
Abstract
SB8 is a biosimilar of bevacizumab based on its similarity demonstrated by physicochemical, functional, non-clinical and clinical studies. Supported by the concept of extrapolation, SB8 was authorized and is used in a similar manner across all types of tumors as reference bevacizumab. Furthermore, SB8 offers convenience with prolonged stability compared with reference bevacizumab in diluted form. Although a biosimilar must demonstrate biosimilarity to a reference product with the ‘totality of evidence’ in a stringent regulatory process for marketing authorization, some concerns remain among healthcare practitioners, particularly about extrapolation. This review summarizes the concepts of the totality of evidence and extrapolation in biosimilar development and the role of bevacizumab biosimilars in the management of metastatic colorectal cancer as an extrapolated indication.
Bevacizumab is a biological medicine that inhibits the interaction of VEGF-A with its receptor VEGFR, resulting in downregulation of the VEGF signaling pathway and neovascularization in solid tumors [Citation1,Citation2]. Initially approved in 2004 and 2005 by the US FDA and the EMA, respectively, for the treatment of metastatic colorectal cancer (mCRC), bevacizumab is now licensed for the treatment of a number of solid tumor types.
Despite the significant clinical benefits of bevacizumab [Citation3], access to treatment is still limited for many patients. In 2020, bevacizumab was among the top ten drug expenditures in the US Medicare Part B program [Citation4]. High out-of-pocket costs or lack of reimbursement are predominant barriers to bevacizumab, more so in the US, Eastern European, and emerging market countries [Citation5–7]. Insurance plan coverage often poses an additional limitation, with varying plans in the US either not covering bevacizumab treatment or only in the highest cost-sharing tier [Citation8]. Additional barriers are treatment guidelines that advise against bevacizumab due to (perceived) low cost effectiveness [Citation5,Citation9,Citation10] and restricted supply caused by the restricted number of manufacturers, limited availability of ingredients, and compliance issues [Citation7,Citation11].
In light of the significant expenditures for biologics [Citation12], biosimilars have been developed as an alternative resulting in increased access to lifesaving medications. A biosimilar is highly similar and clinically equivalent to an already marketed biological medicine, the so-called reference product [Citation13–15]. Biologics are produced from a biological source, such as living cells. Due to the natural variability of living organisms and the complex manufacturing process, biologics are inherently heterogenous [Citation15]. These microheterogeneities can exist within or between batches and while the product’s amino acid sequence and biological activity remain unchanged, small variations in post-translational modifications can occur. Accordingly, a biosimilar can never be an identical copy of its reference product [Citation15]. However, a strict framework is in place to ensure that the microheterogeneities have no clinically meaningful effect on biological activity, pharmacokinetics (PK), pharmacodynamics (PD), efficacy and safety [Citation13,Citation15–17].
With the lower cost of biosimilars, they introduce price competition that impacts the reference product and the whole product class [Citation18]. Estimates of cost savings achieved by biosimilars are substantial and range from €52 million for bevacizumab biosimilars in Spain (estimated 80% market share in third year) [Citation19], to €77 million in several Western European countries in case of an infliximab biosimilar (estimated 30% discount) [Citation20], and to £300 million in the United Kingdom (UK) in case of an adalimumab biosimilar [Citation21]. Lower costs expand patients’ access to treatment, mainly but not limited to countries with low prescription rates of the reference product [Citation18]. Ultimately, decreased costs for health insurances allow more patients to be treated [Citation19,Citation22], to offer lower insurance fees [Citation23], or to reinvest into other medicines [Citation24].
SB8 is a bevacizumab biosimilar developed by Samsung Bioepis. In addition to equivalent clinical efficacy and a comparable safety profile, SB8 demonstrated comparable functional, physicochemical, pharmacodynamic, and pharmacokinetic characteristics to that of the reference bevacizumab [Citation25,Citation26]. SB8 was approved in the EU in 2020/2021 (Aybintio®/Onbevzi™) [Citation27,Citation28], in the Republic of Korea (Onbevzi) [Citation29] and in Canada (Aybintio) [Citation30] in 2021, and in Taiwan (Onbevzi) in 2022 [Citation31]. SB8 received approval for the same types of cancer for which reference bevacizumab is approved based on the concept of extrapolation, meaning that safety and efficacy data from patients with metastatic or recurrent NSCLC, in which SB8 has been evaluated, have been extrapolated to other indications for which reference bevacizumab is approved.
The objective of this review is to provide an overview of SB8 regarding the totality of evidence in biosimilar development, the concept of extrapolation, and to summarize the real-world experience of bevacizumab biosimilars, especially in the mCRC indication.
Totality of evidence in biosimilar development
The development and approval of a biosimilar requires extensive scientific and comparative evaluation mandated by regulatory frameworks implemented by EU and US authorities in 2004 and 2010, respectively [Citation32]. Comparability studies, including comprehensive in vitro, non-clinical, and clinical comparisons of the biosimilar with the reference product, are conducted in a stepwise approach to establish the ‘totality of evidence’ [Citation33,Citation34]. The aim is to convincingly demonstrate that the proposed biosimilar is highly similar to the reference product and prove that the previously established safety and efficacy of the reference product also apply to the biosimilar [Citation16,Citation33].
A stepwise approach is advised to produce the data required to demonstrate biosimilarity [Citation16,Citation33,Citation34]. At each step, the obtained data guides the design of the next one in order to dispel any residual uncertainty. Analytical in vitro studies determine the molecular and functional characteristics of the biosimilar and prove its similarity to the reference product in terms of physicochemical properties (i.e. structure, biological activity, post-translational modifications, thermal stability and purity). In the next step, non-clinical animal studies are conducted to address any residual analytical uncertainty and ensure safe use in humans. These studies mainly focus on pharmacology, including PK and PD, and only determine toxicity in certain cases. Finally, clinical studies in humans aim to confirm biosimilarity and address any remaining uncertainties. The regulatory pathway is not identical to that of new chemical or biological entities as biosimilars are not required to ascertain safety and efficacy that have already been established for the reference product, or to demonstrate superiority versus a comparator. They rather aim to exclude any clinically meaningful differences between the proposed biosimilar and its reference product. By relying on the safety and efficacy data of the reference product, fewer clinical data are required while maintaining the quality and safety of the product, thereby reducing the development costs of a biosimilar.
Based on the concept of the totality of evidence, equivalence of SB8 and reference bevacizumab has been demonstrated in several studies. Initial in vitro studies showed that SB8 and reference bevacizumab have similar physicochemical characteristics and functional properties [Citation35]. As part of the clinical studies for demonstrating biosimilarity, a phase I, randomized, single-dose study comparing SB8 and the reference bevacizumab in healthy male subjects demonstrated equivalent PK and comparable safety and immunogenicity profiles, confirming similarity between the two [Citation26]. A phase III, randomized, double-blind, multicenter study was conducted in patients with metastatic or recurrent non-small-cell lung cancer (NSCLC) [Citation25]. A total of 763 patients were treated with intravenous SB8 (n = 379) or reference bevacizumab (n = 384), co-administered with paclitaxel and carboplatin, every 3 weeks for up to six cycles. The primary end point was best overall response rate (ORR) by 24 weeks. In the full analysis set, the best ORR risk ratio (1.11; 90% CI 0.975–1.269) was within the prespecified equivalence margin (0.737–1.357). In the per protocol set, the best ORR risk difference was 5.3% (95% CI: -2.2% to 12.9%) and thus, the lower margin was within, but the upper margin was slightly above the prespecified equivalence margin (-12.5%–12.5%). Sensitivity analyses supported the robustness of the primary analysis, using age groups (<70, ≥70 years), gender (male, female), geographic region (EU, non-EU) and treatment groups as covariates. Additional efficacy end points, such as PFS and OS, safety, PK, and immunogenicity were comparable between SB8 and reference bevacizumab. In summary, the totality of evidence demonstrates the biosimilarity of SB8 to its reference bevacizumab.
Extrapolation
When a proposed biosimilar has met the criteria of biosimilarity in one therapeutic indication, further indications approved for the reference product can be licensed without direct studies in those indications based on scientific justification. The EMA and FDA grant approval via the process of extrapolation when scientific justification is provided that the mode of action of the reference product does not differ across the approved indications [Citation16,Citation33]. Extrapolation is not unique to biosimilar medicines but rather is an established and reliable regulatory process applied to medicines to avoid unnecessary clinical trials, shorten the development and approval process, and provide timely access to treatment for patients [Citation36]. Importantly, extrapolation must be supported by the totality of evidence demonstrating no anticipated differences in the mode of action as well as in PK, PD, and immunogenic or toxic effects across extrapolated indications for the reference product, and thus for the biosimilar.
Since the approval of the first biosimilar in the EU more than 15 years ago, no safety and efficacy concerns have been reported for biosimilars, neither in originally tested nor in extrapolated indications [Citation37]. Nevertheless, several surveys conducted over the last years among healthcare providers and pharmacists worldwide revealed remaining concerns regarding the use of biosimilars, especially in extrapolated indications. A survey of managed care and specialty pharmacy professionals revealed a higher tendency of prescribing biosimilars for the original indication (84%) than for extrapolated indications (54%) [Citation38]. A similar observation was made among healthcare providers in rheumatology, dermatology, and gastroenterology [Citation39]. Healthcare providers (including physicians specialized in gastroenterology, oncology, hematology, and nurses, consultants and pharmacists) that hesitate to use biosimilars state the concepts of extrapolation and interchangeability as main concerns and give potential new/additional adverse events and immunogenicity as reasons [Citation38,Citation40–44]. These concerns are mainly due to poor understanding of the development and regulatory approval process of biosimilars including the concept of extrapolation, as a study of the European Society for Medical Oncology revealed [Citation45]. Interestingly, there was a higher rate of use in Asia than in Europe, potentially due to several Asian regulatory agencies intensively investing in accelerated licensing of biosimilars and, consequently, a better understanding of the process by healthcare providers.
To date, several biosimilars including SB8 with a phase III equivalence study in one indication have been licensed for the same types of cancer approved for the reference bevacizumab based on extrapolation [Citation46]. Comparability studies that aim to demonstrate biosimilarity are conducted using an equivalence design to establish statistical evidence that the proposed biosimilar is neither inferior nor superior to the reference product. The phase III study of SB8 was conducted, similar to several other licensed or proposed bevacizumab biosimilars [Citation46], in patients with metastatic or recurrent NSCLC [Citation25] rather than mCRC patients based on multiple scientific considerations. The selection of a sensitive study population and an appropriate clinical end point capable of detecting potential differences between biosimilar and reference product are critical for designing clinical trials of biosimilars. In contrast to registration trials of novel anticancer therapeutics where survival-based end points (such as PFS, disease-free survival [DFS] or OS) are preferred to demonstrate clinical benefit and superiority, short-term surrogate end points (such as ORR) measured at a defined time point are considered more appropriate for detecting potential clinically relevant differences in comparability studies [Citation47]. Survival-based end points can be sensitive to factors other than product-related differences [Citation16], usually require larger patient populations with longer follow-up, and may not be amenable to assess the impact of switching between the biosimilar candidate and reference product. Furthermore, historical phase III studies in mCRC with bevacizumab demonstrated superiority of bevacizumab in combination with chemotherapy compared with chemotherapy alone in terms of survival, yet, a clinically meaningful difference was not detected in the response rates [Citation25,Citation48]. Taking the above factors into consideration and based on communications with the FDA and EMA, patients with previously untreated advanced NSCLC are considered a sensitive population for comparability studies of bevacizumab biosimilars to measure differences in response rates. A number of studies of bevacizumab biosimilars in NSCLC patients with ORR as primary end point has been registered, including SB8 [Citation46,Citation49]. The phase III trial conducted in patients with metastatic or recurrent NSCLC demonstrated comparable ORR between bevacizumab and SB8 [Citation25] and thus, extrapolation of SB8 to all cancer indications of reference bevacizumab was considered acceptable as similarity between the two has been demonstrated [Citation1,Citation27].
Scientific justification for extrapolation
The scientific justification for extrapolation is based on an extensive repertoire of clinical studies of bevacizumab in multiple indications and includes data on the mechanism of action, PK, efficacy, safety and immunogenicity.
The mode of action of bevacizumab is inhibition of binding of VEGF-A to its receptor VEGFR on endothelial cells and thereby preventing its pro-angiogenic effects. Solid tumors rely on a nascent vasculature for growth and survival and VEGF is overexpressed in the majority of solid tumors [Citation50]. The expression of VEGF correlates with prognostic indicators across solid tumors in which bevacizumab has been studied and therefore it can be hypothesized that its proposed mode of action, namely the binding of VEGF-A, is considered to be identical across all approved indications [Citation50].
Population pharmacokinetics were analyzed from eight phase I-III clinical studies with reference bevacizumab from patients with solid tumors including colorectal, hormone refractory prostate, metastatic breast, and non-small-cell lung cancer [Citation51]. Bevacizumab was either used as a single therapeutic or in combination with chemotherapy. Bevacizumab doses ranged from 0.1 to 20 mg/kg given every 1 to 3 weeks. Population pharmacokinetics were described with a two-compartment model. Estimated clearance (CL) was 0.207 L/day and volume of distribution (Vc) was 2.39 L for a typical female. The terminal half-life (t1/2) was approximately 20 days for both women and men (). Population pharmacokinetics extracted from 15 phase I-IV clinical studies considering a broader variety of solid tumors analyzed the factors contributing to variability in PK parameters [Citation52]. Factors significantly associated with bevacizumab PK were similar between both studies and gender, body weight, albumin and baseline alkaline phosphatase (BALP) mainly influence CL (increased in males and with higher body weight, decreased with increasing albumin and decreasing BALP). Yet, PK parameters were comparable across all tumor types and with other therapeutic monoclonal antibodies [Citation52].
Table 1. PK parameters of bevacizumab across indications.
The significant clinical benefit of bevacizumab has been consistently demonstrated in several phase II and III studies across multiple indications (summarized in ) [Citation1], resulting in the authorization of bevacizumab in each indication. Six studies conducted in mCRC evaluated the efficacy of bevacizumab in combination with irinotecan plus fluorouracil/leucovorin (IFL), fluorouracil/leucovorin (FU/LV), FOLFOX4, or XELOX chemotherapies as first- or second-line treatment [Citation48,Citation53–57]. Two studies conducted in mBC evaluated the efficacy of bevacizumab in combination with paclitaxel, capecitabine, taxane or anthracycline-based chemotherapies as first-line treatment [Citation58,Citation59]. Three studies conducted in NSCLC evaluated the efficacy of bevacizumab in combination with cisplatin, gemcitabine, erlotinib, or paclitaxel chemotherapy [Citation60–62]. Two studies conducted in mRCC evaluated the efficacy of bevacizumab in combination with IFN-alpha-2a or erlotinib [Citation63,Citation64]. Two studied conducted in epithelial ovarian, fallopian tube or primary peritoneal cancer evaluated the efficacy of bevacizumab in combination with carboplatin and paclitaxel as first-line treatment [Citation65,Citation66]. Three studies conducted in recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer evaluated the efficacy of bevacizumab in combination with carboplatin and paclitaxel or paclitaxel, topotecan, and pegylated liposomal doxorubicin (PLD) as first-line treatment in the recurrent setting [Citation67–69]. One study conducted in cervical cancer evaluated the efficacy of bevacizumab in combination with paclitaxel and cisplatin or paclitaxel and topotecan as first-line treatment [Citation70]. All studies, except for the ones conducted in mRCC, which showed no or only a small difference, demonstrated superiority of bevacizumab combination therapy compared with chemotherapy alone [Citation1].
Table 2. Clinical studies of bevacizumab monitoring efficacy across indications.
Bevacizumab is associated with a number of treatment-related toxicities, such as hypertension, neutropenia, hemorrhage, wound healing issues, and gastrointestinal perforation (). While the types of adverse events are comparable across indications, their incidence differs in different patient populations. For example, while neutropenia is more common in ovarian cancer, thrombotic events occur more often in patients with mCRC. However, these side effects are predictable within the different indications and are thus expected to be comparable with bevacizumab biosimilars.
Table 3. Overview of adverse events reported for bevacizumab across indications.
While toxicity is well established across all bevacizumab indications, clinical data on immunogenicity of bevacizumab across multiple indications is not available. In clinical trials of mCRC, 14 of 2233 patients (0.63%) tested positive for treatment-emergent anti-bevacizumab antibodies and three for neutralizing antibodies against bevacizumab. These results suggest that bevacizumab is inherently non-immunogenic with a low incidence of both binding and neutralizing antibodies.
Overall, the mode of action, PK, efficacy, safety and immunogenicity of bevacizumab are comparable across indications and thus, extrapolation of biosimilars to all indications of reference bevacizumab is scientifically justified.
Bevacizumab biosimilar usage in mCRC indication
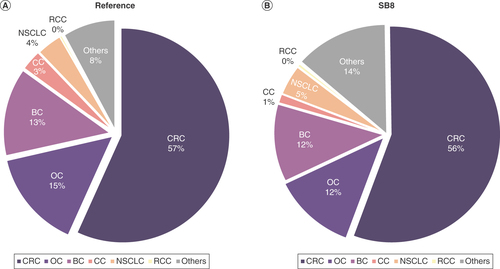
During the last decades, the death rate from CRC has decreased continuously. This can be attributed to the decreasing rate of diagnosed cases due to regular preventive medical checkups, lifestyle changes, and emerging treatment options, especially for mCRC. Bevacizumab has been instrumental in improving OS of patients with mCRC and a number of other solid tumors [Citation73]. Undoubtedly, biosimilars of bevacizumab play an important role in providing treatment to patients. Data on usage of reference bevacizumab and SB8 in France, Germany, Italy, Spain and the UK show that the biosimilar is used similarly to the reference across all tumor types. Both SB8 and reference bevacizumab were mostly used in CRC at 55.7% and 56.7% of total sales, respectively, followed by 12.3% and 14.9% in OC, 11.5% and 13.2% in BC, 4.7% and 3.7% in NSCLC, 1.2% and 3.1% in CC, and 0.4% and 0.2% in RCC for SB8 and reference bevacizumab, respectively (, cumulated data from Q2 2019 to Q1 2022). Furthermore, sales data of reference bevacizumab and a number of biosimilars demonstrate that the absolute number of sold vials (400 mg/16 ml) of all bevacizumab products increased from approximately 200,000 in 2019 to 250,000 in 2022 while the sales values (USD) of all bevacizumab products decreased from approximately 250,000 to 190,000 US dollar in the same period (), emphasizing that indeed biosimilars facilitate patient access to treatment and lower the average price per unit. All sales data was sourced from IQVIA MIDAS® Sales by Disease (from Q2 2019 to Q1 2022, licensed in September 2022), and the number of sold vials (400 mg/16 ml) was calculated (by the authors) from the milliliters of drug sold. IQVIA national audits and MIDAS reflect local industry standard source of pack prices, which might be list price or average invoice price, depending upon the country and the available information; they do not reflect net prices realized by the manufacturers. Sales values reflected in these IQVIA audits are calculated by applying such relevant pricing to the product volume data collected for, and reflected in, such audits. In addition, to allow the national audit sales values to be viewed at a common sales level, MIDAS applies a single average industry margin to the locally reported values. In addition, real-world experience in the US shows that the biosimilars bevacizumab-awwb and bevacizumab-bvzr are actually used in the clinic [Citation74–76], predominantly in mCRC patients [Citation75,Citation76], and that safety and efficacy of the reference and biosimilar are indeed comparable in the real-world setting () [Citation77].
Distribution of sales of (A) Bevacizumab and (B) SB8 across indications in Europe (France, Germany, Italy, Spain, and UK) from second quarter of 2019 to first quarter of 2022. Internal analysis by the authors using data from the following source: IQVIA MIDAS® Sales by Disease quarterly data reflecting estimates of real-world activity. Copyright IQVIA. All rights reserved.
BC: Breast cancer; CC: Cervical cancer; CRC: Colorectal cancer; NSCLC: Non-small-cell lung cancer; OC: Ovarian cancer; RCC: Renal cell carcinoma; UK: United Kingdom.
(A) Sold vials and (B) sales values (in million USD) of reference bevacizumab and biosimilars (including Aybintio, Alymsys/Oyavas, Mvasi and Zirabev) in Europe (France, Germany, Italy, Spain and UK) from second quarter of 2019 to first quarter of 2022. Internal analysis by the authors using data from the following source: IQVIA MIDAS® Sales by Disease quarterly data reflecting estimates of real-world activity. Copyright IQVIA. All rights reserved.
Q: Quarter; UK: United Kingdom; USD: United States Dollar.
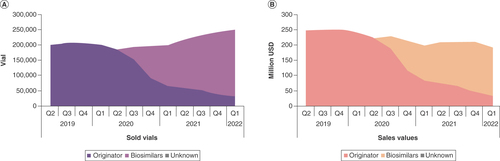
Table 4. Real-world data of bevacizumab biosimilars in mCRC.
Although all approved bevacizumab biosimilars were evaluated in NSCLC patients in clinical trials, the experience of the last years shows that biosimilars are used in multiple indications and predominantly in mCRC patients, similar to reference bevacizumab. Indeed, bevacizumab biosimilars have successfully entered the clinic and now make up 87% of sold vials in several European countries (France, Germany, Italy, Spain and UK), highlighting the requirement for affordable and efficacious cancer therapies ().
Further convenience of SB8
The efficacy of therapeutic monoclonal antibodies depends on their structural integrity. They are usually available in liquid form in single-use vials that require dilution prior to administration. The prescribing information of Avastin (Roche, Genentech) recommends storage of diluted product at 2–8°C for up to 8 h [Citation79], yet, physical in-use stability has been demonstrated at 2–8°C for 30 days plus an additional 48 h at 2–30°C [Citation1]. The prolonged stability of biologics is of utmost importance to decrease drug waste and ensure persistent efficacy in case the diluted product needs to be stored or transported. The stability of several bevacizumab biosimilars has been analyzed (). After extended storage of unopened (at 2–8°C for 36 months and subsequently at 28–32°C for 1 month), opened (at 2–8°C for 72 h), and diluted product (at 2–8°C for 45 days and subsequently at 28–32°C for 3 days) in an environmentally controlled environment, SB8 showed no detriment in protein recovery, activity or purity [Citation80]. These findings can support the definition of increased shelf-life which may eventually reduce drug waste and support healthcare providers faced with exceptional situations. Therefore, the prolonged stability of SB8 adds another benefit to the use of SB8 among bevacizumab and its biosimilars.
Table 5. Stability of bevacizumab biosimilars.
Conclusion
The humanized monoclonal antibody bevacizumab targeting VEGF is a biologic medicine successfully used for the treatment of a number of solid tumors. Since the patent expiration of bevacizumab in 2019 and 2022 in the US and EU, respectively, several bevacizumab biosimilars, including SB8, have been licensed. Biosimilars are evaluated using a sound scientific and regulatory procedure. SB8 was found to be highly similar to reference bevacizumab based on comparative non-clinical and clinical studies in NSCLC patients. SB8 has been approved as an equivalent therapeutic option by health authorities in the EU, Korea, Canada and Taiwan for the same types of cancers as reference bevacizumab based on the scientific principle of extrapolation. Despite remaining concerns regarding biosimilars among healthcare providers and patients, real-world experience of bevacizumab biosimilars in mCRC patients provides evidence that they are comparable in efficacy and safety to reference bevacizumab, that they have been integrated into clinical practice, and that they have the potential to significantly decrease treatment costs and increase patients’ access to treatment.
Future perspective
By 2022, nine bevacizumab biosimilars have been approved in the EU and four in the US [Citation84,Citation85], with several more in the pipeline [Citation46]. All approved bevacizumab biosimilars were evaluated in pivotal trials in NSCLC patients [Citation46] but are licensed for use in the same indications as the reference product based on the concept of extrapolation [Citation27,Citation28]. Indeed, real-world experience showed that the majority of patients using bevacizumab biosimilars have been diagnosed with mCRC [Citation75,Citation76]. However, there is a remaining concern among healthcare providers that leaves them hesitant to use biosimilars in general and in extrapolated indications in particular [Citation38,Citation40,Citation41]. Many of those concerns are based on a lack of knowledge of the scientific and regulatory process of biosimilar development and approval which need to be addressed by educational efforts. Regulatory authorities have established strict and clear guidelines that demand extensive and sound non-clinical and clinical data that demonstrate biosimilarity of the candidate and reference product and justify the extrapolation to all indications [Citation16,Citation33,Citation47]. Since the approval of the first biosimilar in the EU more than 15 years ago, the number of licensed biosimilars is ever rising. Especially the EMA endorses the development and approval of biosimilars, signified by the recommendation to approve 88 biosimilars by 2022 [Citation84]. In comparison, the FDA approved only 36 biosimilars to date [Citation85]. Biosimilars are an important addition to therapeutic options. Considering the high costs of biologics [Citation12], the availability of more cost-effective alternatives significantly increases patient access to affordable treatment [Citation18]. With the continued extension of treatment options and the increased awareness and acceptance among healthcare providers and patients, the market share of biosimilars will expand in the years to come and help to provide treatment benefit to many patients.
Totality of evidence
The authorization of biosimilars is based on the totality of evidence, in which comparability studies establish the biosimilarity of the candidate to its reference product in a stepwise approach.
In vitro, non-clinical and clinical data demonstrate that the biosimilar and its reference product are equivalent in terms of efficacy, safety, pharmacokinetic, pharmacodynamic and immunogenic properties.
SB8 has been approved as a biosimilar of bevacizumab based on the provided totality of evidence.
Extrapolation
Extrapolation is a scientific and regulatory principle that enables approval of a biosimilar for use in an indication held by the reference product without conducting additional comparability studies.
Prerequisites are that the mode of action of the reference product is the same in all indications and that the totality of evidence supports that there are no clinically meaningful differences between the reference product and the biosimilar.
Extrapolation is one of the main concerns that prevents healthcare providers from the use of biosimilars; consequently, educational efforts are advised to address these concerns.
Multiple bevacizumab biosimilars, including SB8, have been approved for all indications of the reference based on extrapolation while their comparability studies were predominantly conducted in non-small-cell lung cancer (NSCLC) patients which proved to be a sensitive population.
Real-world usage in metastatic colorectal cancer indication
SB8 is used similarly to bevacizumab reference across all indications in the real-world setting with colorectal cancer making up more than 50% of all sales.
The absolute number of sold vials increased since introduction of bevacizumab biosimilars, while the total cost spending on treatment with bevacizumab has been reduced.
Real-world experience shows that bevacizumab biosimilars have successfully entered the clinic.
Further convenience of SB8
Structural integrity is a prerequisite for the efficacy of therapeutic monoclonal antibodies.
Prolonged storage of undiluted and diluted product may cut drug waste and facilitate treatment in challenging circumstances.
SB8 provides extended structural integrity of up to 36 months at 2–8°C and subsequently at 28–32°C for 1 month of unopened and up to 45 days at 2–8°C plus an additional 3 days at 28–32°C of diluted product under ambient and in-use conditions.
Author contributions
M Peeters, H-P Lipp and D Arnold contributed to conception of the review. M Park and Y Chan Yoon contributed to acquisition and analysis of references and data. All authors contributed to the interpretation of available evidence and critically reviewing the manuscript for important intellectual content. D Arnold and M Park acted as coordinators and provided important intellectual contributions to the review.
Financial & competing interests disclosure
The project was sponsored by Samsung Bioepis. M Peeters is member of the advisory boards of Amgen, Bayer, Bimini, Ipsen, Merck, MSD, Qurin, Remedus, Sanofi, Sirtex, and Terumo, received speaker fees from Amgen, Bayer, BMS, Merck, MSD, Roche, Sanofi, Servier, and Sirtex, and received scientific grants from Amgen (Inst.), Bayer (Inst.), BMS (Inst.), Ipsen (Inst)., Novartis (Inst.), and Roche (Inst.). H-P Lipp is member of the advisory boards of Hexal/Sandoz, MSD, Pfizer, and Samsung Bioepis and received speaker fees from Amgen, medac, MSD, and Pfizer. D Arnold is member of the advisory boards of Astra Zeneca, Bayer, BMS, Boston Scientific, Eli Lilly, Merck Serono, MSD, Roche, Sanofi, Seagen, Servier, Sirtex, and Terumo, received speaker fees from Amgen, Astra Zeneca, Bayer, BMS, Boston Scientific, GSK, Merck Serono, Roche, Samsung, Sandoz, Sanofi, Seagen, Servier, Sirtex, Terumo, Viatris, Art Tempi, PriME Oncology, and TRM Oncology, received travel support from Astra Zeneca, BMS, MSD, Roche, and Sanofi, received research funding to the institute from Astra Zeneca, InCyte, MSD, Roche, and Sanofi, and received support for non-remunerated activities from Mologen and Oncolytics. M Park and Y Chan Yoon are employees of Samsung Bioepis. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing support was provided by Marietta Hartl, SFL Regulatory Affairs & Scientific Communications, Switzerland, and funded by Samsung Bioepis.
Acknowledgments
The statements, findings, conclusions, views, and opinions contained and expressed in this article are based in part on data obtained under license from the following IQVIA Ltd information service: IQVIA MIDAS® Sales by Disease quarterly data for the period Q2 2019 to Q1 2022. These statements, findings, conclusions, views and opinions are not necessarily those of IQVIA Ltd or any of its affiliated or subsidiary entities.
Additional information
Funding
References
- Summary of product characteristics – Avastin. www.ema.europa.eu/en/documents/product-information/avastin-epar-product-information_en.pdf ( August 15, 2022).
- Ferrara N . VEGF as a therapeutic target in cancer. Oncology69, 11–16 (2005).
- Garcia J , HurwitzHI, SandlerABet al. Bevacizumab (Avastin) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat. Rev.86, 102017 (2020).
- Medicare Part B Spending by Drug. https://data.cms.gov/summary-statistics-on-use-and-payments/medicare-medicaid-spending-by-drug/medicare-part-b-spending-by-drug ( August 16, 2022).
- Monk BJ , LammersPE, CartwrightT, JacobsI. Barriers to the access of bevacizumab in patients with solid tumors and the potential impact of biosimilars: a physician survey. Pharmaceuticals (Basel)10(1), 19 (2017).
- Li E , SchleifR, EdelenB. Hospital management of outpatient oncology treatment decisions: a survey to identify strategies and concerns. J. Oncol. Pract.9(5), e248–e254 (2013).
- Cherny N , SullivanR, TorodeJ, SaarM, EniuA. ESMO European Consortium Study on the availability, out-of-pocket costs and accessibility of antineoplastic medicines in Europe. Ann. Oncol.27(8), 1423–1443 (2016).
- ACS CAN examination of cancer drug coverage and transparency in the health insurance marketplaces. www.fightcancer.org/sites/default/files/National%20Documents/QHP%20Formularies%20Analysis%20-%202017%20FINAL.pdf ( August 16, 2022).
- Nadler E , EckertB, NeumannPJ. Do oncologists believe new cancer drugs offer good value?The Oncologist11, 90–95 (2006).
- Bevacizumab in combination with oxaliplatin and either fluorouracil plus folinic acid or capecitabine for the treatment of metastatic colorectal cancer. www.nice.org.uk/guidance/ta212/resources/bevacizumab-in-combination-with-oxaliplatin-and-either-fluorouracil-plus-folinic-acid-or-capecitabine-for-the-treatment-of-metastatic-colorectal-cancer-pdf-82600246301893 ( August 17, 2022).
- Li E , SubramanianJ, AndersonS, ThomasD, MckinleyJ, JacobsIA. Development of biosimilars in an era of oncologic drug shortages. Drug Des. Devel. Ther.9, 3247–3255 (2015).
- Makurvet FD . Biologics vs. small molecules: drug costs and patient access. Medicine in Drug Discovery9, 100075 (2021).
- Biosimilar and interchangeable products. www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products ( August 19, 2022).
- Guidelines on evaluation of similar biotherapeutic products (SBPs), Annex 2, TRS No 977. https://cdn.who.int/media/docs/default-source/biologicals/trs_977_annex_2.pdf?sfvrsn=e2389a69_3&download=true ( August 16, 2022).
- Biosimilars in the EU – Information guide for healthcare professionals. www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf ( August 15, 2022).
- Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf ( August 17, 2022).
- Considerations in demonstrating interchangeability with a reference product – guidance for industry. www.fda.gov/media/124907/download ( August 19, 2022).
- The impact of biosimilar competition in Europe. www.medicinesforeurope.com/wp-content/uploads/2017/05/IMS-Biosimilar-2017_V9.pdf ( August 17, 2022).
- Calleja MA , AlbanellJ, ArandaEet al. Budget impact analysis of bevacizumab biosimilars for cancer treatment in adult patients in Spain. Eur. J. Hosp. Pharm.doi:10.1136/ejhpharm-2021-002955 (2021).
- Jha A , UptonA, DunlopWC, AkehurstR. The budget impact of biosimilar Infliximab (Remsima(R)) for the treatment of autoimmune diseases in five European countries. Adv. Ther.32(8), 742–756 (2015).
- NHS set to save record £300 million on the NHS’s highest drug spend. www.england.nhs.uk/2018/11/nhs-set-to-save-record-300-million-on-the-nhss-highest-drug-spend/ ( August 17, 2022).
- Kvien TK , PatelK, StrandV. The cost savings of biosimilars can help increase patient access and lift the financial burden of health care systems. Semin. Arthritis Rheum.52, 151939 (2022).
- Mulcahy AW , HlavkaJP, CaseSR. Biosimilar cost savings in the United States. Rand Health Quarterly7(4), 3 (2018).
- Delivering on the potential of biosimilar medicines. www.medicinesforeurope.com/wp-content/uploads/2016/03/IMS-Institute-Biosimilar-Report-March-2016-FINAL.pdf ( August 17, 2022).
- Reck M , LuftA, BondarenkoIet al. A phase III, randomized, double-blind, multicenter study to compare the efficacy, safety, pharmacokinetics, and immunogenicity between SB8 (proposed bevacizumab biosimilar) and reference bevacizumab in patients with metastatic or recurrent nonsquamous non-small cell lung cancer. Lung Cancer146, 12–18 (2020).
- Shin D , LeeYJ, ChoiJ, LeeD, ParkM, PetkovaM. A phase I, randomized, single-dose pharmacokinetic study comparing sb8 (bevacizumab biosimilar) with reference bevacizumab in healthy volunteers. Cancer Chemother. Pharmacol.86(4), 567–575 (2020).
- Summary of product characteristics – Aybintio. www.ema.europa.eu/en/documents/product-information/aybintio-epar-product-information_en.pdf ( August 16, 2022).
- Summary of product characteristics – Onbevzi. /www.ema.europa.eu/en/documents/product-information/onbevzi-epar-product-information_en.pdf ( August 16, 2022).
- Assessment summary information for biosimilar IPRP biosimilars WG (Onbevzi). www.mfds.go.kr/com/file/down.do?dnCd=eng&fileNm=PASIB_Onbevzi(MFDS).pdf&filePath=contents/ ( August 16, 2022).
- Drug submission status Aybintio. www.health.gov.on.ca/en/pro/programs/drugs/drug_submissions/subm_stat_reports/pdf/aybintio.pdf ( August 16, 2022).
- Onbevci – License details. https://info.fda.gov.tw/MLMS/H0001D.aspx?Type=Lic&LicId=60001193 ( September 29, 2022).
- Wang J , ChowSC. On the regulatory approval pathway of biosimilar products. Pharmaceuticals (Basel)5(4), 353–368 (2012).
- Scientific considerations in demonstrating biosimilarity to a reference product. www.fda.gov/media/82647/download ( August 18, 2022).
- Markus R , LiuJ, RamchandaniM, LandaD, BornT, KaurP. Developing the totality of evidence for biosimilars: regulatory considerations and building confidence for the healthcare community. BioDrugs31(3), 175–187 (2017).
- Assessment report – Aybintio. www.ema.europa.eu/en/documents/assessment-report/aybintio-epar-public-assessment-report_en.pdf ( November 3, 2022).
- Weise M , KurkiP, Wolff-HolzE, BielskyMC, SchneiderCK. Biosimilars: the science of extrapolation. Blood124(22), 3191–3196 (2014).
- Kurki P , BarryS, BourgesI, TsantiliP, Wolff-HolzE. Safety, immunogenicity and interchangeability of biosimilar monoclonal antibodies and fusion proteins: a regulatory perspective. Drugs81(16), 1881–1896 (2021).
- Greene L , SinghRM, CardenMJ, PardoCO, LichtensteinGR. Strategies for overcoming barriers to adopting biosimilars and achieving goals of the biologics price competition and innovation: a survey of managed care and speciality pharmacy professionals. J. Manag. Care Spec. Pharm.25(8), 904–912 (2019).
- Teeple A , EllisLA, HuffLet al. Physician attitudes about non-medical switching to biosimilars: results from an online physician survey in the United States. Curr. Med. Res. Opin.35(4), 611–617 (2019).
- Aladul MI , FitzpatrickRW, ChapmanSR. Healthcare professionals’ perceptions and perspectives on biosimilar medicines and the barriers and facilitators to their prescribing in UK: a qualitative study. BMJ Open8(11), e023603 (2018).
- Leonard E , WascovichM, OskoueiS, GurzP, CarpenterD. Factors affecting health care provider knowledge and acceptance of biosimilar medicines: a systematic review. J. Manag. Care Spec. Pharm.25(1), 102–112 (2019).
- Park SK , MoonW, KimES, ParkSH, ParkDI. Knowledge and viewpoints on biosimilar monoclonal antibodies among Asian physicians: comparison with European physicians. Korean J. Gastroenterol.74(6), 333–340 (2019).
- Hadoussa S , BouhlelM, SoussiMA, DriraC, HadoussaM, KhroufMR. Perception of hematologists and oncologists about the biosimilars: a prospective Tunisian study based on a survey. J. Oncol. Pharm. Pract.26(1), 124–132 (2020).
- Sarnola K , MerikoskiM, JyrkkaJ, Hameen-AnttilaK. Physicians’ perceptions of the uptake of biosimilars: a systematic review. BMJ Open10(5), e034183 (2020).
- Giuliani R , TaberneroJ, CardosoF, McgregorKH, VyasM, DeVries EGE. Knowledge and use of biosimilars in oncology: a survey by the European Society for Medical Oncology. ESMO Open4(2), e000460 (2019).
- Biosimilars of bevacizumab. https://gabionline.net/biosimilars/general/Biosimilars-of-bevacizumab ( August 22, 2022).
- Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf ( August 22, 2022).
- Saltz LB , ClarkeS, Diaz-RubioEet al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J. Clin. Oncol.26(12), 2013–2019 (2008).
- Melosky B , ReardonDA, NixonAB, SubramanianJ, BairAH, JacobsI. Bavacizumab biosimilars: scientific justification for extrapolation of indications. Future Oncol.14(24), 2507–2520 (2018).
- Background information for the Oncologic Drugs Advisory Committee 13 July 2017 – Biologics License Application for ABP215. https://fda.report/media/106549/Amgen-Briefing-Information-for-the-July-13--2017-Meeting-of-the-Oncologic-Drugs-Advisory-Committee.pdf ( August 22, 2022).
- Lu JF , BrunoR, EpplerS, NovotnyW, LumB, GaudreaultJ. Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother. Pharmacol.62(5), 779–786 (2008).
- Han K , PeyretT, MarchandMet al. Population pharmacokinetics of bevacizumab in cancer patients with external validation. Cancer Chemother. Pharmacol.78(2), 341–351 (2016).
- Hurwitz HI , FehrenbachL, NovotnyWet al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for metastatic colorectal cancer. N. Engl. J. Med350, 2335–2342 (2004).
- Kabbinavar F , HurwitzHI, FehrenbacherLet al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J. Clin. Oncol.21(1), 60–65 (2003).
- Kabbinavar FF , SchulzJ, MccleodMet al. Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results of a randomized phase II trial. J. Clin. Oncol.23(16), 3697–3705 (2005).
- Giantonio BJ , CatalanoPJ, MeropolNJet al. Bevacizumab in combination with Oxaliplatin, Fluorouracil, and Leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol.25(12), 1539–1544 (2007).
- Bennouna J , SastreJ, ArnoldDet al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. The Lancet Oncology14(1), 29–37 (2013).
- Gray R , BhattacharyaS, BowdenC, MillerK, ComisRL. Independent review of E2100: a phase III trial of bevacizumab plus paclitaxel versus paclitaxel in women with metastatic breast cancer. J. Clin. Oncol.27(30), 4966–4972 (2009).
- Robert NJ , DierasV, GlaspyJet al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J. Clin. Oncol.29(10), 1252–1260 (2011).
- Sandler AB , GrayR, PerryMCet al. Paclitaxel-Carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med355, 2542–2550 (2006).
- Leighl NB , ZatloukalP, MezgerJet al. Efficacy and safety of bevacizumab-based therapy in elderly patients with advanced or recurrent nonsquamous non-small cell lung cancer in the phase III BO17704 Study (AVAiL). J. Clin. Oncol.5(12), 1970–1976 (2010).
- Seto T , KatoT, NishioMet al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. Lancet Oncol.15(11), 1236–1244 (2014).
- Escudier B , PluzanskaA, KoralewskiPet al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet370(9605), 2103–2111 (2007).
- Bukowski RM , KabbinavarFF, FiglinRAet al. Randomized phase II study of erlotinib combined with bevacizumab compared with bevacizumab alone in metastatic renal cell cancer. J. Clin. Oncol.25(29), 4536–4541 (2007).
- Tewari KS , BurgerRA, EnserroDet al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J. Clin. Oncol.37(26), 2317–2328 (2019).
- Perren TJ , SwartAM, PfistererJet al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med365(26), 2484–2496 (2011).
- Aghajanian C , BlankSV, GoffBAet al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol.30(17), 2039–2045 (2012).
- Coleman RL , BradyMF, HerzogTJet al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol.18(6), 779–791 (2017).
- Pujade-Lauraine E , HilpertF, WeberBet al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J. Clin. Oncol.32(13), 1302–1308 (2014).
- Tewari KS , SillMW, LongHJ3rdet al. Improved survival with bevacizumab in advanced cervical cancer. N. Engl. J. Med.370(8), 734–743 (2014).
- Miller K , WangM, GralowJet al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med357, 2666–2676 (2007).
- Burger RA , BradyMF, BookmanMAet al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med365, 2473–2483 (2011).
- Rosen LS , JacobsIA, BurkesRL. Bevacizumab in colorectal cancer: current role in treatment and the potential of biosimilars. Target Oncol.12(5), 599–610 (2017).
- Rhodes W , DeclueRW, AccorttNet al. Real-world use of bevacizumab-awwb, a bevacizumab biosimilar, in US patients with metastatic colorectal cancer. Future Oncol.17(36), 5119–5127 (2021).
- Jin R , MahtaniRL, AccorttN, LawrenceT, SandschaferD, Loaiza-BonillaA. Clinical and treatment characteristics of patients treated with the first therapeutic oncology biosimilars bevacizumab-awwb and trastuzumab-anns in the US. Ther. Adv. Med. Oncol.13, 17588359211041961 (2021).
- Yang J , KeltonJM, ThompsonJ, JimenezJM, MaculaitisMC, ShelbayaA. Real-world usage of bevacizumab-bvzr biosimilar in US Oncology Practice. AJMC28(4), 160–166 (2022).
- Pham C , NiuF, DelateTet al. Real-world outcomes of biosimilar bevacizumab-awwb versus reference bevacizumab in patients with metastatic colorectal cancer. ASCO Annual Meeting I17(36), 5119–5127 (2022).
- Jin R , OgbomoAS, AccorttNet al. Real-world outcomes among patients with metastatic colorectal cancer treated first line with bevacizumab-awwb. ASCO Annual Meeting I40(Suppl. 16), e15581–e15581 (2022).
- Prescribing information Avastin. www.gene.com/download/pdf/avastin_prescribing.pdf ( August 17, 2022).
- Park D , KimJ, YunJ, ParkSJ. Evaluation of the physico-chemical and biological stability of SB8 (Aybintio), a proposed biosimilar to bevacizumab, under ambient and in-use conditions. Adv. Ther.37(10), 4308–4324 (2020).
- Goldschmidt J , HanesV. The totality of evidence and use of ABP 215, a biosimilar to bevacizumab. Oncol. Ther.9(1), 213–223 (2021).
- Arvinte T , PalaisC, PoirierEet al. Part 2: physicochemical characterization of bevacizumab in 2mg/ml antibody solutions as used in human i.v. administration: Comparison of originator with a biosimilar candidate. J. Pharm. Biomed. Anal.176, 112802 (2019).
- Abdel-Tawab M , WassmuthM, GegenfurtnerFet al. Short-term study on in-use stability of opened bevacizumab biosimilar PF-06439535 vials. Eur. J. Hosp. Pharm.doi:10.1136/ejhpharm-2021-003198 (2022).
- Biosimilars approved in Europe. www.gabionline.net/biosimilars/general/biosimilars-approved-in-europe ( August 26, 2022).
- FDA-approved biosimilar products. www.fda.gov/drugs/biosimilars/biosimilar-product-information ( August 26, 2022).