
Abstract
Current treatment guidelines recommend once-daily, low-dose acetylsalicylic acid (ASA; aspirin) for secondary prevention of cardiovascular events. However, the anti-thrombotic benefits of traditional ASA formulations may not extend over a 24-h period, especially in patients at high risk for a recurrent cardiovascular event. A next-generation, extended-release ASA formulation (ER-ASA) has been developed to provide 24-h anti-thrombotic coverage with once-daily dosing. The pharmacokinetics of ER-ASA indicates slower absorption and prolonged ASA release versus immediate-release ASA, with a favorable safety profile. ER-ASA minimizes systemic ASA absorption and provides sustained antiplatelet effects over a 24-h period.
Cardiovascular disease (CVD), including heart disease and stroke, is one of the leading causes of death and disability in the USA [Citation1,Citation2]. Coronary heart disease (48%) and stroke (16%) are the most common causes of CVD-related mortality [Citation1]. In 2011, approximately 85.6 million adults (more than one in three) in the USA were reported to have CVD, and the incidence is anticipated to increase through 2030 [Citation1]. CVD is among the leading medical conditions resulting in functional disability, and causes approximately $125 billion annually in lost worker productivity [Citation1]. Annual direct cost of CVD (e.g., healthcare provider visits, hospital services, medication) in 2011 was approximately $196 billion. Total direct medical costs of CVD are estimated to increase to $918 billion by 2030 [Citation1].
Patients with CVD are at increased risk of secondary cardiovascular (CV) event occurrences. Approximately 15% of patients with CVD and up to 45% of patients with CVD and comorbid diabetes experience a secondary CV event [Citation3–5], and these higher-risk patient cohorts have an increased risk of CVD-related mortality versus individuals without these conditions [Citation6]. The American College of Cardiology Foundation, in collaboration with the American Heart Association and the American Diabetes Association, has recommended long-term, uninterrupted therapy with low-dose (75–162 mg) acetylsalicylic acid (ASA; aspirin) for the secondary prevention of CV events [Citation7,Citation8]. Unfortunately, the recommended once-daily, low-dose ASA may not provide adequate prevention in a substantial percentage of patients [Citation9]. Therefore, additional therapeutic options are needed to maximize risk reduction. In the current article, we review the role of traditional oral ASA formulations in prevention of secondary CV events, and discuss the potential advantages of a new extended-release (ER)-ASA formulation that is currently undergoing US regulatory review for secondary prevention of CV events.
Overview of traditional immediate-release ASA
• ASA pharmacology
The antipyretic and analgesic effects of salicin, the compound in willow bark that provides a therapeutic benefit, have been known since antiquity [Citation10]. In 1897, the acetylated form of salicylic acid (SA) was synthesized [Citation10], and during the 1950s, it was discovered that ASA had anti-thrombotic effects [Citation10]. In 1971, the mechanism underlying the cardioprotective effects of ASA (i.e., inhibition of prostaglandin [PG] synthesis) was elucidated [Citation11].
Platelets may be activated by a variety of stimuli including shear, epinephrine, ADP, thrombin and adhesion to exposed collagen and von Willebrand factor at the site of the injured vessel wall [Citation12]. Upon activation, platelets release positive feedback mediators that facilitate platelet activation, as well as transmitters that impact the vessel wall and other cells and contribute to vasoconstriction, increased capillary permeability and coagulation [Citation13].
Platelet activation and aggregation are the most critical factors in the generation of ischemic event occurrences [Citation12–14]. ASA selectively, rapidly and irreversibly acetylates a serine residue in the cyclooxygenase (COX)-1, and thereby prevents binding of arachidonic acid to the catalytic site and reduces production of PGH2 [Citation15]. Under ‘normal’ conditions, PGH2 is converted to a variety of PGs (e.g., PGI2 [prostacyclin]) and thromboxane A2 (TxA2) through the actions of tissue-specific synthases [Citation15]. Reduced production of PGH2, attributable to ASA inhibition of the COX-1 enzyme, results in lower levels of prothrombotic TxA2 and subsequent TxA2-induced platelet aggregation [Citation15]. Although ASA inhibits both COX-1 and COX-2, the affinity of ASA for COX-1 is 166 times greater than for COX-2; thus COX-1 inhibition is primarily responsible for ASA-induced reductions in TxA2 [Citation15].
Multiple formulations of ASA (single entity and combination products) have been developed, including immediate-release (IR) ASA, which disintegrates in and is rapidly absorbed by the stomach and upper small intestines, and enteric-coated ASA, which breaks down and is absorbed primarily within the duodenum. In humans, a single dose of IR-ASA provides a peak plasma concentration within 40 min and inhibition of platelets within 1 h postdose [Citation15]. Enteric-coated ASA has a slower dissolution profile than IR-ASA, which extends the time to absorption up to 5 h [Citation16]. The overall bioavailability of ASA also differs between the two formulations: the bioavailability with IR-ASA (50%) is greater than that of enteric-coated ASA [Citation17–19]. Once absorbed, ASA is rapidly deacetylated to salicylic acid by carboxylesterases within the liver and the GI tract (i.e., within the portal circulation) [Citation20]; thus, ASA in plasma has a short half-life of approximately 15–20 min, regardless of delivery formulation [Citation17]. Inhibition of platelet activity was observed before systemic levels of ASA were detected, suggesting that inhibition of the COX-1 enzyme of platelets occurs in the prehepatic circulation [Citation12].
The antiplatelet effect of ASA is critically dependent on the concentration of unmetabolized ASA in circulation and platelet generation and turnover [Citation21]. Platelets are continuously generated throughout the day [Citation22] (∼10% of the total platelet population is regenerated during a 24-h period [Citation23]) and have a typical life span of 8–10 days [Citation23,Citation24]. The irreversible acetylation of COX-1 ensures enzyme inhibition for the entirety of a platelet’s lifespan; however, this anti-thrombotic effect is negatively impacted over time as new platelets with uninhibited COX enzymes are continually released into systemic circulation [Citation25].
The pharmacodynamic effects of ASA occur quickly (within hours) after administration [Citation19], as might be expected from its original development as an analgesic aimed at providing immediate pain relief [Citation10]. Serum thromboxane B2 (TxB2; a stable metabolite of TxA2) levels in healthy volunteers decline within the first 20 min of ASA administration and remain low (∼100 ng/ml), despite reductions in plasma ASA concentrations [Citation19]. The significantly lower level of TxB2 correlates with the inhibition of arachidonic acid-induced platelet aggregation as measured by ex vivo assays [Citation12]. Repeated administration of low-dose ASA in healthy individuals results in cumulative and selective inactivation of platelet COX-1 activity [Citation26]. Upon ASA withdrawal, COX-1 activity recovers within approximately 9 days, in accordance with platelet lifespan [Citation26]. However, reemergence of functional platelet COX-1 in circulation may occur as quickly as within 24 h in patients with a higher rate of platelet turnover, such as those with CVD [Citation27], metabolic syndrome [Citation28], or diabetes [Citation29], or after cardiac surgery [Citation12,Citation30].
(A) Arachidonic acid-induced platelet aggregation and (B) TxB2 production in healthy volunteers and patients with cardiovascular disease after a single dose of IR-ASA 75 mg. Platelet aggregation was measured using the Multiplate® Analyzer (Roche Diagnostics International LDT, Rotkreuz, Switzerland), which detects differences in electric resistance between two electrodes in aggregated whole blood. Platelet aggregation was stimulated by arachidonic acid 1.0 mM and collagen 3.2 µg/ml. Serum TxB2 concentrations were measured using an ELISA (Cayman Chemical, Ann Arbor, MI, USA). Platelet aggregation and TxB2 production were significantly greater 24 h after ASA dosing compared with 1 h post-treatment.
ASA: Acetylsalicylic acid; CAD: Coronary artery disease; ST: Stent thrombosis; TxB2: Thromboxane B2.
Reproduced with permission from [Citation61].
![Figure 1. Recovery of platelet aggregation and thromboxane A2 production after immediate-release acetylsalicylic acid administration. (A) Arachidonic acid-induced platelet aggregation and (B) TxB2 production in healthy volunteers and patients with cardiovascular disease after a single dose of IR-ASA 75 mg. Platelet aggregation was measured using the Multiplate® Analyzer (Roche Diagnostics International LDT, Rotkreuz, Switzerland), which detects differences in electric resistance between two electrodes in aggregated whole blood. Platelet aggregation was stimulated by arachidonic acid 1.0 mM and collagen 3.2 µg/ml. Serum TxB2 concentrations were measured using an ELISA (Cayman Chemical, Ann Arbor, MI, USA). Platelet aggregation and TxB2 production were significantly greater 24 h after ASA dosing compared with 1 h post-treatment.ASA: Acetylsalicylic acid; CAD: Coronary artery disease; ST: Stent thrombosis; TxB2: Thromboxane B2.Reproduced with permission from [Citation61].](/cms/asset/cb1e80ab-4de8-47bf-8058-5ca779ffb3ac/ifca_a_12325583_f0001.jpg)
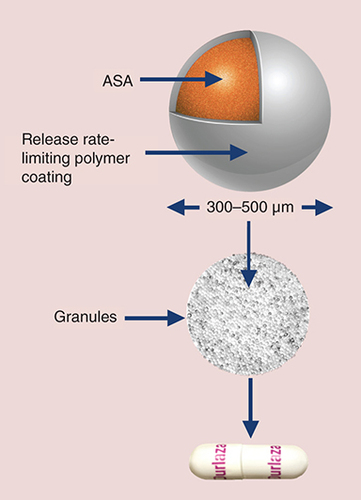
Each extended-release capsule contains a core of release rate-limiting, film-coated microcapsules containing 162.5 mg of ASA.
ASA: Acetylsalicylic acid.
(A) Mean concentration time profiles of single-dose ASA for ER-ASA and IR-ASA; and (B) single-dose mean concentration time profiles of SA for ER-ASA and IR-ASA.
ASA: Acetylsalicylic acid; ER: Extended release; IR: Immediate release; SA: Salicylic acid.
Data taken from [Citation75].
![Figure 4. Pharmacokinetics of extended-release acetylsalicylic acid. (A) Mean concentration time profiles of single-dose ASA for ER-ASA and IR-ASA; and (B) single-dose mean concentration time profiles of SA for ER-ASA and IR-ASA.ASA: Acetylsalicylic acid; ER: Extended release; IR: Immediate release; SA: Salicylic acid.Data taken from [Citation75].](/cms/asset/833e28d9-5c8f-495f-b110-ccc3063e94c5/ifca_a_12325583_f0004.jpg)
(A) Mean percent inhibition of TxB2 production and (B) 11-dehydro-TxB2 after single-dose exposure to ER-ASA 162 mg and IR-ASA 81 mg in healthy volunteers. ER-ASA provides 24-h reduction of TxB2 production.
ASA: Acetylsalicylic acid; ER: Extended release; IR: Immediate release; TxB2: Thromboxane B2.
Data taken from [Citation75].
![Figure 5. Antiplatelet effects of a single dose of extended-release acetylsalicylic acid. (A) Mean percent inhibition of TxB2 production and (B) 11-dehydro-TxB2 after single-dose exposure to ER-ASA 162 mg and IR-ASA 81 mg in healthy volunteers. ER-ASA provides 24-h reduction of TxB2 production.ASA: Acetylsalicylic acid; ER: Extended release; IR: Immediate release; TxB2: Thromboxane B2.Data taken from [Citation75].](/cms/asset/a95235d5-a396-423e-8509-a361c0192b44/ifca_a_12325583_f0005.jpg)
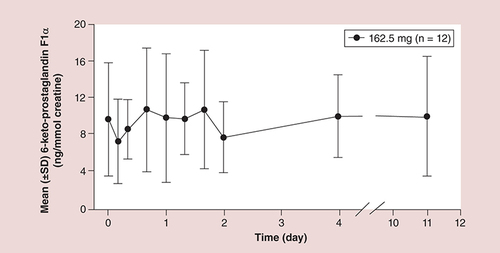
Mean vascular prostacyclin levels were not reduced after multiple extended-release acetylsalicylic acid administrations.
†Mean urine excretion level of 6-keto prostaglandin F1α is a surrogate marker for vascular prostacyclin level
• ASA reduces risk of CV events in high-risk patients
A large body of data has demonstrated that ASA reduces the risk of secondary CV events and mortality. A meta-analysis of 17,000 high-risk patients (i.e., patients with a previous CV event) from 16 clinical trials indicated a significant reduction in the risk of CV events in patients who received antiplatelet therapy versus those who did not [Citation31]. This risk reduction occurred regardless of predisposing medical conditions, including carotid disease and diabetes [Citation32]. In a meta-analysis of 13 studies that examined the efficacy of ASA for the prevention of secondary CV events in patients with diabetes, ASA significantly reduced the risk of all-cause mortality compared with no ASA treatment (relative risk: 0.82; 95% CI: 0.69–0.98; p = 0.03) [Citation33]. The extent of risk reduction for CV-related mortality and for occurrence of a secondary CV event was not related to ASA dose; higher ASA doses (up to 1300 mg/day) did not appear to confer additional risk reduction [Citation32,Citation33]. Moreover, high-dose (150–325 mg/day) ASA has been associated with increased risk of bleeding compared with low-dose (75–149 mg/day) ASA in at least one high-risk population (i.e., patients receiving medical treatment for acute coronary artery syndrome) [Citation34].
• ASA resistance
Current treatment guidelines for secondary prevention of CV events recommend once-daily, low-dose (75–325 mg/day) ASA in high-risk patients [Citation7–8,Citation35]. It has been widely reported that a substantial percentage of patients treated with low-dose ASA exhibit laboratory ASA resistance [Citation36], but the relationship of this phenomenon to clinical outcomes is controversial. A reliable and specific laboratory method to identify ASA resistance has not yet been uniformly defined. Laboratory point-of-care methods, including assays that use platelet agonists such as ADP, collagen, shear and epinephrine (i.e., ‘COX-1 nonspecific’ methods) to stimulate platelets, reflect multiple signaling pathways, not solely COX-1 activity [Citation36,Citation37]. The best definition of ASA resistance or ASA nonresponse is one based on the specific observation of residual COX-1 activity [Citation36]. Assays that measure serum TxB2 and arachidonic acid-induced platelet aggregation are the most specific methods for assessing COX-1 activity [Citation38]; however, other assays that employ arachidonic acid in evaluation of ASA responsiveness are also commonly reported in the literature [Citation38]. With adequate doses of ASA, ASA nonresponse based on COX-1 activity alone occurs in <5% of patients [Citation38]. However, up to 37% of patients with a history of CVD and up to 42% of patients with diabetes, many of whom also have metabolic syndrome [Citation39], show a reduced response to ASA [Citation40–42]. The higher prevalence of ASA resistance, as reported in the latter studies, is due to use of ‘COX-1 nonspecific’ methods (i.e., ADP-, and collagen-induced platelet aggregation). Other nonmethodologic factors (e.g., BMI, aging, ASA formulation, ASA dose, ASA drug–drug interactions, patient adherence and alterations in platelet characteristics [e.g., reactivity and turnover]) may also contribute to ASA nonresponse [Citation43].
• Platelet turnover & reactivity impact ASA response
Patients at high risk for CV events (e.g., patients with coronary artery disease [CAD], diabetes, or metabolic syndrome, smokers and patients undergoing cardiac surgery) tend to have increased platelet turnover [Citation28–30,Citation44–46] and reduced ASA responsiveness [Citation28,Citation40,Citation42,Citation47–54]. Markers of increased platelet turnover (e.g., increased mean platelet volume [MPV], immature platelet fraction) are present in high-risk patients [Citation27,Citation55–56] and have been shown to be predictive of worse adverse cardiac outcomes [Citation57] and CV-related death [Citation58,Citation59].
Studies in patients with disease states associated with high platelet turnover have observed substantial recovery of TxA2 production and platelet aggregation (i.e., nonresponse to ASA) in the presence of low-dose ASA [Citation60,Citation61]. Overall TxA2 synthesis must be inhibited by ≥95% before any clinical effects of ASA are observed [Citation12]. Adequate inhibition of TxA2 may require inhibition of alternative TxA2 production pathways, such as production by thromboxane synthase, which is unaffected by ASA [Citation12]. In addition, uninhibited COX-2 may be rapidly resynthesized by endothelial cells and monocytes/macrophages and may feed TxA2 intermediates (e.g., PGH2) into the thromboxane synthase pathway [Citation12]. This alternate mechanism of TxA2 production may be particularly important at sites where monocytes and macrophages accumulate with platelets (e.g., sites of plaque rupture) [Citation12]. Under normal conditions, COX-2 expression is generally low; however, in patients with high platelet turnover, such as patients who have undergone coronary artery bypass surgery, COX-2 expression may increase to as much as 60% and contribute to high TxA2 concentrations despite COX-1 inhibition by low-dose (81 mg/day) ASA [Citation12]. Würtz et al. [Citation61] demonstrated significant recovery of platelet aggregation (A) and increased TxA2 production (B) within 24 h of administration of ASA 75 mg/day, not only in high-risk patients (i.e., those with previous stent thrombosis or stable CAD) but also in healthy individuals. Although once-daily ASA doses of 162 and 325 mg have been shown to reduce COX-dependent platelet aggregation and TxA2 production in high-risk patients (e.g., patients with diabetes and CAD) [Citation62], a more substantial reduction in these parameters has been observed with twice-daily dosing [Citation63]. Twice-daily dosing of ASA provides a second opportunity or window of time during which COX-1 enzymes within newly generated platelets become inhibited. This additional inactivation period may be particularly relevant for patients with higher rates of platelet turnover.
Accelerated platelet turnover results in an increase in the proportion of immature platelets with COX-2 in systemic circulation [Citation10,Citation64] and increases the rate at which platelets with ASA-inhibited COX-1 are replaced by new, uninhibited platelets [Citation10]. Immature platelets have a larger overall volume [Citation10,Citation65], are more adherent to endothelial surfaces and fibrin meshwork [Citation10], and can synthesize prothrombotic factors (e.g., fibrinogen and adhesion factors) [Citation66]. Immature platelets are also highly reactive, with augmented production of TxA2 and platelet aggregation compared with mature platelets [Citation67,Citation68]. Indices of immature platelets have been associated with nonresponse to ASA [Citation25,Citation67,Citation69]. A significant association between ASA nonresponse and platelet turnover was observed when nonresponse was stratified by platelet volume in healthy volunteers [Citation67]. In patients with stable CAD, a reduced response to ASA 75 mg/day was associated with increased platelet turnover (as assessed by larger MPV and higher IPC) [Citation25]. Data also have shown a trend toward increased platelet turnover and IPC in patients with previous stent thrombosis who were receiving ASA 75 mg/day, although this did not reach statistical significance [Citation69].
• Unmet need for ER-ASA
The COX-1 enzyme in platelets is irreversibly inhibited by ASA, and is not replaced in mature, anucleate platelets [Citation10]. Because of this, the anti-thrombotic effects of ASA extend beyond its short half-life and are assumed to be maintained throughout a 24-h period, supporting the recommendation of once-daily dosing for prevention of secondary CV events [Citation7–8,Citation35]. However, the effects of ASA decline over time as newly generated platelets with uninhibited COX-1 enzymes enter circulation [Citation10,Citation60–61,Citation70]. For patients who take their ASA dose in the morning, ASA concentrations are higher at the time of day during which CV events are most common (i.e., upon awakening) [Citation71,Citation72]. Unfortunately, this protective antiplatelet effect likely declines during the evening hours as inhibited platelets are replaced; therefore, a continuous concentration of ASA is needed to ensure adequate anti-thrombotic protection over a 24-h dosing period. Twice-daily dosing of ASA provides an additional window of time for inhibition of platelet activation in patients with high platelet turnover, which may improve response [Citation63]. Unfortunately, only 65% of patients report adherence to their current secondary preventive ASA regimen [Citation73]; a more complicated dosing regimen may further reduce adherence, potentially affecting overall efficacy of preventative measures [Citation74].
Extended-release ASA
• Extended-release technology
An ER-ASA was initially developed by Flamel Technologies (Flamel of Pessac, France) using ER microcapsule technology. This technology has been successfully applied to other CVD drugs (e.g., carvedilol [Coreg CR®; GlaxoSmithKline, Research Triangle Park, NC]) to provide 24-h exposure with once-daily dosing. In contrast to conventional IR-ASA formulations, ER-ASA (NHP-554C) was designed to release ASA over the entire 24-h period with once-daily dosing. In addition, this formulation delivers ASA into the hepatic circulation in a manner that allows metabolism of ASA and reduces circulating ASA levels, thereby sparing peripheral endothelial prostacyclin levels.
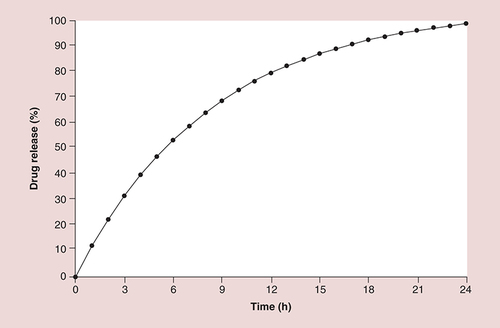
Each soft gelatin ER capsule contains a core of individually film-coated, 300–500 µm, ASA microcapsules (). The proprietary, release rate-limiting film coating of the microcapsules acts as a semipermeable membrane to allow diffusion of ASA throughout the GI tract, thereby slowing absorption and extending the availability of ASA for a prolonged antiplatelet effect. This diffusion-based drug delivery formulation displays a characteristic diffusion profile () in which approximately 30% of the total ASA dose is delivered within the first 3 h after administration, followed by sustained release for the remainder of the 24-h period.
• ER-ASA clinical development program
The overall clinical development program of ER-ASA focused on creating an ASA formulation that inhibited >90% of circulating TxA2 during a 24-h period. Multiple studies (∼15 clinical studies in addition to investigator-initiated studies) have been conducted in healthy individuals and patients with CVD to establish the pharmacokinetics, pharmacodynamics and safety of ER-ASA. As the efficacy of ASA for secondary prevention is well established, only two clinical pharmacology studies (a dose–response study [Citation75] and a food effect study [flamel technologies, venissieux, france, data on file]) were required for US FDA approval, and ER-ASA is currently undergoing regulatory review.
• Pharmacokinetics of ER-ASA
A single-dose study of the pharmacokinetics of ER-ASA 20–325 mg versus IR-ASA 5–81 mg was undertaken in healthy volunteers in a Phase I, open-label, US registration trial [Citation75]. As anticipated, the pharmacokinetic profile of ER-ASA differed from that of IR-ASA. Dose-normalized mean maximum plasma concentrations of ASA and SA, a stable metabolite of ASA, were approximately sixfold and approximately two- to three-fold higher, respectively, with IR-ASA versus ER-ASA [Citation75]. However, the dose-normalized area under the time-concentration curve from time 0 to the time of the last quantifiable concentration for SA was similar with both ASA formulations [Citation75], suggesting that an overall comparable amount of active drug was delivered. Consistent with its ER pharmacokinetic profile, time to maximal plasma ASA concentration occurred later with ER-ASA (2–4 h postadministration) than with IR-ASA (1–2 h postadministration) (A) [Citation75]. In addition, peak serum concentrations of SA were delayed with ER-ASA compared with IR-ASA and remained detectable during a 24-h period (B) [Citation75]. The prolonged release of ASA from the ER-ASA formulation provides a greater time frame during which platelets may be exposed to intact ASA, thereby extending the window of time for platelet inactivation. Plasma concentrations of ASA with ER-ASA administration were quantifiable throughout an 8-h observation period, whereas ASA was undetectable 6 h after administration of IR-ASA. In a separate food effects study, the overall exposure of ER-ASA did not vary when administered with food; however, absorption was delayed fivefold [flamel technologies, venissieux, france, data on file].
• Anti-thrombotic activity
Pharmacodynamic properties of ER- versus IR-ASA were examined in the same Phase I, single-dose study of healthy volunteers [Citation75]. Maximal inhibition of TxB2 production with a single dose (mean percentage reduction ±SD: 90.5% ± 9.8% and 96.5% ± 4.1% with ER-ASA 162.5 mg and IR-ASA 81 mg, respectively) occurred within 8 h after drug administration for both formulations [Citation75]. Marked inhibition of TxB2 production (A) and mean inhibition of urinary 11-dehydro-TxB2 excretion (B) were maintained during a 24-h period after administration of single-dose ER-ASA 162.5 mg and single-dose IR-ASA 81 mg [Citation75]. In addition, arachidonic acid-induced platelet aggregation 24 h postdose was reduced by 28% with single-dose ER-ASA 162.5 mg and 60% with single-dose IR-ASA 81 mg [Citation75]. It should be noted that this study did not assess steady-state pharmacodynamic effects, and time to reach maximum PD effects was not evaluated. In a comprehensive pharmacodynamic study in 40 patients with Type II diabetes and multiple CVD risk factors (NCT02370680), 162.5 mg ER-ASA (NHP-554C) provided sustained antiplatelet effects over 24 h following 10–14 days of maintenance treatment [data on file].
• Safety & tolerability of ER-ASA
Published data on the long-term safety and tolerability of ER-ASA is relatively limited. However, there is extensive long-term safety and tolerability data available for IR-ASA [Citation76], and no additional safety concerns are anticipated with ER-ASA. Systemic exposure to ASA is limited with ER-ASA; therefore, prostacyclin production is spared () [flamel technologies, venissieux, france, data on file]. Based on data from two clinical studies (one in high-risk patients with a history of atherosclerotic disease and one in healthy volunteers), ER-ASA and IR-ASA appear to have similar safety profiles [Citation77,Citation78]. No tolerability issues or safety concerns were noted in patients with atherosclerosis who were switched from a previous low-dose ASA regimen to 162.5 mg ER-ASA or 150 mg or 75 mg IR-ASA for 28 days [Citation77]. Two of 104 patients (2%) who were enrolled and completed the run-in period of the study were discontinued from the study: one patient in the ER-ASA 162.5 mg group because of nausea and one patient in the IR-ASA 75 mg group because of loin pain [Citation77]. In a randomized, double-blind, crossover study in healthy volunteers, endoscopically assessed erosions and petechiae were significantly lower with single-dose ER-ASA versus enteric-coated ASA 75 mg in healthy volunteers (p < 0.001 for both) [Citation78].
• Current status
A new drug application for ER-ASA was submitted to the FDA following completion of the clinical development program. The indications proposed for ER-ASA include:
Reduction of the combined risk of death and nonfatal stroke in patients who have had ischemic stroke or transient ischemia of the brain due to fibrin platelet emboli.
Reduction of the combined risk of death and nonfatal myocardial infarction (MI) in patients with a previous MI or unstable angina pectoris.
Reduction of the combined risk of MI and sudden death in patients with chronic stable angina pectoris.
For use in patients who have undergone revascularization procedures (i.e., coronary artery bypass graft, percutaneous transluminal coronary angioplasty, or carotid endarterectomy) when there is a preexisting condition for which ASA is already indicated.
Because of the altered pharmacokinetics of ER-ASA, it should not be given in situations where a rapid onset of action is necessary (e.g., acute treatment of MI).
Conclusion
CVD is a serious health concern faced by millions of patients. ASA is a mainstay therapy for secondary prevention of CV events, yet up to 60% of patients experience inadequate preventative coverage (i.e., display laboratory testing resistance to ASA and remain at increased risk of a CV event), especially those at high risk for experiencing a second CV event [Citation9,Citation40–42]. This inadequate coverage is at least partially related to biologic factors (e.g., increased platelet turnover), which may attenuate the anti-thrombotic efficacy of low-dose ASA over a 24-h time period [Citation60–61,Citation70]. The pharmacokinetic profile of ER-ASA indicates a longer window of time during which platelet inhibition may occur. Antiplatelet effects (e.g., reduction of TxA2 production) are consistent with the extended pharmacokinetics of ER-ASA, thus ER-ASA provides a unique formulation of ASA that may potentially overcome some of the limitations of IR-ASA by providing 24-h coverage. The potential benefit of the antiplatelet effects provided by ER-ASA has yet to be evaluated in a large-scale clinical outcomes trial.
Future perspective
IR-ASA is the bedrock of antiplatelet therapy for CV risk prevention and has been for the last four decades [Citation12], but the optimal dose and duration of aspirin therapy in high-risk patients are still elusive. It is not surprising that clinicians have been somewhat less than diligent in recommending and/or monitoring IR-ASA for secondary prevention. Thus far, ASA has had a long history of use with little advancement in delivery formulation, and clinicians have had little ability to assess antiplatelet response to aspirin or monitor adherence with IR-ASA regimens. With increased prevalence of both diabetes and obesity [Citation79], treating these patients with optimal doses is very challenging in the absence of any proven pharmacodynamic or clinical benefits. Patients with CVD have multiple risk factors that amplify CV risk, such as vasculopathy (e.g., endothelial dysfunction) [Citation80], higher-than-normal platelet turnover [Citation46] and high on-therapy platelet reactivity [Citation60,Citation61]. Until now, these phenomena were rarely taken into account when considering optimal treatment regimens. As our knowledge regarding the pathophysiology of CVD increases, the overall approach to management of CVD – and IR-ASA’s place within that scheme – is likely to change. The proposed strategies include increasing IR-ASA dose or use of twice-daily IR-ASA dosing in high-risk patients to achieve improved antiplatelet benefits. The position of an ER-ASA formulation in future CVD treatment regimens is under investigation. Recent demonstrations of ER-ASA prolonging plasma ASA concentrations and reports of the 24-h antiplatelet effects provided by this formulation [Citation75] are promising. Treatment with ER-ASA based on assessment of antiplatelet responsiveness may facilitate ‘guided’ strategies in the near future.
Mechanism of acetylsalicylic acid cardioprotection
Acetylsalicylic acid (ASA) prevents cardiovascular thrombus formation by reducing platelet production of thromboxane A2 (TxA2), a vasoconstrictor and platelet activator.
ASA irreversibly inactivates COX-1, the main enzyme responsible for TxA2 production, in platelets as they travel through portal circulation.
Limitations of immediate-release ASA
Immediate-release (IR) ASA has a short half-life of approximately 15–20 min, which provides a limited window of time during which platelet COX-1 inactivation can occur.
Although ASA inactivates platelet COX-1 enzymes for the entire life span of the platelet, platelets are constantly regenerated (∼4 billion new platelets per hour); therefore, ASA levels throughout a 24-h period maintain consistent COX-1 inhibition, especially in patients with higher than normal platelet turnover (e.g., patients with diabetes).
Extended-release ASA formulation
Extended-release (ER) ASA was formulated to provide 24-h ASA exposure with once-daily dosing.
Each ER-ASA capsule contains an inner core of individually coated microcapsules containing 165.2 mg ASA.
The proprietary release rate-limiting film coating of the microcapsules acts as a semipermeable membrane to allow 24-h distribution of ASA into the GI tract.
Pharmacokinetics & pharmacodynamics of ER-ASA
Consistent with its ER design, ER-ASA provides ASA and salicylic acid over a longer time period than IR-ASA.
Maximal plasma concentration of ASA after a single oral dose was lower with ER-ASA than IR-ASA, supporting reduced systemic exposure.
Platelet aggregation and inhibition of COX-1 activity (as measured by thromboxane B2 production) was maintained throughout a 24-h period after a single dose of ER-ASA.
ER-ASA safety
The long-term safety of ASA is well established, and no additional safety concerns are anticipated with ER-ASA.
Conclusion
ER-ASA is a unique formulation of ASA that was developed to deliver low-dose aspirin throughout the 24-h dosing interval for platelet inhibition.
Financial & competing interests disclosure
J Patrick and AT Pennell are supported by and are employees of New Haven Pharmaceuticals, Inc. PA Gurbel reports serving as a consultant for Daiichi Sankyo/Lilly, Bayer, AstraZeneca, New Haven Pharmaceuticals, Inc., Accumetrics, Merck, Medtronic, Janssen, CSL, and Haemonetics; receiving grants from the NIH, Daiichi Sankyo, Lilly, CSL, AstraZeneca, Haemonetics, Harvard Clinical Research Institute, and Duke Clinical Research Institute; receiving payment for lectures, including service on speakers’ bureaus, from Lilly, Daiichi Sankyo, and Merck; and receiving payment for development of educational presentations from Merck, the Discovery Channel, and Pri-Med. PA Gurbel also is holding stock or stock options in Merck, Medtronic, and Pfizer; and holding patents in the area of personalized antiplatelet therapy and interventional cardiology. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing assistance was utilized in the production of this manuscript. Technical editorial and medical writing support, under direction of the authors, was provided by Mary Beth Moncrief, PhD, and Jillian Gee, PhD, Synchrony Medical Communications, LLC, West Chester, PA, with support from New Haven Pharmaceuticals, Inc.
Additional information
Funding
References
- Mozaffarian D , BenjaminEJ, GoASet al. Heart disease and stroke statistics-2015 update: a report from the American Heart Association . Circulation131 ( 4 ), e29 – e322 ( 2015 ).
- Centers for Disease Control and Prevention . Heart disease and stroke prevention address the nation’s leading killers ( 2011 ). www.cdc.gov/chronicdisease/resources/publications/aag/pdf/2011/heart-disease-and-stroke-aag-2011.pdf .
- Giorda CB , AvogaroA, MagginiMet al. Recurrence of cardiovascular events in patients with Type 2 diabetes: epidemiology and risk factors . Diabetes Care31 ( 11 ), 2154 – 2159 ( 2008 ).
- van der Heijden AA , Van’t RietE, BotSDet al. Risk of a recurrent cardiovascular event in individuals with Type 2 diabetes or intermediate hyperglycemia: the Hoorn Study . Diabetes Care36 ( 11 ), 3498 – 3502 ( 2013 ).
- Haffner SM , LehtoS, RonnemaaT, PyoralaK, LaaksoM . Mortality from coronary heart disease in subjects with Type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction . N. Engl. J. Med.339 ( 4 ), 229 – 234 ( 1998 ).
- Cubbon RM , AbbasA, WheatcroftSBet al. Diabetes mellitus and mortality after acute coronary syndrome as a first or recurrent cardiovascular event . PLoS ONE3 ( 10 ), e3483 ( 2008 ).
- American Diabetes Association . Position statement Section 8: cardiovascular disease and risk management . Diabetes Care38 ( Suppl. ), S49 – S57 ( 2015 ).
- Smith SC Jr , BenjaminEJ, BonowROet al. The World Heart Federation and the Preventive Cardiovascular Nurses Association. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation . Circulation124 ( 22 ), 2458 – 2473 ( 2011 ).
- Gasparyan AY , WatsonT, LipGY . The role of aspirin in cardiovascular prevention: implications of aspirin resistance . J. Am. Coll. Cardiol.51 ( 19 ), 1829 – 1843 ( 2008 ).
- Grove EL . Antiplatelet effect of aspirin in patients with coronary artery disease . Dan. Med. J.59 ( 9 ), B4506 ( 2012 ).
- Vane JR . Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs . Nat. New Biol.231 ( 25 ), 232 – 235 ( 1971 ).
- Tantry US , BlidenKP, GurbelPA . Overestimation of platelet aspirin resistance detection by thrombelastograph platelet mapping and validation by conventional aggregometry using arachidonic acid stimulation . J. Am. Coll. Cardiol.46 ( 9 ), 1705 – 1709 ( 2005 ).
- Freynhofer MK , BrunoV, WojtaJ, HuberK . The role of platelets in athero-thrombotic events . Curr. Pharm. Des.18 ( 33 ), 5197 – 5214 ( 2012 ).
- Gurbel PA , MahlaE, AntoninoMJ, TantryUS . Response variability and the role of platelet function testing . J. Invasive Cardiol.21 ( 4 ), 172 – 178 ( 2009 ).
- Tantry US , MahlaE, GurbelPA . Aspirin resistance . Prog. Cardiovasc. Dis.52 ( 2 ), 141 – 152 ( 2009 ).
- Patrignani P , TacconelliS, PiazueloEet al. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action . J. Thromb. Haemost.12 ( 8 ), 1320 – 1330 ( 2014 ).
- Patrono C , García RodríguezLA, LandolfiR, BaigentC . Low-dose aspirin for the prevention of atherothrombosis . N. Engl. J. Med.353 ( 22 ), 2373 – 2383 ( 2005 ).
- Cox D , MareeAO, DooleyM, ConroyR, ByrneMF, FitzgeraldDJ . Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers . Stroke37 ( 8 ), 2153 – 2158 ( 2006 ).
- Pedersen AK , FitzGeraldGA . Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase . N. Engl. J. Med.311 ( 19 ), 1206 – 1211 ( 1984 ).
- Tang M , MukundanM, YangJet al. Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases, and clopidogrel is transesterificated in the presence of ethyl alcohol . J. Pharmacol. Exp. Ther.319 ( 3 ), 1467 – 1476 ( 2006 ).
- Patrono C , CiabattoniG, PatrignaniPet al. Clinical pharmacology of platelet cyclooxygenase inhibition . Circulation72 ( 6 ), 1177 – 1184 ( 1985 ).
- Behnke O , ForerA . From megakaryocytes to platelets: platelet morphogenesis takes place in the bloodstream . Eur. J. Haematol. Suppl.61, 3 – 23 ( 1998 ).
- Eikelboom JW , HirshJ, SpencerFA, BaglinTP, WeitzJI . Antiplatelet drugs: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines . Chest141 ( Suppl. 2 ), Se89 – Se119S ( 2012 ).
- Burch JW , StanfordN, MajerusPW . Inhibition of platelet prostaglandin synthetase by oral aspirin . J. Clin. Invest.61 ( 2 ), 314 – 319 ( 1978 ).
- Grove EL , HvasAM, MortensenSB, LarsenSB, KristensenSD . Effect of platelet turnover on whole blood platelet aggregation in patients with coronary artery disease . J. Thromb. Haemost.9 ( 1 ), 185 – 191 ( 2011 ).
- Patrignani P , FilabozziP, PatronoC . Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects . J. Clin. Invest.69 ( 6 ), 1366 – 1372 ( 1982 ).
- Grove EL , HvasAM, KristensenSD . Immature platelets in patients with acute coronary syndromes . Thromb. Haemost.101 ( 1 ), 151 – 156 ( 2009 ).
- Vaduganathan M , AlviarCL, ArikanMEet al. Platelet reactivity and response to aspirin in subjects with the metabolic syndrome . Am. Heart J.156 ( 5 ), 1002 – 1002 ( 2008 ).
- Mijovic R , KovacevicN, ZarkovM, StosicZ, CabarkapaV, MiticG . Reticulated platelets and antiplatelet therapy response in diabetic patients . J. Thromb. Thrombolysis40 ( 2 ), 203 – 210 ( 2015 ).
- Arazi HC , DoinyDG, TorciviaRSet al. Impaired anti-platelet effect of aspirin, inflammation and platelet turnover in cardiac surgery . Interact. Cardiovasc. Thorac. Surg.10 ( 6 ), 863 – 867 ( 2010 ).
- Baigent C , BlackwellL, CollinsRet al. Antithrombotic Trialists’ (ATT) Collaboration. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials . Lancet373 ( 9678 ), 1849 – 1860 ( 2009 ).
- Antithrombotic Trialists’ Collaboration . Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients . BMJ324 ( 7329 ), 71 – 86 ( 2002 ).
- Simpson SH , GambleJM, MereuL, ChambersT . Effect of aspirin dose on mortality and cardiovascular events in people with diabetes: a meta-analysis . J. Gen. Intern. Med.26 ( 11 ), 1336 – 1344 ( 2011 ).
- Berger JS , SallumRH, KatonaBet al. Is there an association between aspirin dosing and cardiac and bleeding events after treatment of acute coronary syndrome? A systematic review of the literature . Am. Heart J.164 ( 2 ), 153 – 162 ( 2012 ).
- Becker RC , MeadeTW, BergerPBet al. The primary and secondary prevention of coronary artery disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition) . Chest133 ( Suppl. 6 ), S776 – S814 ( 2008 ).
- Gurbel P , TantryUS . Monitoring of antiplatelet therapy . In : Platelets . MichelsonAD ( Eds ), Elsevier, Waltham, MA, USA, 602 – 612 ( 2013 ).
- Gurbel PA , BlidenKP, DiChiaraJet al. Evaluation of dose-related effects of aspirin on platelet function: results from the Aspirin-Induced Platelet Effect (ASPECT) study . Circulation115 ( 25 ), 3156 – 3164 ( 2007 ).
- Tantry US , GesheffM, LiuF, BlidenKP, GurbelPA . Resistance to antiplatelet drugs: what progress has been made?Expert. Opin. Pharmacother.15 ( 17 ), 2553 – 2564 ( 2014 ).
- Hanley AJ , KarterAJ, WilliamsKet al. Prediction of Type 2 diabetes mellitus with alternative definitions of the metabolic syndrome: the Insulin Resistance Atherosclerosis Study . Circulation112 ( 24 ), 3713 – 3721 ( 2005 ).
- Tasdemir E , ToptasT, DemirC, EsenR, AtmacaM . Aspirin resistance in patients with Type II diabetes mellitus . Ups. J. Med. Sci.119 ( 1 ), 25 – 31 ( 2014 ).
- Cetin M , KiziltuncE, CetinZGet al. Acetylsalicylic acid resistance in patients with Type 2 diabetes mellitus, prediabetes & non-diabetic coronary artery disease . Pak. J. Med. Sci.30 ( 3 ), 539 – 544 ( 2014 ).
- Hovens MM , SnoepJD, EikenboomJC, van der BomJG, MertensBJ, HuismanMV . Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review . Am. Heart J.153 ( 2 ), 175 – 181 ( 2007 ).
- Rocca B , PetrucciG . Variability in the responsiveness to low-dose aspirin: pharmacological and disease-related mechanisms . Thrombosis2012, 376721 ( 2012 ).
- Fuster V , ChesebroJH, FryeRL, ElvebackLR . Platelet survival and the development of coronary artery disease in the young adult: effects of cigarette smoking, strong family history and medical therapy . Circulation63 ( 3 ), 546 – 551 ( 1981 ).
- Winocour PD , LaiminsM, ColwellJA . Platelet survival in streptozotocin-induced diabetic rats . Thromb. Haemost.51 ( 3 ), 307 – 312 ( 1984 ).
- Larsen SB , GroveEL, HvasAM, KristensenSD . Platelet turnover in stable coronary artery disease – influence of thrombopoietin and low-grade inflammation . PLoS ONE9 ( 1 ), e85566 ( 2014 ).
- Gum PA , Kottke-MarchantK, PoggioEDet al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease . Am. J. Cardiol.88 ( 3 ), 230 – 235 ( 2001 ).
- Salama MM , MoradAR, SalehMA, SabriNA, ZakiMM, ElSafadyLA . Resistance to low-dose aspirin therapy among patients with acute coronary syndrome in relation to associated risk factors . J. Clin. Pharm. Ther.37 ( 6 ), 630 – 636 ( 2012 ).
- Bednar F , OsmancikP, HlavickaJ, JedlickovaV, PaluchZ, VanekT . Aspirin is insufficient in inhibition of platelet aggregation and thromboxane formation early after coronary artery bypass surgery . J. Thromb. Thrombolysis27 ( 4 ), 394 – 399 ( 2009 ).
- Kammerer I , BachJ, SaggauW, IsgroF . Functional evaluation of platelet aspirin resistance after on-pump coronary bypass grafting using multiple aggregation tests . Thorac. Cardiovasc. Surg.59 ( 7 ), 425 – 429 ( 2011 ).
- Labuz-Roszak B , PierzchalaK, TyrpienK . Resistance to acetylsalicylic acid in patients with Type 2 diabetes mellitus is associated with lipid disorders and history of current smoking . J. Endocrinol. Invest.37 ( 4 ), 331 – 338 ( 2014 ).
- Smith JP , HaddadEV, TaylorMBet al. Suboptimal inhibition of platelet cyclooxygenase-1 by aspirin in metabolic syndrome . Hypertension59 ( 3 ), 719 – 725 ( 2012 ).
- Fateh-Moghadam S , PlockingerU, CabezaNet al. Prevalence of aspirin resistance in patients with Type 2 diabetes . Acta Diabetol.42 ( 2 ), 99 – 103 ( 2005 ).
- Zimmermann N , WenkA, KimUet al. Functional and biochemical evaluation of platelet aspirin resistance after coronary artery bypass surgery . Circulation108 ( 5 ), 542 – 547 ( 2003 ).
- Zaccardi F , RoccaB, PitoccoD, TaneseL, RizziA, GhirlandaG . Platelet mean volume, distribution width, and count in Type 2 diabetes, impaired fasting glucose, and metabolic syndrome: a meta-analysis . Diabetes Metab. Res. Rev.31 ( 4 ), 402 – 410 ( 2015 ).
- Lee EY , KimSJ, SongYJ, ChoiSJ, SongJ . Immature platelet fraction in diabetes mellitus and metabolic syndrome . Thromb. Res.132 ( 6 ), 692 – 695 ( 2013 ).
- Ibrahim H , SchuttRC, HannawiB, DeLaoT, BarkerCM, KleimanNS . Association of immature platelets with adverse cardiovascular outcomes . J. Am. Coll. Cardiol.64 ( 20 ), 2122 – 2129 ( 2014 ).
- Cesari F , MarcucciR, GoriAMet al. Reticulated platelets predict cardiovascular death in acute coronary syndrome patients. Insights from the AMI-Florence 2 Study . Thromb. Haemost.109 ( 5 ), 846 – 853 ( 2013 ).
- Slavka G , PerkmannT, HaslacherHet al. Mean platelet volume may represent a predictive parameter for overall vascular mortality and ischemic heart disease . Arterioscler. Thromb. Vasc. Biol.31 ( 5 ), 1215 – 1218 ( 2011 ).
- Henry P , VermilletA, BovalBet al. 24-hour time-dependent aspirin efficacy in patients with stable coronary artery disease . Thromb. Haemost.105 ( 2 ), 336 – 344 ( 2011 ).
- Würtz M , HvasAM, JensenLOet al. 24-hour antiplatelet effect of aspirin in patients with previous definite stent thrombosis . Int. J. Cardiol.175 ( 2 ), 274 – 279 ( 2014 ).
- DiChiara J , BlidenKP, TantryUSet al. The effect of aspirin dosing on platelet function in diabetic and nondiabetic patients: an analysis from the aspirin-induced platelet effect (ASPECT) study . Diabetes56 ( 12 ), 3014 – 3019 ( 2007 ).
- Capodanno D , PatelA, DharmashankarKet al. Pharmacodynamic effects of different aspirin dosing regimens in Type 2 diabetes mellitus patients with coronary artery disease . Circ. Cardiovasc. Interv.4 ( 2 ), 180 – 187 ( 2011 ).
- Dusse LM , FreitasLG . Clinical applicability of reticulated platelets . Clin. Chim. Acta439, 143 – 147 ( 2015 ).
- Ingram M , CoopersmithA . Reticulated platelets following acute blood loss . Br. J. Haematol.17 ( 3 ), 225 – 229 ( 1969 ).
- Kieffer N , GuichardJ, FarcetJP, VainchenkerW, Breton-GoriusJ . Biosynthesis of major platelet proteins in human blood platelets . Eur. J. Biochem.164 ( 1 ), 189 – 195 ( 1987 ).
- Guthikonda S , LevEI, PatelRet al. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin . J. Thromb. Haemost.5 ( 3 ), 490 – 496 ( 2007 ).
- Mangalpally KKR , Siqueiros-GarciaA, VaduganathanM, DongJF, KleimanNS, GuthikondaS . Platelet activation patterns in platelet size sub-populations: differential responses to aspirin in vitro . J. Thromb. Thrombolysis30 ( 3 ), 251 – 262 ( 2010 ).
- Würtz M , GroveEL, WulffLNet al. Patients with previous definite stent thrombosis have a reduced antiplatelet effect of aspirin and a larger fraction of immature platelets . JACC. Cardiovasc. Interv.3 ( 8 ), 828 – 835 ( 2010 ).
- Perneby C , WallenNH, RooneyC, FitzgeraldD, HjemdahlP . Dose- and time-dependent antiplatelet effects of aspirin . Thromb. Haemost.95 ( 4 ), 652 – 658 ( 2006 ).
- Ridker PM , MansonJE, BuringJE, MullerJE, HennekensCH . Circadian variation of acute myocardial infarction and the effect of low-dose aspirin in a randomized trial of physicians . Circulation82 ( 3 ), 897 – 902 ( 1990 ).
- Muller JE , ToflerGH, StonePH . Circadian variation and triggers of onset of acute cardiovascular disease . Circulation79 ( 4 ), 733 – 743 ( 1989 ).
- Naderi SH , BestwickJP, WaldDS . Adherence to drugs that prevent cardiovascular disease: meta-analysis on 376,162 patients . Am. J. Med.125 ( 9 ), 882 – 887 ( 2012 ).
- Laliberté F , BookhartBK, NelsonWWet al. Impact of once-daily versus twice-daily dosing frequency on adherence to chronic medications among patients with venous thromboembolism . Patient6 ( 3 ), 213 – 224 ( 2013 ).
- Patrick J , DillahaL, ArmasD, SessaW . A randomized trial to assess the pharmacodynamics and pharmacokinetics of a single dose of an extended-release aspirin formulation . Postgrad. Med.127 ( 6 ), 573 – 580 ( 2015 ).
- McQuaid KR , LaineL . Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials . Am. J. Med.119 ( 8 ), 624 – 638 ( 2006 ).
- Brown N , MayJA, WilcoxRGet al. Comparison of antiplatelet activity of microencapsulated aspirin 162.5 mg (Caspac XL), with enteric coated aspirin 75 mg and 150 mg in patients with atherosclerosis . Br. J. Clin. Pharmacol.48 ( 1 ), 57 – 62 ( 1999 ).
- Donnelly MT , StackWA, RichardsonPet al. Microencapsulated aspirin, Asacard, reduces endoscopic damage in healthy volunteers compared with conventional encapsulated aspirin [abstract G0440] . Gastroenterology114, A107 ( 1998 ).
- De Feo P , BorisJM, MaffeisC . Lifestyle modification strategies to counteract the world epidemic growth of obesity and diabetes . Biomed. Res. Int.2014, 640409 ( 2014 ).
- Widmer RJ , LermanA . Endothelial dysfunction and cardiovascular disease . Glob. Cardiol. Sci. Pract.2014 ( 3 ), 291 – 308 ( 2014 ).