
Abstract
Genomic imprinting is an epigenetic feature characterized by parent-specific monoallelic gene expression. The aim of this study was to compare the DNA methylation status of imprinted genes and imprinting control regions (ICRs), harboring differentially methylated regions (DMRs) in a comprehensive panel of 18 somatic tissues. The germline DMRs analyzed were divided into ubiquitously imprinted and placenta-specific DMRs, which show identical and different methylation imprints in adult somatic and placental tissues, respectively. We showed that imprinted genes and ICR DMRs maintain methylation patterns characterized by intermediate methylation levels in somatic tissues, which are pronounced in a specific region of the promoter area, located 200–1500 bp from the transcription start site. This intermediate methylation is concordant with gene expression from a single unmethylated allele and silencing of a reciprocal parental allele through DNA methylation. The only exceptions were seen for ICR DMRs of placenta-specific imprinted genes, which showed low levels of methylation, suggesting that these genes escape parent-specific epigenetic regulation in somatic tissues.
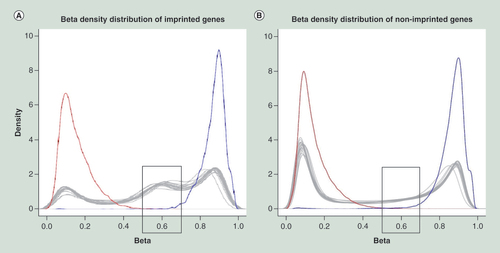
Positive and negative controls are shown by blue and red lines, respectively. Each tissue is shown by a gray line. Imprinted genes have an increased number of intermediately methylated probes with beta-value in 0.5–0.7 range, if compared with nonimprinted genes, as shown in gray boxes.
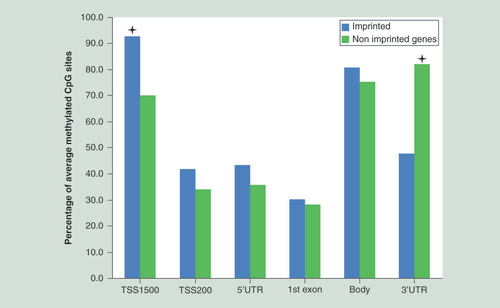
Promoter region TSS1500 has the highest ratio of intermediately methylated probes in imprinted genes, while the smallest difference was detected in TSS200, 5′UTR, the first exon and gene body regions. The asterisks define the differences of 22.8 and 34.2% for TSS1500 and 3′UTR, respectively, being the only significant results for this analysis. The TSS1500 promoter area of imprinted genes contains 22.8% more 0.5–0.7 β-value probes than nonimprinted genes (p = 6.57 × 10-5). The 3′UTR area of imprinted genes has 34.2% less 0.5–0.7 β-value probes than nonimprinted genes (p = 6.2 × 10-13). The differences in the TSS200, 5′UTR, the first exon and gene body were only 7.9, 7.3, 2.2 and 5.7% (p = 0.2128, p = 0.2578, p = 0.7929, and p = 0.338), respectively; being insignificant after multiple correction.
TSS: Transcription start site.
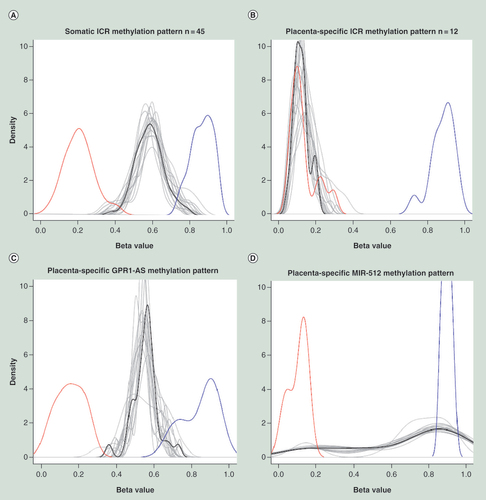
Positive and negative controls are shown by blue and red lines, respectively. Each tissue is shown by a gray line, whereas the mean of all tissues is shown by black line. The pattern specific for ubiquitously imprinted ICR DMRs is associated with increased number of intermediately methylated probes. While the placenta-specific ICR DMRs are associated with dominance of lowly methylated probes, except for GPR1-AS and MIR-512 genes for which unique patterns were observed (as explained in the text).
Genomic imprinting is an epigenetic gene-marking phenomenon that is mostly established in germline and results in monoallelic gene expression according to parental origin [Citation1,Citation2]. The process of genomic imprinting is widely associated with fetal growth and maturation as parent-specific gene activity has predominantly been found in placental tissue. According to the most widely accepted ‘parental conflict hypothesis’, the paternally expressed genes force maternal resources to support the ongoing pregnancy, while the maternally expressed genes tend to distribute the resources equally among all her offspring. Thus, imprinting creates the balance between opposing parental interests and ensures the best reproductive outcome for both genders.
In addition to prenatal expression, postnatal gene imprinting may also occur, mainly in the brain [Citation3], likely influencing personal and parenting behavioral, cognitive and emotional characteristics. Furthermore, some studies have also emphasized the importance of parental inheritance of a risk allele as a possible epigenetic cause of common diseases such as obesity [Citation4] and Type II diabetes [Citation5]. This explains why the focus of research for imprinted genes has shifted from placenta to postnatal somatic tissues, aiming to provide more detailed biological data and evolutionary explanations to parent-specific gene dosage in pre- and postnatal origin of disease.
Previous studies have shown that many imprinted genes are present in clusters [Citation6,Citation7], as in mouse, for instance, the proportion of clustered genes exceeds 80% of total number of imprinted genes [Citation8]. The clustered organization allows cis-regulation of monoallelic expression via imprinted control regions (ICRs) [Citation6,Citation9–10], which possess parental-specific DNA methylation and/or histone modifications [Citation11,Citation12]. These regions have been validated in a series of mouse knock-out experiments and have been shown to occur in human epigenetic diseases, revealing the unique contribution of the respective genes [Citation9,Citation10]. Every ICR harbors at least one differentially methylated region (DMR); the CpG island, which exhibits parent-specific DNA methylation. A sole ICR DMR can regulate the expression of the nearest single imprinted gene or the entire gene cluster.
Imprinted DMRs are classified into two groups – primary DMRs, also called germline DMRs and secondary, also referred to as somatic DMRs. The germline DMRs acquire their methylation status during gametogenesis, are stably maintained through epigenetic reprogramming at oocyte-to-embryo transition, and survive during somatic aging [Citation13–15]. Somatic DMRs, in contrast, acquire their methylation status postfertilization and are regulated by nearby germline ICR DMRs in a hierarchical manner [Citation16–18]. The germline DMRs are further subdivided into ubiquitously imprinted and placenta-specific imprinted genes, which show identical and different methylation imprints in adult somatic and placental tissues, respectively [Citation19].
As imprinted genes have methylation marks only on one parental allele, they show intermediate methylation levels at ICR DMRs [Citation16–17,Citation19–20]. Whether this intermediate methylation pattern is maintained in somatic tissues throughout the life course, including growth, development and aging, remains to be answered. A recent study investigated this question and demonstrated stable DNA methylation imprints for all germline DMRs, which maintained an intermediate methylation level between 35 and 65% in all adult somatic tissues, independent of gene expression [Citation21]. However, the conclusions of this study are somewhat tentative because results were obtained for only a subset of imprinted genes for a restricted number of CpGs. This limitation prompted us to investigate epigenome-wide ICR DMRs on a wider panel of adult somatic tissues.
Recent developments in array and sequencing-based methylation profiling methods have significantly improved our understanding of the global epigenetic regulation of the entire genome and individual genes, and have offered novel tools for studies of imprinted genes and their DMRs. In these analyses, the methylation status of CpG sites are expressed as beta (β)-values, representing the methylation levels of the CpG locus using the ratio of methylated and unmethylated alleles [Citation22]. The assumption of more frequent CpG sites with an intermediate methylation level in imprinted genes led us to postulate that the proportion of ‘intermediate-β-value CpG sites’ in imprinted genes exceeds that of nonimprinted genes, across the entire span of genes, even in adult somatic tissues. We also tested a previously proposed hypothesis that known germline ICR DMRs are persistently inherited across many adult somatic tissues by retaining intermediate methylation levels. Furthermore, we expected minimal variance of methylation levels, as expressed by β-values, for imprinted genes as they have more probes with an intermediate methylation level, representing an equal ratio of unmethylated and methylated alleles of the expressed and silenced alleles, respectively. In contrast, for nonimprinted genes, we presumed greater variance in β-values, as a more random and mixed distribution of the unmethylated and methylated CpG loci is expected, depending on the tissues examined.
Therefore, the aim of the current study was to analyze the methylation status of the imprinted genes and their regulative ICR DMRs in somatic tissues. In order to avoid interindividual variability between tissue samples, a panel of 17 tissues from four autopsy patients was used for DNA separation, bisulfite treatment and subsequent epigenome analysis using the Illumina Infinium HumanMethylation450 BeadChip, covering 486,428 probes. In addition, the tissue panel was complemented with whole-blood samples from 97 healthy adults from Estonian Genome Centre of University of Tartu (EGCUT).
In our study, we focused on the 76 previously established imprinted genes and 59 germline ICR DMRs, as extracted from a search of available databases and literature. The methylation pattern of imprinted genes in somatic tissues was compared with that of nonimprinted genes, in order to highlight whether the gene methylation signature of imprinted and nonimprinted genes could be distinguished in adult somatic tissues. Furthermore, the location of intermediately methylated CpG sites in imprinted and nonimprinted genes was compared will reveal possible critical regulatory regions for gene activity. Finally, we analyzed the methylation status of all known germline-imprinted DMRs in somatic tissues to assess whether the methylation status of these critical genome regions is stably maintained in diverse somatic tissues obtained from the same individual.
Results
Distribution of CpG methylation for imprinted & nonimprinted genes
We compared the distribution of CpG methylation in autopsy tissue panel in 76 imprinted genes from a database search (Supplementary Table 1) and 20,515 nonimprinted genes captured by the Illumina array. We estimated the proportions of different CpG methylation ranges, as categorized by β-values, and performed a one-tailed chi-squared test with one degree of freedom, to determine which region best characterizes ‘intermediate methylation status’ (Supplementary Table 2). Probes over the entire length of a gene were included in this analysis. According to the Illumina annotation, 30.9% of the probes are in promoter areas (13.0 and 17.9% are within 1–200 and 200–1500 nucleotides upstream from the transcription start site, TSS200 and TSS1500, respectively), 4.3 and 15.0% are in 3′UTRs and 5′UTRs, respectively, 7.8% in the first exons and 42.0% in gene bodies. Based on these data, the biggest difference in methylation levels between imprinted and nonimprinted genes was found for the ‘intermediately methylated’ CpGs with the corresponding β-value of 0.5–0.7 (p = 9.33 × 10-187).
Next, we visualized the methylation patterns of all CpGs of imprinted genes, also incorporating well-defined ICR DMRs [Citation19]. When we compared the density plot of methylation levels of all probes across all 17 autopsy tissues between imprinted (A) and nonimprinted (B) genes, we observed many more intermediately methylated probes with β-values of 0.5–0.7 for imprinted genes (25.5%) than for nonimprinted genes (10.0%). Also, the number of probes with smaller β-values (<0.5), indicating the subset of CpG sites with low methylation, was less for imprinted (28.2%) than for nonimprinted genes (48.1%). The number of probes with higher methylation levels (>0.7) showed the smallest difference for imprinted and nonimprinted genes (46.2 and 41.9%, respectively). According to the performed tests, all results were significantly different (p = 2.76 × 10-145) for the three categories of β-values (<0.5, 0.5–0.7 and >0.7) between imprinted and nonimprinted genes.
Similar results were also observed for DNA methylation of blood samples from healthy adults. The proportion of probes with low methylation levels (β-values of <0.5) was 34.9% for imprinted genes and 48.9% for nonimprinted genes (p = 3.93 × 10-25), while the proportion of probes with high methylation levels (β-values >0.7) was lower for nonimprinted genes compared with imprinted genes (39.1 and 44.0%, respectively; p = 0.0079). Intermediately methylated probes demonstrated the biggest difference for these two groups of genes (21.2% for imprinted and 12.0% for nonimprinted genes, p = 1.76 × 10-121).
Equality of variances
We used Levene’s test (levenes.test function in R, version 3.2.1) to compare the variance of β-values of imprinted and nonimprinted genes that were captured by the methylation array. We compared 76 previously validated imprinted genes with 76 randomly selected nonimprinted genes for each of the 17 postmortem tissues analyzed, iterating the procedure 100-times. We also performed the same procedure using whole-blood samples from healthy adults. Supplementary Table 3 presents the average variance of imprinted genes, average variance of 76 randomly selected nonimprinted genes and average p-value from Levene’s test across all comparisons. We found that imprinted genes have smaller variance in β-values than nonimprinted genes (e.g., p = 2.31 × 10-14 for brain tissue and p = 4.22 × 10-12 for nerve tissue). We also observed a similar distribution of methylated probes for blood samples (p = 8.71 × 10-12), corroborating evidence that imprinted genes show less variability in methylation across a broad range of tissue types.
The location of intermediately methylated probes
For all genes captured by the methylation array, we counted the proportion of intermediately methylated probes with β-values in the range 0.5–0.7 for different locations within a gene. The array provides access to six regions of a gene: promoter, 5′UTR, the first exon, gene body and 3′UTR, while the promoter area is further divided into two regions, 200 and 200–1500 nucleotides from TSS of a gene (TSS200 and TSS1500, respectively). We performed a one-tailed chi-squared test to check whether imprinted genes, excluding imprinted DMRs [Citation19], contain more intermediately methylated probes at specific locations when compared with nonimprinted genes (). We discovered that the TSS1500 promoter area of imprinted genes contains 22.8% more 0.5–0.7 β-value probes than nonimprinted genes (p = 6.57 × 10-5). Also the 3′UTR area of imprinted genes has 34.2% less 0.5–0.7 β-value probes than nonimprinted genes (p = 6.2 × 10-13). The differences in the TSS200, 5′UTR, the first exon and gene body were only 7.9, 7.3, 2.2 and 5.7% (p = 0.2128, p = 0.2578, p = 0.7929 and p = 0.338), respectively, and all insignificant after multiple correction. Thus, imprinted genes show a higher percentage of intermediately methylated probes with β-values in the range 0.5–0.7 in an extended promoter region, spanning 1500 bp from the transcription start site.
Analysis of germline ICR DMRs
We used the list of known germline ICR DMRs [Citation19] to test whether the methylation status of these critical genome regions is stably maintained during the formation of multiple types of somatic tissues. From 67 available imprinted DMRs we were unable to normalize two regions for the ZFAT and LIN28B genes, while six DMRs had no Infinium probes after performing quality control. A total of 59 remaining ICR DMRs were thus retained for further analysis, which were divided into two groups – ubiquitously imprinted (n = 45) and placenta-specific imprinted genes (n = 14), as recently suggested [Citation19] (). All ubiquitously imprinted DMRs demonstrated remarkable stability of β-values, varying from 49 to 77%, across all 18 somatic tissues tested. In contrast, placenta-specific DMRs presented low β -values, ranging from 10 to 20% in the somatic tissues tested, except for two genes (GPR1-AS and MIR512). These two DMRs were found to have intermediate methylation levels of 55 and 67%, respectively (Supplementary Table 4), as we observed for ubiquitously imprinted genes. However, visualization demonstrated that only GPR1-AS had the pattern specific to ubiquitously imprinted DMRs, while MIR512 had a unique pattern that was characterized by an increased number of highly methylated probes and a small number of intermediately and lowly methylated probes ().
Discussion
The aim of this study was to compare the DNA methylation of imprinted genes, their ICR DMRs and nonimprinted genes, in a comprehensive panel of somatic tissues. For this analysis, a unique panel of 17 tissues, each obtained from four autopsy patients was used. In addition, the tissue panel was complemented by 97 whole-blood samples obtained from healthy volunteers of the same North-European origin. DNA from all somatic tissue samples was analyzed using the Illumina Infinium HumanMethylation450 BeadChip, providing quantitative methylation data at the single CpG-site level for 486,428 probes. Our results demonstrated that the general pattern of methylation overlaps between all 18 tissue samples, providing assurance that blood samples are a valuable source of DNA for imprinting studies.
The present study is based on analysis of imprinted genes, which were selected based on available databases and verified by a ‘PubMed search’, providing clear evidence of parent-specific monoallelic expression (Supplementary Table 1). For the majority of the 76 imprinted genes, there is overwhelming evidence of monoallelic expression of either of the parental allele in placental and embryonal somatic tissues, and/or in pre- and postnatal brain. In contrast, data to investigate the methylation and expression of imprinted genes in adult somatic tissues are particularly scarce, and are limited to a few reports using a restricted supply of tissues from the same individuals [Citation19,Citation21]. The wide tissue panel obtained from the same autopsy patients is a major advantage of our study, eliminating the effect of interindividual heterogeneity in epigenetic regulation of imprinted genes on study outcome.
Gene imprinting is primarily achieved by the silencing of one parental allele via methylation of CpG-rich islands located at ICRs. Therefore, more intermediately methylated CpG sites are expected in imprinted genes and their ICR DMRs, which correspond to gene expression from the single unmethylated allele and silencing of the reciprocal parental allele through DNA methylation. However, the question of whether imprinted genes also retain the intermediately methylated DNA pattern in somatic tissues of adult humans has, so far, remained unclear.
We observed that imprinted genes possess more CpG loci with intermediate β-values of 0.5–0.7, along the entire length of a gene, in the extensive panel of somatic tissues tested. Our panel covered a wide range of tissues, formed from all three germ layers during embryogenesis: mesodermal (e.g., adipose tissue, bone and lymph nodes), endodermal (e.g., gastric mucosa and tonsils), ectodermal (e.g., coronary artery) and even neuroectodermal (e.g., ischiatic nerve and medulla oblongata) origin. Furthermore, a similar prevalence of intermediately methylated CpG sites was observed in DNA from blood, which is one of the most thoroughly studied tissues in the human body. Data from 17 postmortem tissues provided convincing evidence that imprinted genes more often retain (25.5% of all CpG sites) intermediately methylated CpGs when compared with nonimprinted genes, for which the respective share was only 10%. These results suggest that parental influence on gene expression is not abolished in somatic tissues of adult organisms, and explains the possible association between parent-of-origin effects and risk of common diseases. Parental-specific effects are gaining prominence, as previous studies have highlighted that parental ancestry of inherited alleles plays an important role in the development of complex diseases, like obesity [Citation4] and Type II diabetes [Citation5].
We also hypothesized that the variance of β-values in imprinted genes is smaller than in nonimprinted genes because they have more intermediately methylated CpG probes. This uniformity is likely contrasted by nonimprinted genes, for which expression is highly tissue dependent, resulting in a more stochastic pattern and co-existence of methylated and unmethylated alleles of the same gene. Indeed, our results seem to support this view, as we observed smaller variance in β-values for imprinted genes than for nonimprinted genes, as shown by Levene’s test to compare 76 imprinted genes with 76 nonimprinted genes. In order to strengthen this result, the comparison was performed 100-times, with different sets of randomly selected nonimprinted genes in each permutation. The results of this analysis also suggested that the number of imprinted genes is relatively low, as 100 comparisons of randomly selected nonimprinted genes showed comparable variances for β-values. In conclusion, these results together provide strong evidence that genomic imprinting is maintained across 18 tissues. Unlike many previous studies of imprinted genes, which were limited to a restricted number of tissues, such as placenta or fetal tissues, our study demonstrates clear stability in methylation of the tested imprinted genes in somatic tissues, and highlights blood as an excellent candidate tissue for the study and discovery of novel imprinted genes.
We also studied the locations of intermediately methylated CpG sites with β-values in the range of 0.5–0.7, and found that imprinted genes show a higher concentration in this range in a specific region of promoter area located 200–1500 bp from the transcription start site. This finding is strongly supported by our recent study [Citation23], in which DNA methylome profiling was used to identify tissue-specific differentially methylated regions. In this study, we used the same comprehensive tissue panel from autopsy patients and compared the detected methylation patterns with publicly available gene expression data. We were motivated to exploit publicly available gene expression data because the tissue samples were obtained from autopsy patients, which inevitably led to delay in tissue collection and 4–8 h autolysis time. Although the gene expression patterns before death and postmortem are largely comparable, death may still induce significant changes in a relatively large number of genes [Citation24,Citation25]. Consequently, we compared our DNA methylome data with that of gene expression from public databases, and observed a clear inverse correlation between promoter methylation within CpG islands and gene expression [Citation23]. This finding clearly supports the notion that epigenetic gene regulation is mediated via selective promoter methylation. In the current study, we demonstrated that the highest proportion of intermediately methylated CpG sites is located in promoter regions of imprinted genes, providing a convincing explanation as to why only a single parental allele is expressed, rather than a biallelic mode of activity.
The relationship between low and high promoter methylation with gene expression and silencing, respectively, is not a new idea itself [Citation26–29]. Nevertheless, to our knowledge, the current study is the first to provide compelling evidence that imprinted genes maintain an intermediately methylated status in somatic tissues of adult humans, even when the ICR DMR-related CpGs were excluded. However, whether these ontogenetically conserved epigenetic variations also impact parental-dependent monoallelic expression of those genes in somatic tissues remains an open question. In order to fully understand the parental effect on gene expression, the unequivocal ascertainment of the parental origin of the transcribed allele(s) is required. For example, genome imprinting can be studied by applying whole-transcriptome RNA sequencing of tissue and genotyping of coding DNA variants in family trios, as we recently used for placenta [Citation30]. In this study, parental and child blood DNA genotypes were analyzed by exome SNP genotyping microarrays, mapping the inheritance and estimating the abundance of parental expressed alleles in placental tissue. Imprinted genes showed predominant expression from one of the parental alleles, as demonstrated by the SNP content of sequenced transcripts. However, the use of a similar approach to study imprinting in somatic tissues raises more complex ethical and practical challenges, mostly because of the difficulties in obtaining the tissue samples from family trios.
The current study strongly supports the recently raised hypothesis in the seminal paper of Court et al. [Citation19] that substantiates the distribution of germline ICR DMRs between ubiquitously imprinted and placenta-specific imprinted genes. Placenta-specific imprinted DMRs exist solely in placental tissue and are absent in somatic tissues, including leukocytes, brain, liver, kidney and muscle [Citation19]. Our results, based on a substantially extended list of somatic tissues, corroborated the view of DMRs as divided into two groups, and showed that ubiquitously imprinted DMRs demonstrate an intermediate level of methylation in all somatic tissues. It is likely that this group of genes maintains a similar intermediate methylation of germline imprinted DMRs in all embryonal and extra-embryonal (placental) tissues during prenatal development, as well in most of somatic tissues through adult development and aging. In contrast, placenta-specific imprinted DMRs exhibited, according to our study, low levels of methylation in somatic tissues. It is well known that high levels of gene expression are often associated with low levels of methylation [Citation28,Citation31], leading us to the idea genes found to be imprinted in placental tissue could escape monoallelic expression in adult somatic tissues. Indeed, genes such as GLIS3, DNMT1 and LIN28B support this idea, being regulated by placenta-specific ICRs and monoallelically expressed in placental tissue only [Citation15,Citation32]. We found only two exceptions among the placenta-specific imprinted genes – GPR1-AS and MIR512, which showed an intermediate methylation pattern characteristic to ubiquitously imprinted genes. The placenta only imprinting of these two genes has also been found by other researchers [Citation33,Citation34].
Remarkably, in all placenta-specific DMRs, only the maternal allele is methylated [Citation19]. Intriguingly, placenta-specific imprinted genes do not inherit methylation from the gametes. So, in that sense, by strict definition, the DMRs unique to placenta cannot be classified as germline-originating parental epigenetic marks. However, these epigenetic loci provide evidence for a heritable epigenetic mechanism, which is independent from DNA methylation. Therefore, it has been speculated that during gamete formation, the parental alleles of these genes retain some nonequivalences that are not associated with DNA methylation, but rather with histone modifications, justifying their classification as germline DMRs. As a consequence, the maternally marked alleles will obtain the methylation signals in early embryonal development, when the trophoblast cells are formed, yielding the exclusive imprinted gene methylation profile for placental tissue [Citation19].
In conclusion, we demonstrate that the majority of imprinted genes and ICR DMRs maintain intermediate methylation status in the extended panel of adult somatic tissues obtained from the same individuals investigated in this study. This intermediately methylated pattern is more pronounced in specific regions of promoter areas of imprinted genes when compared with nonimprinted genes. However, the remarkable exception was seen for placenta-specific imprinted genes, which showed low levels of methylation, suggesting that these genes likely escape parental-specific epigenetic regulation in adult somatic tissues. Although our study has extended knowledge of gene imprinting in somatic tissues, future studies are required to provide deeper understanding about the regulation of imprinted genes in somatic tissues throughout the complete human lifespan.
Materials & methods
Sample collection
Seventeen different somatic tissue samples used in the current study were obtained from four autopsy patients – one female (BM419) and three males (KA522, KT538 and SJ600). They were 60, 53, 40 and 54 years of age, respectively, at the time of death. Their causes of death were either cerebellar hemorrhage (BM419 and SJ600) or myocardial infarction with acute cardiovascular insufficiency (KA522 and KT538), being the only severe disease for these patients. Between 4 and 8 h passed between the death of each subject and the collection of tissue samples.
Samples were collected from the following locations: abdominal and subcutaneous adipose tissue; coronary and splenic artery; bone; yellow and red bone marrow; gastric mucosa; joint cartilage; lymph node; tonsils; bladder; gallbladder; abdominal and thoracic aorta; medulla oblongata and ischiatic nerve. All 17 tissues were obtained from every study subject, except in the case of one individual (BM419) for whom the yellow bone marrow and joint cartilage tissues were not available. All samples were snap-frozen in liquid nitrogen and stored at -80°C prior to analysis. The tissue samples were collected at the Pathology Centre within the North Estonia Medical Centre, Estonia. More information about the subjects for tissue-panel study is provided in our previous studies [Citation23,Citation35–36].
Whole blood samples were obtained from 97 healthy adult individuals (Nmale = 48 and Nfemale = 49) selected from EGCUT. All subjects were older than 18 years, with a mean age of 52.7 years, and gave written informed consent for participation in the study.
The study was carried out with the approval of the Research Ethics Committee of the University of Tartu.
DNA extraction & methylation array analysis
DNA was extracted from 25 mg tissue samples using the NucleoSpin® Tissue kit (Macherey-Nagel GmbH, Düren, Germany). DNA from whole blood samples was extracted by the salting-out method using 10 M ammonium acetate. DNA was precipitated in isopropanol, washed in 70% ethanol and finally resuspended in 1× TE buffer. The purity and concentrations of the DNA samples were measured by spectrophotometer (NanoDrop® ND1000; Thermo Fisher Scientific, Inc., MA, USA).
Negative and positive controls for unmethylated and methylated DNAs, respectively, were also represented in this study. For ‘unmethylated control DNA’, the DNA from subcutaneous adipose tissue was amplified using the GenomiPhi kit (GE Healthcare, NJ, USA), while for ‘methylated control DNA’ the universal methylated human DNA standard (Zymo Research Orange, CA, USA) was used. From each sample, 600 ng of genomic DNA was bisulfite modified using the EZ DNA Methylation kit (Zymo Research Orange) according to the manufacturer’s recommendations for the Illumina Infinium Assay.
After purification, 4 μl of each bisulfite-converted DNA sample and controls (total of 165 samples) was used for hybridization on the Infinium Human Methylation 450 BeadChip, following the Illumina Infinium HD Methylation protocol. The original IDAT files were extracted from the HiScan scanner. We also processed two technical (the same DNA was analyzed twice) and two biological (the extraction from the same tissue was performed twice and both of the DNAs were analyzed) replicates. The Pearson correlation coefficients (PCCs) were >0.99 for all the replicates, confirming a good level of reproducibility for the array analysis.
Methylation normalization
Data processing and quality control analyses were performed using the Bioconductor package minfi [Citation37]. Prenormalized raw data was used to convert the intensities from the red and the green channels into methylated (M) and unmethylated (U) signals. β-values for each CpG probe were calculated according to Illumina’s recommendations using [β = M/(M+U+100)].
The difference in the distribution of β-values for type I and type II probes was corrected using SWAN (Subset-quantile Within Array Normalization) normalization [Citation38]. Detection p-values were obtained for every CpG probe in every sample. Failed positions were defined as signal levels lower than background from both methylated and unmethylated channels. Probes with detectable methylation level in <5% of samples (detection p-value <0.01) were excluded. Also, CpG sites on X and Y chromosomes, cross-reactive probes and SNPs inside the probe body or at the nucleotide extension were discarded, which left 353,094 probes for further analysis.
CpG sites location mapping
An Illumina Infinium HumanMethylation450 BeadChip contains more than 485,000 methylation sites, covering 21,231 out of 21,474 UCSC RefGenes, with an average of 17 CpG sites per gene. In order to achieve a comprehensive assessment of gene region methylation, each gene was split into six regions. CpG sites are distributed across the promoter, 5′UTR, the first exon, gene body and 3′UTR regions. Promoter regions were further divided into two mutually exclusive bins of 1–200 bp and 200–1500 bp blocks upstream of the transcription start site. Each CpG site for validated genes was mapped to its exact location in the genome using the Illumina annotation file, build 37.
Equality of variances
In the case of gene imprinting, we expected more intermediately methylated probes, and therefore mid-range β-values, which should lead to smaller variance of β-values among these probes. We used Levene’s test to compare the equality of variances of probes’ β-values between imprinted and nonimprinted genes.
The list of human imprinted genes & ICR DMRs
We used the ‘Catalogue of Parent of Origin Effects Imprinted Genes and Related Effects’ database [Citation39] and ‘Geneimprint’ catalogue [Citation40] as an open scientific source to identify the list of human imprinted genes, with all references provided in Supplementary Table 1. Also we used the ‘PubMed search’ to find articles related to these imprinted genes and to verify that all our imprinted genes are indeed shown to be imprinted in humans. Also, highly isoform-dependent transcripts were excluded from this list. In addition, genes that have previously been found to be imprinted in model organisms and were only predicted to be imprinted in humans based on the conserved sequences between two species were also excluded from this list. In total, we were left with 76 known human imprinted genes for downstream analyses. In all analyses, we compared these genes with the rest of 20,515 nonimprinted genes, captured by Illumina array. The list of germline ICR DMRs was obtained from the previous study [Citation19].
The biggest difference in methylation levels between imprinted and nonimprinted genes was found for intermediately methylated CpGs with corresponding β-values of 0.5–0.7.
Imprinted genes have smaller variance in β-values than nonimprinted genes across 18 human somatic tissues.
Imprinted genes show a higher percentage of intermediately methylated probes with β-values in range 0.5–0.7 in an extended promoter region, spanning 200–1500 bp from the transcription start site.
Imprinting control regions and their differentially methylated regions (DMRs) of ubiquitously imprinted genes demonstrate remarkable stability of intermediate methylation levels, varying from 49 to 77%, across all 18 somatic tissues tested.
The placenta-specific imprinting control regions DMRs present low methylation levels, in a range of 10–20%, in somatic tissues tested; except for two genes (GPR1-AS and MIR512) with intermediate methylation levels of 55 and 67%, respectively.
Visualization demonstrated that only GPR1-AS had the methylation pattern specific to ubiquitously imprinted DMRs, while MIR512 had a unique pattern that was characterized by an increased number of highly methylated probes and a small number of intermediately and lowly methylated probes.
Data access
The data used in this study have been deposited in NCBI’s Gene Expression Omnibus repository and are accessible through GEO Series, accession number GSE50192.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Supplemental Table 1
Download MS Word (122 KB)Supplemental Table 2
Download MS Word (19.6 KB)Supplemental Table 3
Download MS Word (22 KB)Supplemental Table 4
Download MS Word (34.7 KB)Acknowledgements
The authors are grateful to all families who participated in this study. We also thank Neeme Tõnisson for constructive comments and suggestions on the study.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/full/10.2217/epi.16.8
Financial & competing interests disclosure
This research was funded by grants from the European Union’s Horizon 2020 research grants ePerMed and WIDENLIFE; innovation programme (grant 692065); Enterprise Estonia (grant EU30020 and EU48695); Estonian Ministry of Education and Research (grant IUT34-16); EU-FP7 Eurostars Program (grant NOTED, EU41564); EU-FP7 IAPP Project (grant SARM, EU324509); Estonian Research Council (grants IUT20-60 and PUT736); the Development Fund of the University of Tartu (grant SP1GVARENG); EU structural support through Archimedes Foundation (grant 3.2.1001.11-0033); the European Regional Development Fund to the Centre of Excellence in Genomics (EXCEGEN; grant 3.2.0304.11-0312) and Wellcome Trust Senior Fellowship in Basic Biomedical Science (grant WT098017). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the prosduction of this manuscript.
Additional information
Funding
References
- McGrath J , SolterD . Completion of mouse embryogenesis requires both the maternal and paternal genomes . Cell37 ( 1 ), 179 – 183 ( 1984 ).
- Surani MA , BartonSC . Development of gynogenetic eggs in the mouse: implications for parthenogenetic embryos . Science222 ( 4627 ), 1034 – 1036 ( 1983 ).
- Renfree MB , SuzukiS , Kaneko-IshinoT . The origin and evolution of genomic imprinting and viviparity in mammals . Philos. Trans. R. Soc. Lond. B. Biol. Sci.368 ( 1609 ), 20120151 ( 2013 ).
- Hoggart CJ , VenturiniG , ManginoMet al. Novel approach identifies SNPs in SLC2A10 and KCNK9 with evidence for parent-of-origin effect on body mass index . PLoS Genet.10 ( 7 ), e1004508 ( 2014 ).
- Lindsay RS , KobesS , KnowlerWC , HansonRL . Genome-wide linkage analysis assessing parent-of-origin effects in the inheritance of birth weight . Hum. Genet.110 ( 5 ), 503 – 509 ( 2002 ).
- Barlow DP . Genomic imprinting: a mammalian epigenetic discovery model . Annu. Rev. Genet.45 ( 1 ), 379 – 403 ( 2011 ).
- Edwards CA , Ferguson-SmithAC . Mechanisms regulating imprinted genes in clusters . Curr. Opin. Cell Biol.19 ( 3 ), 281 – 289 ( 2007 ).
- MouseBook . www.mousebook.org .
- Peters J , RobsonJE . Imprinted noncoding RNAs . Mamm. Genome.19 ( 7–8 ), 493 – 502 ( 2008 ).
- Ideraabdullah FY , VigneauS , BartolomeiMS . Genomic imprinting mechanisms in mammals . Mutat. Res.647 ( 1–2 ), 77 – 85 ( 2008 ).
- Lalande M . Parental imprinting and human disease . Annu. Rev. Genet ( 1997 ).
- Smith ZD , MeissnerA . DNA methylation: roles in mammalian development . Nat. Rev. Genet.14 ( 3 ), 204 – 220 ( 2013 ).
- Bourc’his D , XuGL , LinCS , BollmanB , BestorTH . Dnmt3L and the establishment of maternal genomic imprints . Science294 ( 5551 ), 2536 – 2539 ( 2001 ).
- Hata K , OkanoM , LeiH , LiE . Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice . Development129 , 1983 – 1993 ( 2002 ).
- Das R , LeeYK , StrogantsevRet al. DNMT1 and AIM1 Imprinting in human placenta revealed through a genome-wide screen for allele-specific DNA methylation . BMC Genomics14 ( 1 ), 685 ( 2013 ).
- Lopes S . Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions . Hum. Mol. Genet.12 ( 3 ), 295 – 305 ( 2003 ).
- Coombes C , ArnaudP , GordonEet al. Epigenetic properties and identification of an imprint mark in the Nesp-Gnasxl domain of the mouse Gnas imprinted locus . Mol. Cell. Biol.23 ( 16 ), 5475 – 5488 ( 2003 ).
- Kagami M , MatsuokaK , NagaiTet al. Paternal uniparental disomy 14 and related disorders: placental gene expression analyses and histological examinations . Epigenetics7 ( 10 ), 1142 – 1150 ( 2012 ).
- Court F , TayamaC , RomanelliVet al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of the human imprintome and suggests a germline methylation independent establishment of imprinting . Genome Res.24 ( 4 ), 554 – 569 ( 2014 ).
- Kobayashi H , YamadaK , MoritaSet al. Identification of the mouse paternally expressed imprinted gene Zdbf2 on chromosome 1 and its imprinted human homolog ZDBF2 on chromosome 2 . Genomics93 ( 5 ), 461 – 472 ( 2009 ).
- Woodfine K , HuddlestonJE , MurrellA . Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue . Epigenetics Chromatin4 ( 1 ), 1 ( 2011 ).
- Bibikova M , BarnesB , TsanCet al. High density DNA methylation array with single CpG site resolution . Genomics98 ( 4 ), 288 – 295 ( 2011 ).
- Lokk K , ModhukurV , RajashekarBet al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns . Genome Biol.15 ( 4 ), R54 ( 2014 ).
- Franz H , UllmannC , BeckerAet al. Systematic analysis of gene expression in human brains before and after death . Genome Biol.6 ( 13 ), R112 ( 2005 ).
- Sluimer JC , KistersN , CleutjensKBet al. Dead or alive: gene expression profiles of advanced atherosclerotic plaques from autopsy and surgery . Physiol. Genomics30 ( 3 ), 335 – 341 ( 2007 ).
- Vaniushin BF . DNA methylation and epigenetics . Genetika42 ( 9 ), 1186 – 1199 ( 2006 ).
- Newell-Price J , ClarkAJ , KingP . DNA methylation and silencing of gene expression . Trends Endocrinol. Metab.11 ( 4 ), 142 – 148 ( 2000 ).
- Ehrlich M . DNA methylation in cancer: too much, but also too little . Oncogene21 ( 35 ), 5400 – 5413 ( 2002 ).
- Li Y , SasakiH . Genomic imprinting in mammals: its life cycle, molecular mechanisms and reprogramming . Cell Res.21 ( 3 ), 466 – 473 ( 2011 ).
- Metsalu T , ViltropT , TiiratsAet al. Using RNA sequencing for identifying gene imprinting and random monoallelic expression in human placenta . Epigenetics9 ( 10 ), 1397 – 1409 ( 2014 ).
- Li E , BeardC , JaenischR . Role for DNA methylation in genomic imprinting . Nature366 ( 6453 ), 362 – 365 ( 1993 ).
- Barbaux S , Gascoin-LachambreG , BuffatCet al. A genome-wide approach reveals novel imprinted genes expressed in the human placenta . Epigenetics7 ( 9 ), 1079 – 1090 ( 2012 ).
- Kobayashi H , YanagisawaE , SakashitaAet al. Epigenetic and transcriptional features of the novel human imprinted lncRNA GPR1AS suggest it is a functional ortholog to mouse Zdbf2linc . Epigenetics8 , 635 – 645 ( 2013 ).
- Noguer-Dance M , Abu-AmeroS , Al-KhtibMet al. The primate-specific microRNA gene cluster (C19MC) is imprinted in the placenta . Hum. Mol. Genet.19 ( 18 ), 3566 – 3582 ( 2010 ).
- Krjutškov K , KoltšinaM , GrandKet al. Tissue-specific mitochondrial heteroplasmy at position 16,093 within the same individual . Curr. Genet.60 ( 1 ), 11 – 16 ( 2014 ).
- Žilina O , KoltšinaM , RaidR , KurgA , TõnissonN , SalumetsA . Somatic mosaicism for copy-neutral loss of heterozygosity and DNA copy number variations in the human genome . BMC Genomics16 ( 1 ), 703 ( 2015 ).
- Aryee MJ , JaffeAE , Corrada-BravoHet al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays . Bioinformatics30 ( 10 ), 1363 – 1369 ( 2014 ).
- Makismovic J , GordonL , OshlackA . SWAN: subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips . Genome Biol.13 ( 6 ), R44 ( 2012 ).
- Catalogue of Parent of Origin Effects Imprinted Genes and Related Effects . http://igc.otago.ac.nz/Search.html .
- Geneimprint . www.geneimprint.com/ .