg18 for the primary analysis. Custom genome dot plots (also known as Manhattan plots) were generated from the ANOVA results. For hierarchical clustering and gene ontologies from Enrichr [Citation30] a stringency of p < 0.005 was used. For Partek Pathways a gene list with a p < 0.05 stringency was used. The subset-quantile within array normalization (SWAN) algorithm [Citation31] from the minifi bioconductor package was used within Partek for a secondary analysis to compare across batches [Citation32].
Confirmation by pyrosequencing
Using the same bisulfite converted buccal swab DNA from the discovery cohort, EpigenDx performed pyrosequencing on the PSQ96 HS System (Qiagen) following the manufacturer’s instructions, using custom assays [Citation33], and a gradient of controls with known methylation levels. This allowed for the quantification of the absolute percent methylation [Citation34] of each CpG at select loci using QCpG software (Qiagen). The sequencing reads also allowed for the analysis of SNPs known to be within or close to the CpGs of the 450K probes.
Comparative mouse model epigenomics
All the significant (p < 0.01) annotated peaks from the mouse model Differential Enrichment Peak (DEP) analysis were extracted and combined for peak filtering. Peaks from the MeDIP CpG Island tilling arrays are represented as unique stretches of assembled contiguous probes showing significant alterations to DNA methylation. This near base-pair resolution data was previously generated from the mouse model using a number of bioinformatic scripts and packages [Citation8]. The filtering criteria was set as peaks within both top 20% PeakScore and top 20% PeakDMValue. Alignment of reference genomes was done using the UCSC genome browser [Citation35].
Results
Neurodevelopmental alcohol exposure causes differential genome-wide DNA methylation
Using a stringent but not false discovery rate (FDR) corrected CpG list (children p < 0.005 and mice p <0.01) genome-wide differential methylation was analyzed in: mice brains subjected to neurodevelopmental alcohol; buccal swab DNA of FASD patients and matched controls; and also a replication cohort (). In each case, the heat maps show that mice with FASD phenotypes and children diagnosed with FASD clearly group together and are distinct from matched non-FASD controls. The results have also allowed for the identification of specific genes that are concordant between the three independent sets of results. The experiments involving mice were described earlier and are based on a MeDIP-CHIP tilling array that analyzes CpG islands with greater than 2 CpGs affected per a 50–75 base pair probe and then tilled into larger contiguous sequences [Citation8]. For the children, we used the 450K methylation array, which analyzes alterations to single CpGs via sodium bisulfite conversion and an epigenotyping approach. From theses arrays a one-way ANOVA was created to generate a list of CpGs and corresponding genes that showed significant differential methylation between FASD children and matched controls (after extensive preprocessing and data normalization). The results of the discovery and replication cohorts were analyzed separately in order to avoid any batch effects and confounding differences from sample heterogeneity. These effects appeared to reduce the statistical power more substantially than the limits of sample size in this experimental design. Therefore, in order to accommodate for some of the limitations of sample size the list for the discovery cohort was filtered down to a stringent, but not FDR corrected, 269 significant (p < 0.005) differentially methylated CpG sites (Supplementary Data). Interestingly, of the sites showing an increase in methylation, 21 represent sites in the regulatory features of the clustered protocadherins genes located on 5q31 (D & & ). This preferential methylation of the protocadherin cluster is repeated in the mice as well as the replication samples with some variations. Furthermore, alterations to imprinted ncRNA from the SNRPN-UBE3A locus (Snord 116 [HBII-85] [p < 0.005] and Snord 115 [HBII-52] [p < 0.01]) were present in the human buccal DNA, which was the most ubiquitous marker for prenatal alcohol exposure in our mouse models.
Finding signal(s) in the noise
We used differentially methylated genes (with a less stringent statistical cut-off, p < 0.05) from the discovery cohort that represented established maternal drinking and uniform manifestation of FASD (developmental delays, hyperactivity/poor impulse control, and mental deficits) in an analysis towards identification of specific biological processes, functions, and pathways affected as a result of changes in methylation. While the less stringent cut-off for this gene list doesn’t account for multiple testing, the downstream ontology and pathways analysis do as they function on the principles of independent component analysis. The results () show that the major biological processes affected include nervous (p = 0.00000001), anatomical (p = 0.00000008) and skeletal (p = 0.006) development, cell adhesion (p = 0.00002), and cell projection assembly (p = 0.002). These processes appear to be affected by significant alterations (p < 0.05) to major molecular functions including neuropeptide binding and receptors, protein interactions, and channel activity. These genes and their associated molecular functions are also involved in major canonical pathways including hippo signaling, glutamatergic synapses, serotonergic synapses, and cholinergic synapses as well as axon guidance, focal adhesion, and apoptosis. Interestingly, the top three enriched canonical pathways are hippo signaling (p = 0.0002), glutamatergic synapse (p = 0.001), and calcium signaling (p = 0.005).
The FASD methylation footprint includes protocadherin genes
shows the Genemania network generated using the stringent gene list (269 CpG sites, p < 0.005) that showed significant altered methylation in patients with FASD. The results reveal involvement of members of the α, β, and γ family of protocadherin genes (D). & include probes showing significant differential methylation from children (p < 0.005) and mice (p < 0.01) in clustered protocadherins. & also include corresponding protocadherin genes that were differentially methylated in adult brains following neurodevelopmental alcohol exposure in mice. A includes a plot of the raw Illumina β values from significant (p < 0.005) CpGs in the differentially methylated region of the protocadherin γ cluster and how they align with alternate transcripts choice. Protocadherins are involved in the generation of synaptic complexity in the developing brain and evolved primarily by tandem duplications and divergence. This complex locus contains three clusters (α, β, and γ) of protocadherin gene families that are transcriptionally complex and very similar between humans and mice (B). The clustered protocadherin locus generates comparable transcripts in the two species and has relatively conserved CpG sites. More importantly, a number of the CpG sites/islands are specifically sensitive to increased methylation following alcohol exposure in both species.
The top canonical pathways are related to neurodevelopment & synaptic signaling
The top canonical pathways identified in this analysis involve hippo signaling, glutamatergic synapses, and calcium signaling, all of which are fundamental pathways related to neurodevelopment. Hippo signaling (A) had 25 out of 120 genes affected, was the most significant (p = 0.0002) canonical pathway, and is a highly conserved signaling pathway studied in cancer that is involved in the control of cell growth and proliferation, apoptosis, and organ size control. It is also interconnected to the TGFβ and Wnt signaling pathways. Interestingly, hippo signaling has recently been linked to chromatin modification by recruiting a histone methyltransferase complex [Citation36] and regulates the growth of niche stem cell precursors [Citation37]. The glutamatergic synapse pathway (B) was the top synaptic pathway and showed a significant (p = 0.001) enrichment with 18 out of 77 genes belonging to it showing significant (less stringent list, p < 0.05) alterations to CpG methylation. A number of glutamate receptors are G-protein coupled receptors and are involved in secondary messenger signaling. Also of importance is the significant (p = 0.005) alteration to the calcium signaling pathway, with 23 out of 102 genes affected. Calcium signaling has numerous fundamental and diverse roles in (neural) signaling. The relevance of thesepathways toneuralevents is an unexpected result of easilyobtainedbuccal swabs.
The results of discovery cohort are shared by the replication cohort
The replication cohort (R) included six patients (3–10 years old) and six matched controls (B) and their cheek swab DNA was used in genome-wide DNA methylation profiles (C). They were recruited from the same clinic and diagnosed by the same Pediatrician (J Kapalanga). However, the replication cohort was also much more heterogeneous compared with the discovery cohort with respect to sex, medication exposure, environment, and socioeconomic status. In order to compare the cohorts across batches a different preprocessing algorithm was selected: the subset-quantile within array normalization (SWAN) algorithm [Citation31] from the minifi [Citation38] bioconductor package. When the two cohorts were preprocessed separately by the Illumina algorithm or together with the SWAN algorithm, they produced genome-wide results that had similarities but were not identical to the discovery cohort. There was a trend of decreased significance in the replication cohort and greater levels of variation. Despite these differences, however, the main similarities between the two cohorts were significant alterations in ontologies related to glutamate and the canonical glutamatergic synapse pathway. Additionally, there was a consistent and prominent protocadherin signal on the Manhattan plot, as well as replication of alterations to imprinted regions also seen in the mouse models and the discovery cohort.
Next we reanalyzed the replication cohort results following removal of females and children on medication (B; R1 and R4). Also, four controls of the replication cohort were removed to better match age range and medical history. The selected scans were then preprocessed using the Illumina algorithm. Upon removal, the groups cluster much more closely and the signal is almost identical to what was observed in the discovery cohort. Glutamatergic synapses, cell-adhesion, neuroactive ligand receptor interactions, MAPK signaling, axon guidance, and a number of other pathways were all significant (p < 0.005) even though the ranking of specific pathways varied slightly. The results argue that although DNA methylation is altered in patients with FASD, other factors, including sex, age, and medication history, may affect the final outcome.
Confirmations via pyrosequencing
Next we sought to confirm the microarray results using an independent technology that does not depend on the matched controls for modeling. For this we chose probes with different performances from the carefully matched discovery cohort. First we chose the only CpG to pass multiple testing at the FDR filtering level in this relatively small cohort. The analysis identified an enhancer of COLEC11 (FDR p = 1.93 × 10-7). Pyrosequencing analysis of the affected CpG (cg15730644) revealed that the COLEC11 CpG shows a significant increase (p = 0.002) with an average methylation of 94.2% in FASD compared with 79.8% in matched controls (). It also shows that all children affected with FASD consistently have higher (~94% of cells) methylation at this CpG site compared with their matched controls (~80% of cells). The sequencing run also examined SNP rs182514706 (T>G) and found that it was not present in any members of either the exposed or control groups and thus would not interfere with the confidence in the performance of this probe. COLEC11 is involved in fundamental developmental processes and serves as a guidance cue for neural crest cell migration where aberrations are known to produce spectrum disorders with similar endophenotypes to FASD known as 3MC syndrome [Citation39].
The next two probes examined did not pass a FDR cut-off and are representative of the gene list used for higher level analysis, including heatmaps, ontologies, and pathways. HTT (Huntington) is a major hub in neurological disorders and showed a 1.5-fold increase (p = 0.003) in methylation in FASD patients. Pyrosequencing analysis of the affected CpG (cg26128129) revealed that the CpG in a enhancer of HTT shows a significant increase (p = 0.001) with an average methylation of 87.6% in FASD and an average of 50.7% in matched controls. Interestingly, there is variation among the control samples as compared with the FASD patients (). This CpG is known to contain a SNP [rs362313 (C>T)] that causes a loss of the CpG, as well as another SNP near the probe [rs147422679 A>G]; however, pyrosequencing confirmed neither SNP was present in any of the individuals examined. The final probe assessed by pyrosequencing was PCDHB18, which is a member of the protocadherin β cluster. The affected CpG (cg27086874) is in a north CpG shore that is located in a reprogramming-specific differentially methylated region (DMR). The results show an identical pattern to the arrays and the mouse models (results not shown). The available results support the suggestion that CpG specific DNA methylation is altered in the cheek swab DNA of children affected with FASD.
Discussion
Ethanol is a classic teratogen capable of inducing a wide range of developmental abnormalities. It interferes with a number of developmental processes at many levels. Of interest to this report is that PAE interferes with one-carbon metabolism [Citation40], which is essential for DNA methylation, histone modifications, and DNA synthesis. As such, it is implicated in complex gene-environment interactions that alter gene expression patterns, especially during neurodevelopment [Citation18,Citation41–44]. Much of this understanding has come from animal models that have successfully generated phenotypes relevant to FASD [Citation6,Citation7]. The mouse results support involvement of a number of developmental pathways in the etiology of FASD [Citation7]. Also, the underlying mechanisms may involve epigenetic remodeling that persists into adulthood [Citation8] even though the molecular targets that could be considered as causal and ‘driving’ are not known. Furthermore, although the animal studies are informative, the literature lacks replication of such results in humans, which forms the primary objective of this report. The analysis of human children has also implicated a fundamental and well-known player in alcohol research: the glutamatergic synapse (B). Additionally, the identification of hippo-signaling (A) offers a very promising candidate for the mechanism given its role in regulating stem cells and controlling organ size. However, not much is currently known about this pathway in a context outside of cancers, which are diseases of cell differentiation, and developmental fly biology. Thus, the focus for remainder of this report is a single genomic locus, which is consistently affected in children born with FASD and in PAE mice that may also ‘drive’ psychiatric phenotypes, and has not yet been implicated in the literature.
Increased methylation in protocadherin genes can result in FASD phenotypes
The protocadherins are the largest subgroup of the cadherin gene superfamily of homophilic cell-adhesion proteins [Citation45,Citation46]. In humans, they are clustered on 5q31 and show a concerted increase in methylation in FASD (D & A). They are primarily expressed in the developing nervous system [Citation47] where they are stochastically involved in a neuronal self-avoidance that allows neurons to interact with the same neuronal subtype but not themselves. Indeed, protocadherins have been observed mediating cell-to-cell interactions in dendrites, axons, synapses, growth cones, and neuronal soma [Citation48–54]. In vertebrates, the majority of protocadherin genes are organized in three clusters: α, β, and γ (B). Intriguingly, the cell specific variation in transcripts created by this locus parallels the complexity of the immune system. Unlike the immune system, which relies on genetic recombination, the protocadherin complexity is realized by epigenetic mechanisms that parallel the act of recombination via alternative transcript choice. The transcriptional initiation of the α and γ families is dependent on the methylation status of numerous promoters in the variable regions of the family. β, on the other hand, does not contain a shared exon and each gene appears to be under the control of its own promoter. The α and γ gene families form a DNA loop with CTCF, which has methylation sensitive binding and is proposed to bring the isoform specific promoters into the proximity of flanking enhancers and transcriptional machinery. Thus, it appears that CTCF serves as a master transcription factor for this locus and is used to determine isoform expression [Citation55]. Furthermore, the regulation of this stochastic expression is dependent on epigenetic priming by de novo methylation patterns established at promoters by Dnmt3b during early embryonic stages, which goes on to causatively influence isoform expression in subsequent differentiated cells [Citation56].
The isoform diversity created by these clusters creates a molecular signature on the cell surface of individual neurons that is used to interact in trans [Citation54]. This diversity is accomplished by individual neurons expressing different combinations of the protocadherin isoforms on their cell surface and forms even more diversity by creating multimers (in cis). Ultimately, each neuron could have its own unique identity amongst other individuals of the same subtype [Citation57,Citation58]. This is believed to be the biological basis for the specific and yet enormous network formation of precise neuronal connections required for neurodevelopment. Given this essential role underlying neurodevelopment, it comes as no surprise that the protocadherin gene family clusters have been implicated in a number of neurodevelopmental disorders [Citation59]. Also, an increase in methylation in some, most, or all gene promoters ( & ), as observed in human FASD patients and all mouse models, is expected to reduce the isoform diversity, thus restraining brain function.
We also assessed the significance of affected sequences in a transcription factor analysis [Citation30]. It revealed a significant (p < 0.005) enrichment for CTCF insulator binding sites [Citation60], which are involved in establishing chromatin domains and boundaries and result in a large-scale nuclear architecture that is specific to cell type. Another transcription factor enriched for was RAD21, which cooperates with pluripotency transcription factors, including CTCF in the maintenance of embryonic stem cell identity [Citation61]. CTCF is known to be the master regulator of the clustered protocadherins [Citation62] where it influences complex DNA looping (A), and many genes under its transcriptional regulation appear deregulated in our mouse models including genomically imprinted genes [Citation8].
The increased methylation of protocadherin genes in the cheek swab DNA of FASD patients are backed by comparable results in the brains of adult mice prenatally exposed to alcohol (B). Among the most common is Pcdhb18, which was shown to have a significant (FDR corrected p < 0.05) decrease (40%) in adult brain gene expression in one of our mouse models of FASD [Citation7]. Such results have the potential to cause down-regulation of specific gene products depending on the specificity of methylated promoters in neural cells of mice as well as humans. Furthermore, such changes are expected to affect function because Pcdhb18 has been demonstrated to control axon growth and arborization in zebrafish [Citation63]. Finally, it has not escaped our attention that this rather synchronous alteration in DNA methylation of a specific gene family with a critical role in brain functioning in children with FASD may open the door to early diagnosis of this common and complex disorder as well as offer novel strategies for its amelioration.
Conclusion & future perspective
While the analysis was theoretically limited by small sample size, it also had the practical and unquantifiable added statistical power of extremely stringent sample matching and concordance with our multiple mouse models, which showed almost identical alterations. Furthermore, the changes could be representative of alterations to individual cellular populations, particularly neurons or glia populations, but nonetheless these observations would still be meaningful as both buccal cells and neural precursor cells are related at the ectoderm primary germ layer. The connection via neuroepithelial cells also suggests a role for the developmental events of epithelial–mesenchymal transition (EMT) and the inverse mesenchymal–epithelial transition (MET), which are used in neural crest migration and neural tube formation. Thus, despite the potential caveats, we suggest that the results included may have a number of implications. First, the methylomic profile ( & ) may serve as a diagnostic feature of FASD, including early diagnosis from an easily accessible peripheral tissue. Second, the identification of target loci and molecules, including protocadherins, may serve as a foundation for the development of novel strategies of treatment and application of any corrective therapy, which are most effective if applied as early as possible during neurodevelopment [Citation64].
Table 1. Clinical features of patients from the discovery and replication cohorts with the diagnosis of fetal alcohol spectrum disorders.
Table 2. Nucleotide-specific analysis of CpGs in the protocadherin gene family clusters showing significant (p < 0.005) alterations to CpG methylation in human buccal epithelial DNA from children of the discovery sample (with fetal alcohol spectrum disorders compared to matched controls).
Table 3. Tiling-based analysis of protocadherin gene clusters showing significant (p < 0.01) increases in methylation in adult brain tissue from a mouse model of fetal alcohol spectrum disorders.
Table 4. Gene ontology and KEGG-based canonical pathway analysis of significant differences in CpG methylation from the discovery cohort.
Table 5. Pyrosequencing results for COLEC11 and HTT.
First analysis of alterations to genome-wide DNA methylation in human children with fetal alcohol spectrum disorders (FASD).
Results of buccal swabs from human children with FASD have similar DNA methylation patterns at candidate loci when compared with prenatal alcohol exposure (PAE) adult mouse brains.
Results are replicated but not identical in a much more heterogeneous human FASD cohort.
Common candidates include the clustered protocadherin locus, glutamatergic synapse pathways, and the hippo signaling pathway.
The regions identified are not limited to ubiquitous environmentally responsive imprinted regions or one-carbon metabolic pathways.
The candidates will be of use for future development of molecular therapies, such as somatic epigenome editing, which involves modified genome editing systems inducing precision epigenetic alterations without altering sequence.
The experiments were conceived and designed by BI Laufer, J Kapalanga, and SM Singh. Children were previously diagnosed by J Kapalanga and under his care swabs were collected and matched to controls. Human DNA isolation and purification was done by BI Laufer, CA Castellani, and EJ Diehl. All bioinformatic analysis was done by BI Laufer. L Yan designed and oversaw the pyrosequencing assays. BI Laufer and SM Singh wrote the manuscript with intellectual contributions from J Kapalanga and edits from CA Castellani, EJ Diehl, J Kapalanga, and L Yan. The entire project was under the supervision of SM Singh.
Disclaimer
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical conduct of research
Male and female C57BL/6J (B6) mice were originally obtained from Jackson Laboratories (Bar Harbor, ME) and maintained at the Health Sciences Animal Care Facility at the University of Western Ontario (London, ON, Canada). The Animal Use Subcommittee of the University of Western Ontario approved all procedures undertaken on the animals and can viewed in detail in Laufer et al. 2013. All human samples were collected under the care of J Kapalanga at Grey Bruce Health Services and ethical approval from Grey Bruce Health Services, the University of Western Ontario, and the parents/guardians was obtained for buccal swab collection and anonymous diagnostic information. The authors state that they have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Supplemental Information
Download MS Excel (50.9 KB)Acknowledgements
The authors thank I Craig for his expertise in editing some of the figures for clarity, the TCAG for expertise in array hybridization and scanning, and A Meyer from EpigenDx for technical expertise with pyrosequencing.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/full/10.2217/epi.15.60
Financial & competing interests disclosure
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) scholarship to BI Laufer, a scholarship from NSERC to EJ Diehl, and grants from NSERC and the Canadian Institute of Health Research (CIHR) to SM Singh. L Yan is the Founder and President of EpigenDx and offered their services at a substantial collaborative discount. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Guerri C , BazinetA , RileyEP . Foetal alcohol spectrum disorders and alterations in brain and behaviour . Alcohol Alcohol.44 ( 2 ), 108 – 114 ( 2009 ).
- Lee S , ChoiI , KangS , RivierC . Role of various neurotransmitters in mediating the long-term endocrine consequences of prenatal alcohol exposure . Ann. NY Acad. Sci.1144 , 176 – 188 ( 2008 ).
- Bosco C , DiazE . Placental hypoxia and foetal development versus alcohol exposure in pregnancy . Alcohol Alcohol.47 ( 2 ), 109 – 117 ( 2012 ).
- Kleiber ML , WrightE , SinghSM . Maternal voluntary drinking in C57BL/6J mice: advancing a model for fetal alcohol spectrum disorders . Behav. Brain Res.223 ( 2 ), 376 – 387 ( 2011 ).
- Kleiber ML , LauferBI , WrightE , DiehlEJ , SinghSM . Long-term alterations to the brain transcriptome in a maternal voluntary consumption model of fetal alcohol spectrum disorders . Brain Res.1458 , 18 – 33 ( 2012 ).
- Mantha K , KleiberM , SinghS . Neurodevelopmental timing of ethanol exposure may contribute to observed heterogeneity of behavioral deficits in a mouse model of Fetal Alcohol Spectrum Disorder (FASD) . J. Behav. Brain Sci.3 , 85 – 99 ( 2013 ).
- Kleiber ML , ManthaK , StringerRL , SinghSM . Neurodevelopmental alcohol exposure elicits long-term changes to gene expression that alter distinct molecular pathways dependent on timing of exposure . J. Neurodev. Disord.5 ( 1 ), 6 ( 2013 ).
- Laufer BI , ManthaK , KleiberML , DiehlEJ , AddisonSM , SinghSM . Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice . Dis. Model. Mech.6 ( 4 ), 977 – 992 ( 2013 ).
- Olney JW , TenkovaT , DikranianK , QinYQ , LabruyereJ , IkonomidouC . Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain . Brain Res. Dev. Brain Res.133 ( 2 ), 115 – 126 ( 2002 ).
- Choi IY , AllanAM , CunninghamLA . Moderate fetal alcohol exposure impairs the neurogenic response to an enriched environment in adult mice . Alcohol. Clin. Exp. Res.29 ( 11 ), 2053 – 2062 ( 2005 ).
- Young C , OlneyJW . Neuroapoptosis in the infant mouse brain triggered by a transient small increase in blood alcohol concentration . Neurobiol. Dis.22 ( 3 ), 548 – 554 ( 2006 ).
- Green ML , SinghAV , ZhangY , NemethKA , SulikKK , KnudsenTB . Reprogramming of genetic networks during initiation of the Fetal Alcohol Syndrome . Dev. Dyn.236 ( 2 ), 613 – 631 ( 2007 ).
- Kajimoto K , AllanA , CunninghamLA . Fate analysis of adult hippocampal progenitors in a murine model of Fetal Alcohol Spectrum Disorder (FASD) . PLoS ONE8 ( 9 ), e73788 ( 2013 ).
- Subbanna S , NagreNN , ShivakumarM , UmapathyNS , PsychoyosD , BasavarajappaBS . Ethanol induced acetylation of histone at G9a Exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice . Neuroscience258 , 422 – 432 ( 2013 ).
- El Shawa H , AbbottCW , 3rd , HuffmanKJ . Prenatal ethanol exposure disrupts intraneocortical circuitry, cortical gene expression, and behavior in a mouse model of FASD . J. Neurosci.33 ( 48 ), 18893 – 18905 ( 2013 ).
- Kleiber ML , DiehlEJ , LauferBIet al. Long-term genomic and epigenomic dysregulation as a consequence of prenatal alcohol exposure: a model for fetal alcohol spectrum disorders . Front. Genet.5 , 161 ( 2014 ).
- Zhou FC . Dissecting FASD through the global transcriptome . Alcohol. Clin. Exp. Res.39 ( 3 ), 408 – 412 ( 2015 ).
- Haycock PC . Fetal alcohol spectrum disorders: the epigenetic perspective . Biol. Reprod.81 ( 4 ), 607 – 617 ( 2009 ).
- Hutson JR , StadeB , LehotayDC , CollierCP , KapurBM . Folic acid transport to the human fetus is decreased in pregnancies with chronic alcohol exposure . PLoS ONE7 ( 5 ), e38057 ( 2012 ).
- Ungerer M , KnezovichJ , RamsayM . In utero alcohol exposure, epigenetic changes, and their consequences . Alcohol. Res.35 ( 1 ), 37 – 46 ( 2013 ).
- Perkins A , LehmannC , LawrenceRC , KellySJ . Alcohol exposure during development: impact on the epigenome . Int. J. Dev. Neurosci.31 ( 6 ), 391 – 397 ( 2013 ).
- Veazey KJ , CarnahanMN , MullerD , MirandaRC , GoldingMC . Alcohol-induced epigenetic alterations to developmentally crucial genes regulating neural stemness and differentiation . Alcohol. Clin. Exp. Res.37 ( 7 ), 1111 – 1122 ( 2013 ).
- Stringer R , LauferB , KleiberM , SinghS . Reduced expression of brain cannabinoid receptor 1 (Cnr1) is coupled with an increased complementary micro-RNA (miR-26b) in a mouse model of fetal alcohol spectrum disorders . Clin. Epigenetics5 ( 1 ), 14 ( 2013 ).
- Mantha K , LauferBI , SinghSM . Molecular changes during Neurodevelopment following second-trimester binge ethanol exposure in a mouse model of Fetal Alcohol Spectrum Disorder: from immediate effects to long-term adaptation . Dev. Neurosci.36 ( 1 ), 29 – 43 ( 2014 ).
- Kleiber ML , LauferBI , StringerRL , SinghSM . Third trimester-equivalent ethanol exposure is characterized by an acute cellular stress response and an ontogenetic disruption of genes critical for synaptic establishment and function in mice . Dev. Neurosci.36 ( 6 ), 499 – 519 ( 2014 ).
- Laufer BI , DiehlEJ , SinghSM . Neurodevelopmental epigenetic etiologies: insights from studies on mouse models of fetal alcohol spectrum disorders . Epigenomics5 ( 5 ), 465 – 468 ( 2013 ).
- Hard ML , AbdolellM , RobinsonBH , KorenG . Gene-expression analysis after alcohol exposure in the developing mouse . J. Lab. Clin. Med.145 ( 1 ), 47 – 54 ( 2005 ).
- Sur M , LeameyCA . Development and plasticity of cortical areas and networks . Nat. Rev. Neurosci.2 ( 4 ), 251 – 262 ( 2001 ).
- Hirano S , TakeichiM . Cadherins in brain morphogenesis and wiring . Physiol. Rev.92 ( 2 ), 597 – 634 ( 2012 ).
- Chen EY , TanCM , KouYet al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool . BMC Bioinformatics14 , 128–2105–14–128 ( 2013 ).
- Maksimovic J , GordonL , OshlackA . SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips . Genome Biol.13 ( 6 ), R44–2012–13–6-r44 ( 2012 ).
- Pidsley R , Y WongCC , VoltaM , LunnonK , MillJ , SchalkwykLC . A data-driven approach to preprocessing Illumina 450K methylation array data . BMC Genomics14 , 293–2164–14–293 ( 2013 ).
- EpigenDx . www.EpigenDx.com .
- Lim A , CandiloroI , WongNet al. Quantitative methodology is critical for assessing DNA methylation and impacts on correlation with patient outcome . Clin. Epigenetics6 ( 1 ), 22 ( 2014 ).
- The UCSC Genome Browser database ( 2015 ). www.genome.ucsc.edu .
- Oh H , SlatteryM , MaL , WhiteKP , MannRS , IrvineKD . Yorkie promotes transcription by recruiting a histone methyltransferase complex . Cell. Rep.8 ( 2 ), 449 – 459 ( 2014 ).
- Sarikaya DP , ExtavourCG . The hippo pathway regulates homeostatic growth of stem cell niche precursors in the drosophila ovary . PLoS Genet.11 ( 2 ), e1004962 ( 2015 ).
- Aryee MJ , JaffeAE , Corrada-BravoHet al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays . Bioinformatics30 ( 10 ), 1363 – 1369 ( 2014 ).
- Rooryck C , Diaz-FontA , OsbornDPet al. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome . Nat. Genet.43 ( 3 ), 197 – 203 ( 2011 ).
- Halsted CH , VillanuevaJA , DevlinAM , ChandlerCJ . Metabolic interactions of alcohol and folate . J. Nutr.132 ( 8 Suppl. ), S2367 – S2372 ( 2002 ).
- Sathyan P , GoldenHB , MirandaRC . Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium . J. Neurosci.27 ( 32 ), 8546 – 8557 ( 2007 ).
- Haycock PC , RamsayM . Exposure of mouse embryos to ethanol during preimplantation development: effect on DNA methylation in the H19 imprinting control region . Biol. Reprod.81 ( 4 ), 618 – 627 ( 2009 ).
- Liu Y , BalaramanY , WangG , NephewKP , ZhouFC . Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation . Epigenetics4 ( 7 ), 500 – 511 ( 2009 ).
- Kaminen-Ahola N , AholaA , MagaMet al. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model . PLoS Genet.6 ( 1 ), e1000811 ( 2010 ).
- Hirano S , TakeichiM . Cadherins in brain morphogenesis and wiring . Physiol. Rev.92 ( 2 ), 597 – 634 ( 2012 ).
- Chen WV , ManiatisT . Clustered protocadherins . Development140 ( 16 ), 3297 – 3302 ( 2013 ).
- Sano K , TaniharaH , HeimarkRLet al. Protocadherins: a large family of cadherin-related molecules in central nervous system . EMBO J.12 ( 6 ), 2249 – 2256 ( 1993 ).
- Kohmura N , SenzakiK , HamadaSet al. Diversity revealed by a novel family of cadherins expressed in neurons at a synaptic complex . Neuron20 ( 6 ), 1137 – 1151 ( 1998 ).
- Wang X , WeinerJA , LeviS , CraigAM , BradleyA , SanesJR . Gamma protocadherins are required for survival of spinal interneurons . Neuron36 ( 5 ), 843 – 854 ( 2002 ).
- Kallenbach S , KhantaneS , CarrollPet al. Changes in subcellular distribution of protocadherin gamma proteins accompany maturation of spinal neurons . J. Neurosci. Res.72 ( 5 ), 549 – 556 ( 2003 ).
- Phillips GR , TanakaH , FrankMet al. Gamma-protocadherins are targeted to subsets of synapses and intracellular organelles in neurons . J. Neurosci.23 ( 12 ), 5096 – 5104 ( 2003 ).
- Junghans D , HeidenreichM , HackI , TaylorV , FrotscherM , KemlerR . Postsynaptic and differential localization to neuronal subtypes of protocadherin beta16 in the mammalian central nervous system . Eur. J. Neurosci.27 ( 3 ), 559 – 571 ( 2008 ).
- Lefebvre JL , KostadinovD , ChenWV , ManiatisT , SanesJR . Protocadherins mediate dendritic self-avoidance in the mammalian nervous system . Nature488 ( 7412 ), 517 – 521 ( 2012 ).
- Chen WV , AlvarezFJ , LefebvreJLet al. Functional significance of isoform diversification in the protocadherin gamma gene cluster . Neuron75 ( 3 ), 402 – 409 ( 2012 ).
- Golan-Mashiach M , GrunspanM , EmmanuelR , Gibbs-BarL , DiksteinR , ShapiroE . Identification of CTCF as a master regulator of the clustered protocadherin genes . Nucleic Acids Res.40 ( 8 ), 3378 – 3391 ( 2012 ).
- Toyoda S , KawaguchiM , KobayashiTet al. Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity . Neuron82 ( 1 ), 94 – 108 ( 2014 ).
- Bonn S , SeeburgPH , SchwarzMK . Combinatorial expression of alpha- and gamma-protocadherins alters their presenilin-dependent processing . Mol. Cell. Biol.27 ( 11 ), 4121 – 4132 ( 2007 ).
- Schreiner D , WeinerJA . Combinatorial homophilic interaction between gamma-protocadherin multimers greatly expands the molecular diversity of cell adhesion . Proc. Natl Acad. Sci. USA107 ( 33 ), 14893 – 14898 ( 2010 ).
- Kalmady SV , VenkatasubramanianG . Evidence for positive selection on Protocadherin Y gene in Homo sapiens: implications for schizophrenia . Schizophr. Res.108 ( 1–3 ), 299 – 300 ( 2009 ).
- Kim TH , AbdullaevZK , SmithADet al. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome . Cell128 ( 6 ), 1231 – 1245 ( 2007 ).
- Nitzsche A , Paszkowski-RogaczM , MatareseFet al. RAD21 cooperates with pluripotency transcription factors in the maintenance of embryonic stem cell identity . PLoS ONE6 ( 5 ), e19470 ( 2011 ).
- Guo Y , MonahanK , WuHet al. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice . Proc. Natl Acad. Sci. USA109 ( 51 ), 21081 – 21086 ( 2012 ).
- Biswas S , EmondMR , DuyPQ , Hao leT , BeattieCE , JontesJD . Protocadherin-18b interacts with Nap1 to control motor axon growth and arborization in zebrafish . Mol. Biol. Cell25 ( 5 ), 633 – 642 ( 2014 ).
- Paley B , O’ConnorMJ . Intervention for individuals with fetal alcohol spectrum disorders: treatment approaches and case management . Dev. Disabil. Res. Rev.15 ( 3 ), 258 – 267 ( 2009 ).