
Abstract
GEN-1 is a gene-based immunotherapy, comprising a human IL-12 gene expression plasmid and a synthetic plasmid delivery system, delivered intraperitoneally (ip.) to produce local and persistent levels of a pleiotropic immunocytokine, IL-12, at the tumor site in patients with advanced ovarian cancer. The goal of local and persistent IL-12 delivery is to remodel the highly immunosuppressive tumor microenvironment to favor immune stimulation while avoiding serious systemic toxicities, a major limitation of recombinant IL-12 therapy. Safe and sustained local production of IL-12 and related immunocytokines at the tumor site could produce potentially more favorable immunological changes in the tumor microenvironment and antitumor responses than a bolus systemic delivery of recombinant IL-12. Treatment safety, clinical benefits and biological activity of GEN-1 ip. in patients with ovarian cancer and in representative animal models are described.
Ovarian cancer unmet need
Ovarian cancer is the fifth leading cause of cancer deaths among women in the USA, with over 14,000 deaths expected in 2018 [Citation1]. The 5-year relative survival for localized ovarian cancer is high, at 93%, but only 15% of patients are diagnosed with localized disease [Citation1]. The remaining patients are diagnosed with either regional or distant disease (advanced ovarian cancer), where 5-year relative survival rates drop to 73% and 29%, respectively [Citation1]. Of 7000 women with advanced ovarian cancer enrolled in the California Cancer Registry, only 19% survived at least 10 years [Citation2]. Therefore, there is an urgent need for more effective ovarian cancer therapies producing durable responses and prolonged survival for women with advanced disease.
The management of advanced ovarian cancer (stages III–IV) has primarily consisted of cytoreductive surgery followed by adjuvant chemotherapy, but in patients considered poor candidates for surgery or those unlikely to have all visible disease surgically removed (R0), surgery may be preceded by neoadjuvant chemotherapy to improve the chances of successful resection [Citation3]. In two Phase III randomized controlled neoadjuvant chemotherapy trials in stage III or IV epithelial ovarian cancer, the median progression-free survival (PFS; defined as the length of time from assignment in a clinical trial to disease progression or death from any cause) was only 12 months and median overall survival (OS) was 24–30 months [Citation4,Citation5]. Despite some improvement in front-line treatment options and response rates up to 80% to platinum-based chemotherapy, most patients will subsequently recur [Citation6], primarily in the peritoneal cavity [Citation7]. This underscores the need for treatment modalities that can control local disease progression in the peritoneal cavity [Citation2].
Immunogenicity & the tumor microenvironment
Clinical research suggests that ovarian cancer is an immunogenic malignancy, able to provoke a response from an individual’s immune system. Spontaneous antitumor responses driven by T-cell lymphocytes or antibodies have been reported in patients with ovarian cancer [Citation8]. Cytotoxic T cells have been isolated from primary tumor, ascites and blood [Citation9]. The presence of CD8+ tumor-infiltrating lymphocytes (TILs) has been linked to better prognosis in patients with late-stage ovarian cancer. For instance, a study of 186 patients with ovarian cancer who responded to chemotherapy showed that the presence or absence of TILs was independently correlated with OS (median OS: 50.3 months for TIL+ tumors vs 18.0 months for TIL– tumors; p < 0.001) [Citation9]. A subsequent meta-analysis confirmed the prognostic significance of these TILs [Citation10]. Another group of T cells, called immunosuppressive Tregs, also associated, albeit negatively, with survival in ovarian cancer [Citation11,Citation12]. Tregs limit antitumor immunity through secretion of nonspecific, inhibitory immunocytokines like IL-10 and TGF-β [Citation13].
These immune cells and immunocytokines are present within the tumor microenvironment, part of a dynamic milieu of both cellular and acellular components, including tumor blood vessels, stromal cells, immune infiltrates and soluble factors (). This environment can be immunosuppressive favoring tumor growth, or immunoactive favoring tumor control by the immune system. The balance of the tumor microenvironment can be shifted between these two opposing scenarios through the secretion of specific immunocytokines and chemokines, which either promote or inhibit tumor survival and proliferation by modulating the immune cell populations in the tumor microenvironment [Citation14]. For this reason, in addition to other forms of immunotherapy, immunocytokine therapy has been an active area of research interest in ovarian cancer.
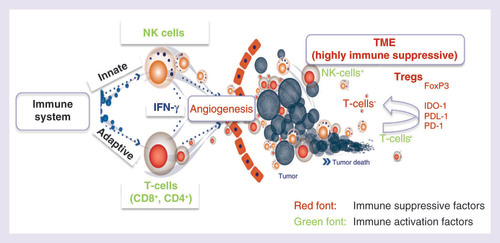
The peritoneal cavity from patients with advanced ovarian cancer represents the primary tumor environment, and is an attractive target for immune modulation. Ascites comprises a variety of malignant and nonmalignant cells, including immune cells [Citation15]. Many immune cells increase in number and exhibit cytolytic activity against tumor cells after stimulation with certain cytokines [Citation15,Citation16].
IL-12 & its activity in ovarian cancer
IL-12 is well characterized as a pleiotropic cytokine and is one of the most potent inducers of antitumor immunity, with multiple action mechanisms including activation of natural killer cells; maturation, proliferation and activation of cytotoxic CD8+ T cells; potentiation of antibody-dependent cell-mediated cytotoxicity; and interference with Treg differentiation [Citation17,Citation18]. In addition to its effects on the immune system, IL-12 also exerts antiangiogenic effects through IFN-γ-inducible protein 10 [Citation19]. Angiostatin, a potent endogenous antiangiogenic agent, has been reported to produce antiangiogenesis in tumors via IL-12 production [Citation20]. Most IL-12 functions are believed to be due to its ability to induce cells to secrete IFN-γ [Citation18]. Production of other immunostimulatory cytokines, such as granulocyte–macrophage colony-stimulating factor and TNF-α, from effector immune cells has also been reported in IL-12 action [Citation18]. Due to the observed stimulatory effect of IL-12 on innate and acquired immune systems and the inhibitory effect on both the immunosuppressive Treg pathway and angiogenesis, IL-12-based therapy is considered a suitable approach for tumor inhibition.
The antitumor and immunological activities of IL-12 and other immunocytokines, such as IFN-α, IFN-γ and IL-2, have been examined in numerous preclinical cancer models [Citation18] and in human cancers including ovarian cancer [Citation21–26], and recombinant human IL-12 (rhIL-12) administered by intravenous (iv.) or subcutaneous (sc.) injection has produced favorable immunological and clinical responses in patients with various malignancies [Citation27–31]. However, treatment-related grade 3 or higher hematological and liver toxicities have been observed following rhIL-12 treatment, attributable to elevated blood IL-12 levels. Progressive desensitization to subsequent rhIL-12 iv. treatment has also been reported in patients with cancer [Citation27]. In patients with peritoneal carcinomatosis of ovarian and gastrointestinal malignancies, rhIL-12 was administered locally by intraperitoneal (ip.) infusion in an attempt to reduce the serious toxicities observed with systemic administration. High doses of rhIL-12 ip. produced dose-limiting toxicity (DLT) in two of four patients [Citation32], prompting a reduction in dose for Phase II testing. Unfortunately, although no DLTs were observed in the Phase II trial at the reduced dose, neither were any objective responses observed [Citation33]. This was likely partially explained by the dosing limitations caused by DLT development. Because of serious DLTs, rhIL-12 therapy has not advanced to approval despite aggressive clinical testing. There is a need for developing alternative IL-12 delivery methods, which can maintain therapeutic IL-12 concentrations in the tumor environment when administered repeatedly without resulting in systemic toxicity or response desensitization.
For optimal efficacy and safety, anticancer cytokine therapies should be present at persistent, therapeutic and nontoxic concentrations. This pharmacokinetic profile is not achievable with rhIL-12 protein. Some alternative approaches to rhIL-12 involve administration of the IL-12 gene via a viral vector, electroporation or implantation of IL-12 gene-modified cells [Citation34–37]. These approaches also have limitations, including the generation of neutralizing antibodies to the viral vector and insertional mutagenesis from chromosomal integration of viral sequences, limitation of electroporation method to solid tissues and local inflammation from the use of an electric current, and a lack of simplicity and versatility of cell-based therapies.
GEN-1 concept & drug design
In order to overcome the shortcomings of rhIL-12 administration, IL-12 should be delivered in such a manner that it facilitates immune stimulation by providing persistent protein levels locally at the tumor site while maintaining a favorable safety profile. GEN-1 has been developed to accomplish this; it is a gene-based IL-12 immunotherapy designed to pleiotropically promote immune stimulation, but to do so in a local and persistent manner, which has not been achieved with previous approaches to local or systemic rhIL-12 administration.
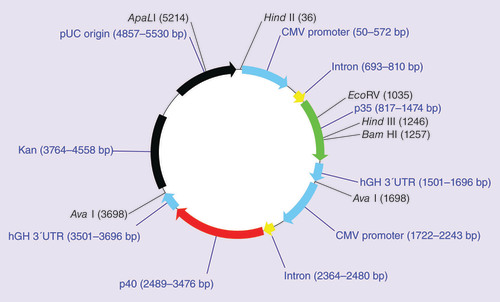
GEN-1 comprises a plasmid vector encoding the p35 and p40 subunits of human IL-12 genes, each under the control of a cytomegalovirus promotor (), and a synthetic lipopolymer delivery system, polyethylene glycol (PEG)-polyethylenimine (PEI)-cholesterol (PPC). The IL-12 plasmid is formulated with PPC under controlled conditions to produce well-defined GEN-1 nanoparticles suitable for gene transfer into mammalian cells. The GEN-1 nanoparticles exhibit several features important for safe and efficient delivery of IL-12 gene. Specifically, the GEN-1 nanoparticles prevent DNA degradation by DNAses [Citation38] and hence improve plasmid availability for cellular uptake. The presence of cholesterol is designed to facilitate uptake by cellular membranes [Citation39], and PEG to improve in vivo stability [Citation40]. The presence of PEI is for condensation of DNA into nanoparticles and escape of nanoparticles from endosomes [Citation41]. GEN-1 is designed to produce IL-12 levels at the administration site for several days after a single injection, making ip. (or intracavitary) injection the preferred delivery method for two reasons. First, local IL-12 delivery may be associated with fewer systemic adverse events (AEs) compared with systemic administration. Second, when IL-12 is administered sc. or iv. as recombinant protein to patients, it has a half-life of only 5–12 h; [Citation28] therefore, a persistent supply of IL-12, such as that produced by localized GEN-1 injection, may improve its therapeutic potential.
GEN-1-proposed mechanisms of action
Since GEN-1 is designed to produce IL-12, its mechanism of action is anticipated to be consistent with IL-12 pharmacology. The evidence of IL-12 production in the GEN-1 mechanism of action has been demonstrated in clinical and preclinical studies [Citation25,Citation38,Citation42,Citation43], demonstrating that the IL-12 plasmid delivered by GEN-1 nanoparticles is transcriptionally active. The plasmid specificity and biological activity of IL-12 from pmIL-12/PPC nanoparticles were verified both in vitro and in vivo by direct comparison with a non-IL-12 coding plasmid formulated into nanoparticles with PPC delivery polymer. The biological activity of IL-12 produced from rhIL-12/PPC transfected cells was verified by IFN-γ production by IL-12-conditioned media in natural killer-92 cells [fewell j et al. unpublished data (2004)]. The increases in IFN-γ and TNF-α show IL-12 produced from gene transfer is biologically active and promotes the downstream immunostimulatory pathways. Additional evidence of immune activation in GEN-1 action comes from changes in immune cell populations in tumor tissue and ascites following GEN-1 ip. administration in combination with neoadjuvant chemotherapy. A shift in immune cell populations favoring immune stimulation over immune suppression was observed in an ongoing study in patients with newly diagnosed ovarian cancer [Citation44].
An important aspect of GEN-1 mechanisms of action is GEN-1’s ability to produce local and durable increases in IL-12 and its downstream cytokines. The ip. administration of pmIL-12/PPC nanoparticles produced significant increases in IL-12 and IFN-γ, which remained above pretreatment levels for approximately 1 week [Citation42], which formed the basis for weekly GEN-1 dosing in human clinical trials. This type of pharmacokinetic profile, in which the increase in IL-12 is local and durable, and is generated from within the body, is anticipated to be different and more effective compared with a bolus infusion of short-lived exogenously administered rhIL-12 protein therapy and may be critical in the remodeling of the tumor microenvironment.
The proposed GEN-1 concept has been investigated in multiple animal and human studies described in this review article. The preclinical studies have been conducted in a mouse model of disseminated ID8 [Citation45] ovarian cancer, and since human IL-12 is not active in mice, a murine IL-12 DNA plasmid formulated with the PPC delivery system (pmIL-12/PPC) was used for preclinical studies [Citation42].
GEN -1 pharmacology
Preclinical pharmacological studies were conducted to optimize GEN-1 composition for gene transfer and examine its biological and anticancer activity in animal models. The PPC composition was optimized by evaluating the effect of increasing amounts of PEG in PPC polymer during synthesis on gene transfer activity. The addition of cholesterol and PEG resulted in an increase in luciferase gene transfer activity in tumor tissue [Citation38,Citation39]. The optimal molar ratio between PEG and PEI was 2:1, at which the gene transfer activity was approximately tenfold higher than from PEI cholesterol. The charge ratio between plasmid DNA and PPC polymer was optimized using a luciferase gene transfer assay in COS-1 cells [Citation38]. At PPC:DNA ratio of 11:1, the GEN-1 nanoparticles exhibited maximum gene transfer with low cytotoxicity. In a DNAse protection assay, free DNA was completely degraded within 1 min of incubation while DNA in complexes with PPC remained stable throughout 60 min incubation, demonstrating better DNA stability in complexes with PPC [Citation38].
Numerous in vivo pharmacology studies have been conducted to characterize the activity of pmIL-12/PPC in the ID8 C57BL/6 mouse model of peritoneally disseminated ovarian cancer, stably expressing VEGF [Citation42]. Mice were ip. injected with tumor cells to establish disease [Citation42]. Approximately 32 days after tumor implantation, when mice had carcinomatosis and significant ascites, pmIL-12/PPC was injected ip. Peak mIL-12 expression in ascites was recorded 1 day after injection, with detectable levels present 7 days after injection (). Murine IFN-γ (mIFN-γ) expression was delayed relative to mIL-12 expression, with peak levels occurring at day 3 and levels at 25% of the peak by day 7 () [Citation42]. This is consistent with the known relationship between IL-12 and IFN-γ, in which IL-12 stimulates lymphoid cells to secrete IFN-γ [Citation18]. Both mIL-12 and mIFN-γ were also detected in serum, but levels were approximately tenfold lower than those measured in ascites [Citation42], suggesting mIL-12 expression is primarily contained in the peritoneal cavity. When a second pmIL-12/PPC injection was administered to mice 1 week after the first injection, the expression kinetics of both mIL-12 and mIFN-γ were similar to those after the initial injection [Citation42].
Mice were implanted intraperitoneally with ID8 cells to induce peritoneal carcinomatosis and ascites formation. After 32 days, mice were administered pmIL-12/PPC intraperitoneally at a dose of 250 μg (DNA). Ascites was collected 1, 2, 3, and 7 days post administration, and protein levels were determined by ELISA. Values are expressed as mean ± standard deviation and normalized to total amount of ascites fluid. (A) mIL-12 expression. (B) Murine IFN-γ expression. (C) VEGF expression.
Reproduced with permission from [Citation42] © John Wiley and Sons, Inc. (2009); permission conveyed through Copyright Clearance Center, Inc.
![Figure 3. Protein expression levels after pmIL-12/PPC intraperitoneal administration to mice with disseminated ovarian cancer.Mice were implanted intraperitoneally with ID8 cells to induce peritoneal carcinomatosis and ascites formation. After 32 days, mice were administered pmIL-12/PPC intraperitoneally at a dose of 250 μg (DNA). Ascites was collected 1, 2, 3, and 7 days post administration, and protein levels were determined by ELISA. Values are expressed as mean ± standard deviation and normalized to total amount of ascites fluid. (A) mIL-12 expression. (B) Murine IFN-γ expression. (C) VEGF expression.Reproduced with permission from [Citation42] © John Wiley and Sons, Inc. (2009); permission conveyed through Copyright Clearance Center, Inc.](/cms/asset/6a96f28d-6435-49ab-933d-d67708c462a0/ifon_a_12332476_f0003.jpg)
Baseline VEGF levels are artificially high in the ascites of this mouse model of ovarian cancer because the ID8 cell line has been engineered to constitutively express VEGF to promote aggressive tumor growth [Citation42]. Nonetheless, pmIL-12/PPC injection in these mice caused a greater than fivefold reduction in VEGF levels observed 1 day after injection and lasting at least 3 days () [Citation42].
In addition to testing protein expression level in ascites and serum in this mouse model, a PCR array of 84 genes involved in immune activation was performed on both ascites and tumor nodules collected 1 day after pmIL-12/PPC injection [Citation42]. Overall, a shift toward a Th1 immune response was noted in both ascites and tumor tissue, consistent with IL-12’s role as a key cytokine responsible for inducing a Th1 response [Citation46].
To examine the efficacy of pmIL-12/PPC in mice with disseminated ovarian cancer, mice were injected with tumor cells to establish advanced disease [Citation42]. A total of four weekly ip. injections of pmIL-12/PPC at escalating doses (10, 25, 50, 100 and 250 μg cohorts) were initiated 18 days after tumor implant. Experiments have been performed previously that have specifically examined the relationship between tumor burden and ascites formation. It has been shown in both pmIL-12/PPC-treated and untreated animals, a strong correlation exists between tumor burden and accumulated ascites and that the accumulation of ascites and resulting weight gain is a useful surrogate marker of disease progression in this animal model [Citation42]. Even in the 10 μg cohort, animals weighed less than untreated tumor-bearing mice (p < 0.05; ). Mean animal weights from the 50 and 250 μg cohorts were significantly less than that of the 10 μg cohort (p < 0.05) and indistinguishable from the mean weight of naive (nontumor bearing) mice, suggesting pmIL-12/PPC administration had a dose-dependent positive impact on disease progression. In support of this, demonstrates that animals in the 50 and 250 μg cohorts had a 53% increase in median survival compared with the untreated tumor-bearing mice.
Animals were given 4 weekly pmIL-12/PPC intraperitoneal administrations starting 20 days after tumor implant. Dosages were adjusted by changes in DNA concentration of the formulation using a fixed injection volume. (A) Comparison of animal weights at 42 days after tumor implant. Values are expressed as mean ± standard deviation with n = 5 for each point. (B) Kaplan–Meier survival curves for various treatment groups (n = 5 for each group).
Reproduced with permission from [Citation42] © John Wiley and Sons, Inc. (2009); permission conveyed through Copyright Clearance Center, Inc.
![Figure 4. Efficacy outcomes of untreated tumor-bearing mice versus pmIL-12/PPC-treated mice.Animals were given 4 weekly pmIL-12/PPC intraperitoneal administrations starting 20 days after tumor implant. Dosages were adjusted by changes in DNA concentration of the formulation using a fixed injection volume. (A) Comparison of animal weights at 42 days after tumor implant. Values are expressed as mean ± standard deviation with n = 5 for each point. (B) Kaplan–Meier survival curves for various treatment groups (n = 5 for each group).Reproduced with permission from [Citation42] © John Wiley and Sons, Inc. (2009); permission conveyed through Copyright Clearance Center, Inc.](/cms/asset/c86f7824-b773-47bd-8877-efef542acacc/ifon_a_12332476_f0004.jpg)
In a subcutaneously implanted mouse colorectal model, pmIL-12/PPC administration at doses that caused disappearance of tumor on one flank also provided protection against tumor rechallenge on the other flank (data not shown), suggesting that these mice had developed systemic immunity against the tumor following local pmIL-12/PPC treatment, such as might occur with the presence of tumor-specific memory T cells [Citation18].
The efficacy of pmIL-12/PPC in combination with a clinically relevant chemotherapy treatment regimen was evaluated in a mouse model of disseminated ovarian cancer. Mice that had been administered the ID8 cell line were divided into four treatment cohorts: untreated, carboplatin plus paclitaxel, pmIL-12/PPC and pmIL-12/PPC plus carboplatin plus paclitaxel [Citation42]. Mice receiving pmIL-12/PPC were administered nine weekly 100 μg injections starting 18 days after tumor implant, and mice receiving chemotherapy were administered four cycles of carboplatin 30 mg/kg iv. and paclitaxel 6 mg/kg iv. (once every 21 days) starting 14 days after tumor implant. Animals’ weight gain in both the carboplatin plus paclitaxel and pmIL-12/PPC cohorts were similar to one another but decreased compared with untreated controls (). The combination therapy cohort (receiving both pmIL-12/PPC and chemotherapy) appeared to have an additional reduction in weight gain compared with the monotherapy groups. demonstrates that the combination therapy cohort had significantly prolonged survival (median, 80 days) compared with all other cohorts (range: 42–64 days; p < 0.02).
Mice were administered intraperitoneally ID8 cells. Animal weights and survival analysis of animals given combination treatment of pmIL-12/PPC with a paclitaxel/carboplatin treatment regimen consisting of carboplatin 30 mg/kg (bodyweight) and paclitaxel (6 mg/kg) given iv. (this treatment regimen has been previously evaluated in the ID8 tumor model and shown to produce therapeutic efficacy without overt toxicity). Treatments started 14 days after tumor implant. The pmIL-12/PPC was administered at a total dose of 100 μg per treatment starting 18 days after tumor implant. (A) Comparison of animal weights across cohorts over time. (B) Survival of animals across cohorts. Significance between each cohort was determined by the Gehan–Breslow nonparametric analysis of variance (n = 10 for each cohort).
Reproduced with permission from [Citation42] © John Wiley and Sons, Inc. (2009); permission conveyed through Copyright Clearance Center, Inc.
![Figure 5. Efficacy outcomes of untreated tumor-bearing mice versus pmIL-12/PPC- and/or chemotherapy-treated mice.Mice were administered intraperitoneally ID8 cells. Animal weights and survival analysis of animals given combination treatment of pmIL-12/PPC with a paclitaxel/carboplatin treatment regimen consisting of carboplatin 30 mg/kg (bodyweight) and paclitaxel (6 mg/kg) given iv. (this treatment regimen has been previously evaluated in the ID8 tumor model and shown to produce therapeutic efficacy without overt toxicity). Treatments started 14 days after tumor implant. The pmIL-12/PPC was administered at a total dose of 100 μg per treatment starting 18 days after tumor implant. (A) Comparison of animal weights across cohorts over time. (B) Survival of animals across cohorts. Significance between each cohort was determined by the Gehan–Breslow nonparametric analysis of variance (n = 10 for each cohort).Reproduced with permission from [Citation42] © John Wiley and Sons, Inc. (2009); permission conveyed through Copyright Clearance Center, Inc.](/cms/asset/8599c899-6934-4366-86e7-72c96fec1d28/ifon_a_12332476_f0005.jpg)
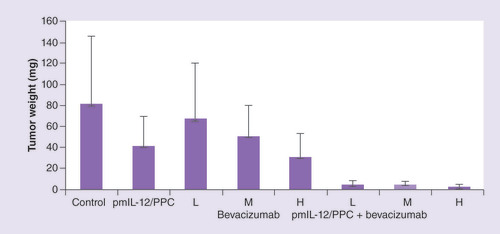
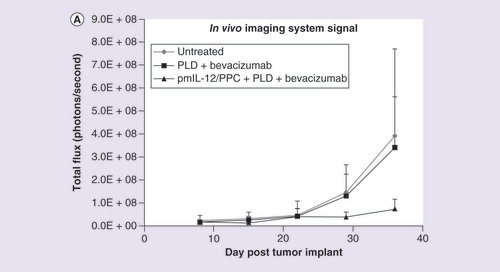
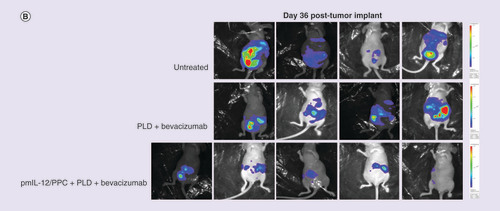
In an SKOV3 nude mouse model, ip. pmIL-12/PPC significantly improved the activity of bevacizumab and pegylated liposomal doxorubicin (PLD) treatment. As shown in , pmIL-12/PPC at 100 μg DNA alone produced 50% inhibition of tumor growth, and in the same study, bevacizumab alone also produced tumor inhibition at all three doses tested (5 [low], 10 [medium] and 20 mg/kg [high]) [Citation47]. Addition of pmIL-12/PPC potentiated bevacizumab activity at all three doses. At the lowest bevacizumab dose, tumor inhibition was only 15%; however, the addition of pmIL-12/PPC improved the antitumor activity to approximately 95%. These results show that good antitumor response can be achieved even with the combination of low-dose bevacizumab and pmIL-12/PPC. In another study, the impact on efficacy of adding pmIL-12/PPC to bevacizumab plus PLD, a standard platinum-resistant ovarian cancer regimen, was explored. In the same mouse model but with cancer cells expressing luciferase gene, combination treatment with PLD (7.5 mg/kg) plus bevacizumab (10 mg/kg) produced marginal tumor response, but the addition of pmIL-12/PPC dramatically improved the response to over 90% () [Citation47]. The addition of pmIL-12/PPC did not significantly influence the animal weight gain, serum chemistry or hematology profile in healthy mice (data not shown). These studies suggest that pmIL-12/PPC can improve antitumor response and prolong survival in mice when administered as monotherapy and can improve the activity of standard cancer therapies for ovarian cancer.
Comparison of tumor weights in peritoneally disseminated SKOV3 human cancer-bearing immunocompromised mice. To induce tumor formation, mice were administered intraperitoneally 7 × 106 SKOV-3 cells. Treatments consisted of pmIL-12/PPC at 100 μg DNA dose, bevacizumab at low (5 mg/kg), medium (10 mg/kg) or high (20 mg/kg) dose, or a combination thereof. Treatments were started 10 days after tumor implant and were given weekly for 4 weeks (pmIL-12/PPC) or 6 weeks (bevacizumab). At 59 days after tumor implant, the animals were euthanized and all visible tumors from the peritoneal cavity and abdominal organs were collected and weighed. The values are presented as mean ± standard error of the mean (n = 6 for each group).
(A) Quantified levels of luciferase bioluminescence using an in vivo imaging system in immunocompromised mice injected with SKOV3 human ovarian cancer cell line (7 × 106 cells) constitutively expressing the luciferase gene. Mice were treated with pegylated liposomal doxorubicin (7.5 mg/kg; two treatments once every 3 weeks) plus bevacizumab (10 mg/kg; five treatments administered weekly) or pegylated liposomal doxorubicin plus bevacizumab plus pmIL-12/PPC (100 mg; four treatments administered weekly). (B) Photographic montage showing the relative luciferase levels in mice with peritoneally disseminated SKOV3 human cancer 36 days after tumor inoculation.
PLD: Pegylated liposomal doxorubicin.
A 28-day good laboratory practice toxicity study was conducted to assess pmIL-12/PPC toxicity when injected ip. for 4 weeks in mice (10, 50 and 250 μg, equivalent to 1.2, 6.0 and 30.0 mg/m2 per animal, respectively). No drug-related deaths or clinical signs of toxicity with ip. administration were observed (data not shown). Histopathologic examination of animals revealed no systemic toxicity, with inflammation noted in organs located in or adjacent to the peritoneal cavity that diminished after 1 month. In addition, a mild granulomatous to pyogranulomatous inflammation reaction was observed at the injection sites. The no-observed adverse effect level of pmIL-12/PPC administered weekly to mice was 50 μg. Based on this safety toxicity data, the starting GEN-1 dose in the first in-human clinical trial was 0.6 mg/m2, which is a tenth of the no-observed adverse effect level.
Taken together, these in vitro and in vivo pharmacology studies provided sufficient evidence of potential GEN-1 efficacy and safety to warrant investigation in clinical trials.
GEN-1 clinical data
GEN-1 has been tested in several advanced ovarian cancer clinical studies, including two monotherapy studies and two in combination with chemotherapy. & present safety and efficacy data, respectively, from these four studies. To date, approximately 75 patients have been treated with GEN-1 alone or in combination with standard chemotherapy in recurrent or newly diagnosed patients with ovarian cancer. Overall, the treatment has been well tolerated and has not been associated with systemic toxicity that is characteristic of recombinant IL-12 therapy. Also, there have been no reports of specific pathological autoimmunity attributable to phIL-12/PPC treatment. The most common AEs that were judged possibly related to phIL-12/PPC treatment included low-grade fever, chills, abdominal pain, gastrointestinal discomfort and nausea. In one patient, a set of AEs characterized by chills, rigor and drop in blood pressure within 24 h after the treatment were judged as cytokine release syndrome. The symptoms were managed by iv. fluid and analgesic and did not require treatment with corticosteroids or cytokine antagonists, which remain the standard of care for cytokine release syndrome observed with some immunotherapy agents [Citation48]. The reason that the hyperactivation of the immune system by some immunotherapy agents [Citation48] has not been observed with GEN-1 is probably due to small changes in blood levels of immunocytokines. The effects of GEN-1 treatment on tumor Response Evaluation Criteria In Solid Tumors (RECIST) or patient survival have been encouraging, as described below, and warrant continued development of GEN-1 for the treatment of ovarian cancer. Clinical studies also demonstrate the evidence of local and durable delivery of IL-12 and production of IFN-γ, a potent central mediator of IL-12 action.
Table 1. Safety from GEN-1 clinical studies in advanced ovarian cancer.
Table 2. Efficacy from GEN-1 clinical studies in advanced ovarian cancer.
While early GEN-1 studies were conducted in patients with recurrent ovarian cancer, the focus of current GEN-1 development is in patients with newly diagnosed disease in combination with neoadjuvant chemotherapy. A brief description of all previous GEN-1 clinical studies in patients with ovarian cancer is described below.
First-in-human Phase I study
The first-in-human study of GEN-1 was GEN-1-101 (NCT00137865), a Phase I dose-escalation trial in 13 patients with platinum-resistant recurrent ovarian cancer [Citation49]. Patients were enrolled into one of four dose-escalating cohorts (0.6, 3, 12 and 24 mg/m2) in which they received four weekly GEN-1 ip. doses. At least 7 days prior to drug treatment, patients received a single iv. dose of antibiotics and the ip. catheter was placed. GEN-1 was administered on days 0, 7, 14 and 21 on an inpatient basis.
All patients had undergone previous surgery and had received two to six prior chemotherapy regimens (median, 3). No dose-related trends in safety measures were apparent and the maximum tolerated dose (MTD) was not reached. The most frequently reported AEs were fever (69%) and abdominal pain (54%). Most AEs were mild to moderate in severity, and five serious AEs were reported in five patients (peritonitis, n = 3; urinary tract infection, n = 1; and abdominal inflammation, n = 1). The serious AE of peritonitis with fever was judged as possibly related to treatment and was considered a DLT. Six patients had at least one grade ≥3 laboratory value after receiving GEN-1, but none of these events were judged as clinically significant.
All post-treatment PF samples contained high levels of IL-12 plasmid (most levels were 104–106 higher than those in blood), and plasmid was detected in only 29% of the blood samples at much lower levels than in corresponding peritoneal fluid (PF) samples collected [Citation49]. Only one patient had measurable IL-12 at a post-treatment time point (in PF 1 day after first dose). For the PF samples collected 1 day after each GEN-1 treatment, IFN-γ levels were elevated above baseline in every patient at one or more of the four time points (median, three elevations), but IFN-γ levels were not detected in any samples 1 week after treatment nor in any blood samples at any time point. The high-dose (12 and 24 mg/m2) cohorts had more IFN-γ-positive samples than the low-dose (0.6 and 3.0 mg/m2) cohorts (50% vs 16%, respectively).
No objective responses were observed 5 weeks after the last GEN-1 dose, but 31% of patients had stable disease (SD), 17% from the low-dose cohorts and 43% from the high-dose cohorts. Mean survival was 25.5 and 19.7 months for the high-dose cohorts (12 and 24 mg/m2, respectively) and 10.1 and 15.5 months for the low-dose cohorts (0.6 and 3.0 mg/m2, respectively).
The IL-12 plasmid distribution results suggest that GEN-1 remained localized within the peritoneal cavity. The lack of detectable IL-12 suggests either that levels were below the level of detection and/or that it was rapidly cleared. In either case, the increase in IFN-γ demonstrates that GEN-1 did produce biologically active IL-12. The activity of GEN-1 was encouraging and appeared to be dose related.
Phase II monotherapy study
A Phase II study of GEN-1 monotherapy sponsored by the Gynecologic Oncology Group (GOG) was GOG-0170Q (NCT01118052), a multicenter trial that enrolled 22 patients with platinum-resistant or persistent epithelial ovarian, fallopian tube or primary peritoneal cancer who had received up to two prior chemotherapy regimens, at least one of which was a platinum regimen [Citation50]. Patients received weekly ip. doses of GEN-1 24 mg/m2 on an outpatient basis; treatment continued until disease progression or intolerable AEs. The primary objective of the study was to evaluate PFS rate at 6 months and objective response rate (ORR). Efficacy was assessed using RECIST criteria. Safety was assessed through AE assessment and laboratory and physical assessments.
Twenty patients were evaluable for safety and received 58 total cycles (one cycle = four weekly GEN-1 doses), with a median of two cycles. Six patients received one cycle or less due to AEs or disease progression. The toxicities in this study were similar to those observed in the Phase I monotherapy study although there were more disease-related toxicity events. Of the 16 patients evaluable for efficacy seven had SD (35%). 6-month PFS rate was 30%, although three of the six patients had gone off study and received additional therapy within the 6-month period. Median PFS was 2.9 months and median OS was 9.2 months. In this study, GEN-1 monotherapy demonstrated some, albeit limited, activity in a primarily platinum-resistant population and was tolerable.
Phase I GEN-1 plus carboplatin/docetaxel study
The GEN-1-201 study (NCT00473954) was a multicenter Phase I dose-escalation trial of GEN-1 (12, 18 and 24 mg/m2 administered every 10–11 days × 4) in combination with carboplatin (area under the curve of five) and docetaxel (75 mg/m2) in 13 patients with platinum-sensitive ovarian cancer (every 21 days × 6) [Citation51]. Clinical and laboratory safety evaluations and AE assessments were performed prior to each dose, and 1 and 5 weeks following the last dose of study drug. Blood and PF were collected prior to and 24 h after each dose for cytokine (IL-12, IFN-γ and TNF-α) analysis. Efficacy was assessed using RECIST criteria.
The most frequently reported AEs for this combination regimen were alopecia (85%), pyrexia (85%), abdominal pain (77%) and nausea (77%). Twenty cases of AEs were judged as definitely related to treatment, most frequently including abdominal pain (n = 10), hypotension (n = 3) and low-grade fever (n = 2). Only three grade 3 AEs were probably related to GEN-1, abdominal pain (n = 2) and cytokine release syndrome with hypotension (n = 1 in cohort 4). Of note, this case of cytokine release syndrome is the only one documented across all patients treated with GEN-1. No deaths nor discontinuations resulted from an AE; there were no DLTs. ORR among 12 evaluable patients was 50% (17% complete responses [CRs] and 33% partial responses [PRs]), and SD rate was 42%. Ten of 12 patients survived >12 months and two patients survived >5 years. Median PFS was 8.8 months and median OS was 16.6 months. Both IFN-γ and TNF-α from post-treatment PF samples showed a positive correlation with GEN-1 dose; the cytokine levels decreased when GEN-1 was administered concurrently with chemotherapy, presumably due to concomitant administration of corticosteroids to reduce hypersensitivity from docetaxel treatment.
In this trial of platinum-sensitive disease, GEN-1 plus standard chemotherapy produced clinical benefit in nearly all patients and the regimen appeared tolerable.
Phase I GEN-1 plus PLD study
GOG-9928 (NCT01489371) was a multicenter Phase I dose-escalation trial of GEN-1 in combination with PLD, a widely approved cancer chemotherapy, in 16 patients with platinum-resistant ovarian cancer [Citation25]. PLD was chosen as a combination drug for GEN-1 because it has monotherapy activity of 8–20% in platinum-resistant disease [Citation52–54]; it can induce immunogenic cell death [Citation55], leading to increased presentation of tumor antigens; and its dosing allows for four weekly GEN-1 treatments. Patients were assigned to one of three DL cohorts: DL1 (GEN-1 24 mg/m2 and PLD 40 mg/m2; n = 4), DL2 (GEN-1 36 mg/m2 and PLD 40 mg/m2; n = 5) and DL3 (GEN-1 36 mg/m2 and PLD 50 mg/m2; n = 7). PLD was administered every 28 days and GEN-1 was administered weekly. Patients continued with weekly GEN-1 until disease progression, intolerable AEs or patient withdrawal.
In addition to weekly AE assessment, blood and PF samples were collected prior to the first two GEN-1 treatments (days 1 and 8) and on days 2, 4, 9, 11 and 15 for cytokine (IL-12, IFN-γ, TNF-α and VEGF) analysis. Efficacy was assessed using RECIST criteria.
Eight patients had one prior chemotherapy regimen, and eight had two or three prior regimens. The most frequently reported AEs for this combination regimen were leukopenia (88%), anemia (88%), neutropenia (81%), fatigue (81%), constipation (63%), nausea (63%) and abdominal pain (63%). Among those events judged to be at least possibly related to GEN-1 treatment, 2 were grade 4 events (neutrophil count decreased, n = 2) and 25 were grade 3 events (neutrophil count decreased, n = 7; white blood cell decreased, n = 6; anemia, n = 4; abdominal pain, n = 2; mucositis oral, n = 2; urinary tract infection, n = 1; platelet count decreased, n = 1; hyperglycemia, n = 1; and hypokalemia, n = 1). No AEs were judged to be DLTs.
PF samples were available from 11 patients. IL-12 levels, quantified with a more sensitive assay than used in previous studies, were undetectable at baseline and increased to 65 pg/100 mg protein at peak. IFN-γ and TNF-α levels increased from baseline by 46- and 13-fold, respectively, after GEN-1 administration. VEGF levels showed a 22% decline after GEN-1 administration. Cytokine changes in plasma were similar in trend to those in ascites but much smaller in magnitude. The fold increases in IFN-γ and TNF-α levels in this study were relatively lower than those in the carboplatin/docetaxel study, which could be due to differences in platinum-sensitive versus platinum-resistant population and type of chemotherapy.
Fourteen of 16 patients were evaluable for response. The overall ORR for the study was 21% and for DL3 was 28%. The overall clinical benefit (PR + SD) for the study was 57% and for DL3 was 86%, indicating a trend in improved outcomes with increasing dose. Median duration of response for the three PRs ranged from 6.7 to 10.3 months. Median PFS was 4.7 months and median OS was 11.1 months.
Discussion
Ovarian cancer is a disease of high unmet need. Despite enormous research in this area, only marginal improvements in treatment outcomes have been realized over the last two decades. Cytotoxic therapy for ovarian cancer is still the predominant treatment approach, which is marred by serious toxicity and drug resistance. New classes of drugs, such as antiangiogenic agents and poly (ADP-ribose) polymerase inhibitors, have only recently gained regulatory approval and have demonstrated marginal survival benefits.
Stimulation of the body’s immune system against cancer has long been considered an effective and curative treatment method. Historically, immunotherapy approaches such as vaccines have not performed well in ovarian and other types of cancers. Recently, the use of a new and emerging class of immunotherapy drugs, the checkpoint inhibitors, have gained credence in the treatment of some cancers, but their impact in ovarian cancer has been marginal [Citation56]. Ovarian cancer is characterized by a highly immunosuppressive environment in which multiple factors play a role in cancer progression and immune suppression. Therefore, failure of agents, such as vaccines or checkpoint inhibitors, that target a single immune mechanism is not surprising; rather, it underscores the need for agents that have a broader impact on the tumor immune microenvironment. Additionally, the reversal of the tumor microenvironment in favor of immune stimulation and consequently the promotion of tumor eradication may require long-term delivery of an immune agent. In this regard, the GEN-1 concept of safe and persistent local delivery of IL-12, a powerful immunostimulatory cytokine with multiple mechanisms of action, offers a treatment modality that could potentially remodel the tumor microenvironment. The available pharmacological data from animal and human studies support the GEN-1 concept for safe, local and durable immune activity.
Clinical experience with GEN-1 ip. in women with recurrent ovarian cancer shows treatment feasibility with good safety. The most common GEN-1-related AEs are local in nature, including gastrointestinal discomfort, nausea and low-grade fever. Across 33 patients treated with GEN-1 monotherapy in two clinical trials in the recurrent setting, most AEs were mild or moderate in severity and only one DLT was observed, even in one of the two trials permitting indefinite treatment, either until disease progression or intolerable toxicity [Citation49,Citation50]. In the first study, two cases of infectious peritonitis were attributed to the ip. catheter [Citation49]. In subsequent studies, the risk of peritonitis was minimized with the use of a percutaneous catheter [Citation51]. In combination trials in which GEN-1 was administered in conjunction with cytotoxic drugs (carboplatin, docetaxel, PLD), no overlapping toxicities were observed [Citation25,Citation51], which demonstrates that GEN-1 combination with cytotoxic drugs is feasible and can be continued in larger trials.
The MTD and optimal biological GEN-1 dose have not been identified from dose-escalation studies conducted in the dose range of 0.6–36 mg/m2. Higher GEN-1 doses are being examined for safety and biological activity in the ongoing OVATION study [Citation43]. As for treatment duration, a total of four weekly treatments (one cycle) were administered in the first human clinical trial. The weekly treatment schedule in the clinic is based on modeling the duration of biological response in preclinical studies [Citation42], and recently supported by clinical data as well [Citation25]. In recent studies, the GEN-1 weekly dosing regimen was extended until intolerable toxicity or progression. Several patients received weekly treatments for several months without intolerable toxicity. A safe, long-term delivery of immunostimulatory cytokine via GEN-1 treatment to remodel the peritoneal immune environment in favor of immune stimulation is a promising and groundbreaking approach to ovarian cancer treatment.
The initial GEN-1 studies were conducted in a monotherapy setting to establish a clean safety profile for GEN-1. Although these small studies have shown encouraging efficacy trends in a difficult-to-treat recurrent population, the combination approaches are more realistic scenarios in the future development of GEN-1. In two combination trials of GEN-1 in conjunction with first- or second-line chemotherapy, the addition of GEN-1 did not exacerbate chemotherapy toxicity, and no DLTs were observed across the 29 patients treated in these two trials [Citation25,Citation51]. In platinum-sensitive disease, GEN-1 plus carboplatin plus docetaxel produced a 50% ORR, 42% SD and 16.6-month median OS. Efficacy was more modest in difficult-to-treat platinum-resistant ovarian cancer, with GEN-1 plus PLD producing a 21% ORR and median OS of 11.1 months.
The current GEN-1 development is focused on patients with newly diagnosed ovarian cancer as an adjunct to neoadjuvant chemotherapy. Patients with newly diagnosed disease are expected to have a healthier and potentially more responsive immune system as compared with patients with heavily pretreated and recurrent disease. Moreover, the availability of primary tumor before and after treatment in a neoadjuvant population provides opportunity for comprehensive immunological studies to better understand the mechanisms of action of GEN-1 and to guide future studies. In an ongoing Phase I study (OVATION) in newly diagnosed advanced ovarian cancer (stage III/IV disease), the safety and biological activity of escalated GEN-1 doses are examined in combination with standard neoadjuvant chemotherapy (carboplatin plus paclitaxel) only prior to interval surgical debulking. In total, 14 patients could be assessed for RECIST response, debulking status and pathological response. One of the 14 patients experienced a complete pathologic response, a micro pathological response was seen in seven patients, and the remaining six patients experienced a macro pathological response [Citation43].
One of the goals of neoadjuvant chemotherapy is to shrink existing tumor to permit maximal removal of tumor at the time of interval debulking surgery. In the 201-14-101 study, a total of 12 patients (86%) demonstrated a response prior to their interval debulking surgery. Nine of 14 patients had an R0 interval debulking response, three had an R1 response and two had an R2 response.
The PFS data from this study are still being collected and will be finalized later. The current estimate of the median PFS in the intent-to-treat population is 17.0 months and in the per-protocol-treated population is 24.5 months (manuscript in preparation). Despite providing eight doses of GEN-1 at up to 79 mg/m2, the MTD has still not been reached. The analysis of primary tumor before and after the combination treatment indicates intriguing treatment-related changes in the cytokine levels and immune cell populations in primary tumor and in ascites.
These promising results have led to the design of the OVATION II trial, a Phase I/II GEN-1 study similar in study design to the OVATION trial, except it uses a randomized trial design with a control arm and patients receive GEN-1 weekly pre- and postinterval surgical debulking, as compared with predebulking only in the OVATION study, along with carboplatin and paclitaxel. It examines GEN-1 in the first-line setting of advanced ovarian cancer in combination with standard neoadjuvant chemotherapy, with the primary end point of PFS and secondary efficacy end points of ORR, pathologic complete response (pCR), CR, OS and surgical response. Target enrollment is 130 patients and is due and was opened in summer of 2018.
Platinum-resistant disease is also a potential path for future GEN-1 development. This is supported by GEN-1 plus PLD Phase I trial results and strong preclinical data of GEN-1 combination with PLD plus bevacizumab, a current standard of care in platinum-resistant disease. Results from the preclinical study, in which the addition of GEN-1 significantly improved the efficacy of low-dose bevacizumab and did not significantly affect hematology or serum chemistry profile, warrants clinical evaluation of GEN-1 combination with bevacizumab at standard or low doses; lowering the bevacizumab dose could lower its toxicity.
Significant development has been made in GEN-1 manufacturing. Clinical utility of DNA formulations with lipid- or polymer-based delivery systems has been historically challenging with respect to particle stability and dosing flexibility [Citation57]. Aggregation of nanoparticles and decomplexation of DNA from delivery carrier resulting from factors affecting particle physical stability have been implicated in this effect [Citation57]. Besides physical factors, changes in chemical structure of the delivery system during storage could also trigger undesirable changes and adversely affect particle stability.
Historically, these gene-based nanodrug products have proven difficult to manufacture at >0.1 mg/ml DNA concentration, restricting the ability to dose escalate by volume. In contrast, the GEN-1 formulation as a lyophilized powder enables a shelf-life >3 years and the ability to reconstitute the drug at the bedside to doses at 0.5 mg/ml, which allows for flexible dosing. Further progress in formulation development has been made to produce up to 5 mg/ml DNA concentration (data not shown). These favorable formulation characteristics may improve GEN-1’s commercial viability. The manufacturing process has been scaled up successfully from bench to clinic while maintaining the product quality.
In summary, GEN-1 is a novel immunotherapy agent delivered ip. to provide IL-12 immunotherapy to patients with ovarian cancer in a locoregional manner, in an attempt to improve on efficacy and safety profile of systemically administered rhIL-12. Several early-stage clinical trials of GEN-1 administered alone or in combination with standard chemotherapy have demonstrated encouraging safety and efficacy results. Responses have been observed both in platinum-sensitive and platinum-resistant disease. Moreover, cytokine analysis from PF has demonstrated a positive immune effect consistent with IL-12 pharmacology. Because the GEN-1 MTD has not yet been reached in any of the studies, higher doses are planned in the upcoming OVATION II study to attempt to extend the efficacy of this novel agent. Safe and durable local delivery of a powerful pluripotent immune cytokine over a long period of time could potentially allow for more favorable remodeling of the tumor microenvironment against sustainable tumor growth and therefore offers a new treatment paradigm in ovarian cancer treatment. An overall shift in tumor microenvironment to one favoring immune stimulation by GEN-1 [Citation44] could also be conducive to activity of other therapeutic entities, including CAR T-cell therapy, checkpoint inhibitors, vaccines and antiangiogenic agents, and provide opportunities for novel combination trials.
Future perspective
Immunotherapy-based interventions are expected to dominate cancer treatment well into the next decade. Additional molecular defects in immune pathways will likely be identified and targeted for development of novel drug concepts. A combination of multiple interventions addressing more than one immune defect to improve the efficacy may take center stage in the immunotherapy world of the future. Development of a GEN-1-like agent that could boost the immune system at multiple levels in a safe and persistent manner could prove useful in combination with already-approved agents or novel therapies.
Ovarian cancer unmet need
Ovarian cancer is a leading cause of cancer deaths among women, with <20% of women with advanced disease surviving at least 10 years, highlighting the urgent need for more effective ovarian cancer therapies.
Immunogenicity & the tumor microenvironment
Ovarian cancer is an immunogenic malignancy, with CD8+ tumor-infiltrating lymphocytes, immunosuppressive Tregs and numerous other immune cells and cytokines playing important roles in the immunogenicity of the tumor microenvironment.
IL-12 & its activity in ovarian cancer
IL-12 is a pleiotropic cytokine and one of the most potent inducers of antitumor immunity, with multiple mechanisms of action, including activation of natural killer cells; maturation, proliferation and activation of cytotoxic tumor-infiltrating lymphocytes; potentiation of antibody-dependent cell-mediated cytotoxicity; interference with Treg differentiation; and inhibition of angiogenesis.
Although recombinant human IL-12 (rhIL-12) injection has produced favorable immunological and clinical responses in patients with various malignancies, treatment-related hematological and liver toxicities have limited its clinical utility.
GEN-1 concept & drug design
To overcome the shortcomings of rhIL-12 administration, IL-12-based therapy should produce persistent IL-12 protein levels locally at the tumor site while maintaining a favorable safety profile.
GEN-1 is a gene-based IL-12 immunotherapy designed to pleiotropically promote immune stimulation in a local and persistent manner when delivered intraperitoneally. GEN-1 comprises a human IL-12 gene expression plasmid that encodes for the IL-12 gene and a synthetic plasmid delivery system that protects plasmid DNA from degradation and facilitates plasmid uptake by cells.
GEN-1 proposed mechanisms of action
GEN-1’s mechanism of action is anticipated to be consistent with endogenous IL-12 pharmacology, both with respect to changes in cytokine levels and immune cell populations. A locoregional and durable IL-12 production from within the body gives a unique advantage in its action mechanisms over rIL-12 therapy.
GEN-1 pharmacology
Preclinical pharmacological studies optimized the composition of GEN-1 for gene transfer.
Mouse models of disseminated ovarian cancer demonstrated GEN-1’s ability to delay disease progression and prolong survival as monotherapy, and to further improve these outcomes when added to chemotherapy.
GEN-1 clinical data
In two monotherapy trials in terminally ill patients with recurrent ovarian cancer who had failed multiple chemotherapy treatments, GEN-1 demonstrated no objective responses, but it did produce stable disease and 6-month progression-free survival in approximately a third of patients, while maintaining a favorable safety profile. These studies also demonstrated primarily local distribution of IL-12 plasmid and increases in downstream cytokines.
A trial examining GEN-1 in combination with chemotherapy in platinum-sensitive disease produced clinical benefit in nearly all patients; combination therapy in platinum-resistant disease yielded a 57% clinical benefit rate and evidence of increasing benefit at higher doses.
Discussion
GEN-1 is a novel immunotherapy agent delivered intraperitoneally to provide IL-12 immunotherapy to women with ovarian cancer in a localized and durable manner, in an attempt to improve on therapeutic outcomes and the safety profile of systemically administered rhIL-12.
Clinical experience with GEN-1 in women with recurrent ovarian cancer shows treatment feasibility with good safety, as evidenced by common GEN-1-related AEs of gastrointestinal discomfort, nausea and low-grade fever.
In two combination trials of GEN-1 in conjunction with chemotherapy, the addition of GEN-1 did not exacerbate chemotherapy toxicity.
Because the maximum tolerated dose of GEN-1 has not been reached in any of the ovarian cancer studies thus far, higher doses are planned in the upcoming OVATION II study in an attempt to extend its safety and activity evaluation.
Financial & competing interests disclosure
PH Thaker received consulting and advisory fees from Celsion; institutional grant and speaker bureau fees from Merck and Tesaro; is a consultant for Abbvie-Stemcentrx. N Borys, J Fewell and K Anwer are employees of Celsion Corporation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing assistance in the preparation of this manuscript was provided by J Klem, Klem Medical Communications, LLC.
References
- Siegel RL , MillerKD, JemalA. Cancer statistics, 2018. CA Cancer J. Clin.68, 7–30 (2018).
- Cress RD , ChenYS, MorrisCR, PetersenM, LeiserowitzGS. Characteristics of long-term survivors of epithelial ovarian cancer. Obstet. Gynecol.126(3), 491–497 (2015).
- National Comprehensive Cancer Network (NCCN) . Clinical practice guidelines in oncology (NCCN Guidelines®): ovarian cancer version 2.2018. https://www.nccn.org/professionals/physician_gls/recently_updated.aspx.
- Kehoe S , HookJ, NankivellMet al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open-label, randomised, controlled, non-inferiority trial. Lancet386(9990), 249–257 (2015).
- Vergote I , TropeCG, AmantFet al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N. Engl. J. Med.363(10), 943–953 (2010).
- McGuire WP 3rd , MarkmanM. Primary ovarian cancer chemotherapy: current standards of care. Br. J. Cancer.89(Suppl. 3), S3–S8 (2003).
- Bhatt A , GlehenO. The role of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy (HIPEC) in ovarian cancer: a review. Indian J. Surg. Oncol.7(2), 188–197 (2016).
- Schlienger K , ChuCS, WooEYet al. TRANCE- and CD40 ligand-matured dendritic cells reveal MHC class I-restricted T cells specific for autologous tumor in late-stage ovarian cancer patients. Clin. Cancer Res.9(4), 1517–1527 (2003).
- Zhang L , Conejo-GarciaJR, KatsarosDet al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med.348(3), 203–213 (2003).
- Hwang WT , AdamsSF, TahirovicE, HagemannIS, CoukosG. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol. Oncol.124(2), 192–198 (2012).
- Curiel TJ , CoukosG, ZouLet al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med.10(9), 942–949 (2004).
- Sato E , OlsonSH, AhnJet al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl Acad. Sci. USA102(51), 18538–18543 (2005).
- Santoiemma PP , PowellDJJr. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol. Ther.16(6), 807–820 (2015).
- Schiavoni G , GabrieleL, MatteiF. The tumor microenvironment: a pitch for multiple players. Front. Oncol.3, 90 (2013).
- Hansen JM , ColemanRL, SoodAK. Targeting the tumour microenvironment in ovarian cancer. Eur. J. Cancer56, 131–143 (2016).
- Freedman RS , LenziR, KudelkaAP, LawrenceDD, RosenblumM, PlatsoucasCD. Intraperitoneal immunotherapy of peritoneal carcinomatosis. Cytokines Cell Mol. Ther.4(2), 121–140 (1998).
- Robertson MJ , RitzJ. Interleukin 12: basic biology and potential applications in cancer treatment. Oncologist1(1 & 2), 88–97 (1996).
- Tugues S , BurkhardSH, OhsIet al. New insights into IL-12-mediated tumor suppression. Cell Death Differ.22(2), 237–246 (2015).
- Sgadari C , AngiolilloAL, TosatoG. Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood87(9), 3877–3882 (1996).
- Albini A , BrigatiC, VenturaAet al. Angiostatin anti-angiogenesis requires IL-12: the innate immune system as a key target. J. Transl. Med.7, 5 (2009).
- Berek JS , HackerNF, LichtensteinAet al. Intraperitoneal recombinant α-interferon for ‘salvage’ immunotherapy in stage III epithelial ovarian cancer: a Gynecologic Oncology Group Study. Cancer Res.45(9), 4447–4453 (1985).
- Berek JS , MarkmanM, StonebrakerBet al. Intraperitoneal interferon-α in residual ovarian carcinoma: a Phase II gynecologic oncology group study. Gynecol. Oncol.75(1), 10–14 (1999).
- Mantia-Smaldone GM , CorrB, ChuCS. Immunotherapy in ovarian cancer. Hum. Vaccin. Immunother.8(9), 1179–1191 (2012).
- Pujade-Lauraine E , GuastallaJP, ColomboNet al. Intraperitoneal recombinant interferon gamma in ovarian cancer patients with residual disease at second-look laparotomy. J. Clin. Oncol.14(2), 343–350 (1996).
- Thaker PH , BradyWE, LankesHAet al. A Phase I trial of intraperitoneal GEN-1, an IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer, administered with pegylated liposomal doxorubicin in patients with recurrent or persistent epithelial ovarian, fallopian tube or primary peritoneal cancers: an NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol.147(2), 283–290 (2017).
- Vlad AM , BudiuRA, LenznerDEet al. A Phase II trial of intraperitoneal interleukin-2 in patients with platinum-resistant or platinum-refractory ovarian cancer. Cancer Immunol. Immunother.59(2), 293–301 (2010).
- Atkins MB , RobertsonMJ, GordonMet al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin. Cancer Res.3(3), 409–417 (1997).
- Bajetta E , Del VecchioM, MortariniRet al. Pilot study of subcutaneous recombinant human interleukin 12 in metastatic melanoma. Clin. Cancer Res.4(1), 75–85 (1998).
- Cebon J , JagerE, ShackletonMJet al. Two Phase I studies of low dose recombinant human IL-12 with Melan-A and influenza peptides in subjects with advanced malignant melanoma. Cancer Immun.3, 7 (2003).
- Gollob JA , MierJW, VeenstraKet al. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-γ induction is associated with clinical response. Clin. Cancer Res.6(5), 1678–1692 (2000).
- Rook AH , WoodGS, YooEKet al. Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood94(3), 902–908 (1999).
- Lenzi R , RosenblumM, VerschraegenCet al. Phase I study of intraperitoneal recombinant human interleukin 12 in patients with Mullerian carcinoma, gastrointestinal primary malignancies, and mesothelioma. Clin. Cancer Res.8(12), 3686–3695 (2002).
- Lenzi R , EdwardsR, JuneCet al. Phase II study of intraperitoneal recombinant interleukin-12 (rhIL-12) in patients with peritoneal carcinomatosis (residual disease <1 cm) associated with ovarian cancer or primary peritoneal carcinoma. J. Transl. Med.5, 66 (2007).
- Barrett JA , CaiH, MiaoJet al. Regulated intratumoral expression of IL-12 using a RheoSwitch Therapeutic System® (RTS®) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther.25, 106–116 (2018).
- Brown DW , BahramiAJ, CantonDAet al. Development of an adaptive electroporation system for intratumoral plasmid DNA delivery. Bioelectrochemistry122, 191–198 (2018).
- Imboden M , ShiF, PughTDet al. Safety of interleukin-12 gene therapy against cancer: a murine biodistribution and toxicity study. Hum. Gene Ther.14(11), 1037–1048 (2003).
- Kang WK , ParkC, YoonHLet al. Interleukin 12 gene therapy of cancer by peritumoral injection of transduced autologous fibroblasts: outcome of a Phase I study. Hum. Gene Ther.12(6), 671–684 (2001).
- Fewell JG , MatarM, SlobodkinGet al. Synthesis and application of a non-viral gene delivery system for immunogene therapy of cancer. J. Control Rel.109(1–3), 288–298 (2005).
- Mahato RI , LeeM, HanS, MahatoRI, LeeM, HanS. Intratumoral delivery of p2CMVmIL-12 using water-soluble lipopolymers. Mol. Ther.4(2), 130–138 (2001).
- Ogris M , BrunnerS, SchullerS, OgrisM, BrunnerS, SchullerS. PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther.6(4), 595–605 (1999).
- Boussif O , Lezoualc’hF, ZantaMAet al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl Acad. Sci. USA92(16), 7297–7301 (1995).
- Fewell JG , MatarMM, RiceJSet al. Treatment of disseminated ovarian cancer using nonviral interleukin-12 gene therapy delivered intraperitoneally. J. Gene Med.11(8), 718–728 (2009).
- Thaker PH , BradleyWH, LeathCAet al. Phase I study of the safety and activity of formulated IL-12 plasmid administered intraperitoneally in combination with standard neoadjuvant chemotherapy in patients with newly diagnosed advanced stage ovarian cancer. Presented at: American Society of Clinical Oncology – Society for Immunotherapy of Cancer. National Harbor, MD, USA, Abstract 155 (2017).
- Anwer K , MatsuzakiJ, BsharaWet al. Immunological changes following intraperitoneal administration of a formulated IL-12 plasmid in combination with standard neoadjuvant chemo in newly diagnosed advanced stage ovarian cancer patients. Presented at: American Association of Cancer Research. Addressing Critical Questions in Ovarian Cancer Research and Treatment. Pittsburgh, PA, USA, Abstract A76 (2017).
- Roby KF , TaylorCC, SweetwoodJPet al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis21(4), 585–591 (2000).
- Hasko G , SzaboC. IL-12 as a therapeutic target for pharmacological modulation in immune-mediated and inflammatory diseases: regulation of T helper 1/T helper 2 responses. Br. J. Pharmacol.127(6), 1295–1304 (1999).
- Fewell JG , MatarMM, RiceJKet al. Interleukin-12 gene therapy in combination with bevacizumab and PEGylated liposomal doxorubicin for treatment of disseminated ovarian cancer. Presented at: American Association for Cancer Research. New Orleans, LA, USA, Abstract 2078 (2016).
- Sriratana P , NortonJ. New immunotherapies in oncology treatment and their side effect profiles. J. Am. Board Fam. Med.31(4), 620–627 (2018).
- Anwer K , BarnesMN, FewellJ, LewisDH, AlvarezRD. Phase-I clinical trial of IL-12 plasmid/lipopolymer complexes for the treatment of recurrent ovarian cancer. Gene Ther.17(3), 360–369 (2010).
- Alvarez RD , SillMW, DavidsonSAet al. A Phase II trial of intraperitoneal EGEN-001, an IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: a gynecologic oncology group study. Gynecol. Oncol.133(3), 433–438 (2014).
- Anwer K , KellyFJ, ChuC, FewellJG, LewisD, AlvarezRD. Phase I trial of a formulated IL-12 plasmid in combination with carboplatin and docetaxel chemotherapy in the treatment of platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol.131(1), 169–173 (2013).
- Colombo N , KutarskaE, DimopoulosMet al. Randomized, open-label, Phase III study comparing patupilone (EPO906) with pegylated liposomal doxorubicin in platinum-refractory or -resistant patients with recurrent epithelial ovarian, primary fallopian tube, or primary peritoneal cancer. J. Clin. Oncol.30(31), 3841–3847 (2012).
- Gordon AN , GranaiCO, RosePGet al. Phase II study of liposomal doxorubicin in platinum- and paclitaxel-refractory epithelial ovarian cancer. J. Clin. Oncol.18(17), 3093–3100 (2000).
- Markman M , GordonAN, McGuireWP, MuggiaFM. Liposomal anthracycline treatment for ovarian cancer. Semin. Oncol.31(6 Suppl. 13), 91–105 (2004).
- Kepp O , GalluzziL, MartinsIet al. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev.30(1), 61–69 (2011).
- Gadducci A , GuerrieriME. Immune checkpoint inhibitors in gynecological cancers: update of literature and perspectives of clinical research. Anticancer Res.37(11), 5955–5965 (2017).
- Molina MD , ArmstrongTK, ZhangY, PatelMM, LentzYK, AnchordoquyTJ. The stability of lyophilized lipid/DNA complexes during prolonged storage. J. Pharm. Sci.93(9), 2259–2273 (2004).