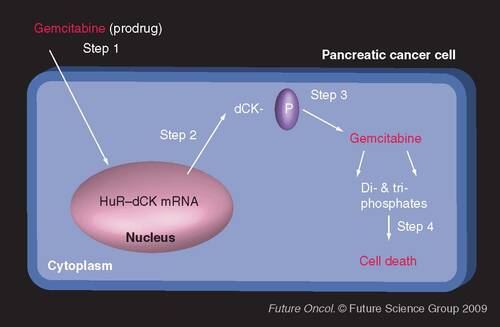
Step 1: Gemcitabine is infused as a prodrug; Step 2: Gemcitabine induces the cytoplasmic accumulation of HuR–dCK mRNA complexes (as well as the accumulation of HuR associated to other mRNAs, yet to be identified); Step 3: Activated and increased HuR enhances the expression of dCK (the enzyme that activates the prodrug gemcitabine), and proteins encoded by other HuR target mRNAs; Step 4: Gemcitabine (prodrug) is metabolized into active metabolites in cancer cells, in turn inhibiting the elongation of the DNA chain and causing cell death.
dCK: Deoxycytidine kinase.
The cancer research community, both researchers and clinicians, share a common vision that targeted and personalized medicine is the future of modern oncology. Many of us believe that identifying a patient’s genetic polymorphism, or detecting a specific protein’s level of expression or activity in a tumor or surrounding stromal cells, will ultimately predict a patient’s response to a specific drug. Thus, someday we hope to be able to simply correlate a molecular signature with a chemotherapeutic cocktail that is just right for each patient. This concept revolutionized the treatment of bacterial infections (specific antibiotic selected for a specific bacterium) and yet there have been only a few such successful examples of this in cancer therapy (e.g., Gleevec® in gastrointestinal stromal tumor/chronic myeloid leukemia and Herceptin® in breast cancer). Currently, large volumes of data are being generated and analyzed to understand what the drug of choice should be for each individual patient or tumor. These endeavors include large-scale cancer genome sequencing projects, knockout gene models and high-throughput drug screens. These efforts are innovative and have already generated landmark findings.
Although current technology and resources allow for such large-scale, high-speed DNA sequencing, there is still a need for understanding pathways involved in the processes of tumorigenesis, drug metabolism and drug resistance. Certainly, discovering genetic mutations in tumor cells will and has identified biochemical pathways that are disrupted in the complex process of tumorigenesis, yet it may not be telling us everything we need to know in order to break de novo or acquired drug resistance by cancer cells (i.e., killing tumor cells). Our recent studies showed that a cellular stress response pathway is activated when cancer cells are exposed to commonly used chemotherapeutics, such as gemcitabine and tamoxifen Citation[1,2]. Understanding such key pathways that are activated upon drug exposure can offer highly informative insights into the mechanisms of cancer cell resistance and sensitivity. To illustrate this notion, we provide an overview of a molecular response activated upon stress in cancer cells, which likely regulates many genes involved in cancer cell susceptibility to drugs.
Cellular stress and environmental factors dramatically impact overall cell function and proliferation. This can occur in the absence of genetic mutations. The proteins that control both the rate of decay of mRNA transcripts and protein translation rates can dramatically impact upon signaling pathways and cell outcomes. Rapid changes in the subset of expressed mRNAs and proteins, some elicited within minutes, occur when cells are treated with chemotherapeutic agents. This is too fast a timeframe to be mediated by gene transcription but can easily arise through post-transcriptional alteration in mRNA stability and/or protein translation rate. Our own work has shown that gemcitabine and tamoxifen treatment of cancer cells can alter the location of key RNA-binding proteins that could dramatically alter gene-expression patterns and hence modulate cellular responses. Furthermore, it suggests that there is a critical need to examine post-transcriptional regulatory processes in order to understand patients’ response to therapy and to help predict which patients would or would not benefit from the currently available arsenal of therapies.
An important RNA-binding protein, HuR associates with collections of target mRNAs and stabilizes and/or modulates their translation, causing key changes in the levels of proteins implicated in cell survival. Proteins encoded by HuR-target mRNAs include c-Myc, p53, deoxycytidine kinase, HIF-1α, VEGF, Bcl-2, estrogen receptor and many cyclins Citation[1–6]. HuR potently affects the abundance of these proteins, akin to the modulation in levels elicited by transcription or epigenetic factors. Importantly, however, HuR can elicit its actions rapidly and transiently. For instance, we found that induction of this pathway most likely occurs within 2 h post-exposure to stress agents such as gemcitabine, while genetic or epigenetic changes may take decades to exert an influence on a cell.
To date, no mutations within the HuR gene have been reported. Yet, we have seen that the subcellular localization of HuR contributes to drug responses. Although the outcomes are different depending on the drug treatment, increased cytoplasmic levels of HuR dramatically increase response or establish resistance to commonly used therapeutics. For example, treatment of cancer cells with gemcitabine or tamoxifen induces HuR to either stabilize the mRNA and alter the protein translation of key genes involved in drug metabolism (deoxycytidine kinase [dCK]) and drug resistance (estrogen receptor and EGF receptor), respectively Citation[1,2,7].
These are but two examples of how the distribution and function of an RNA-binding protein can dramatically impact drug responses. Currently, we are screening for other agents whose actions may be regulated by HuR in a similar manner. Moreover, many other RNA-binding proteins are implicated in stress responsiveness; whether or not they affect cancer cell responses to chemotherapy remains to be identified. The onslaught of research on microRNAs and their capacity to regulate mRNA stability and translation has already exposed both the potential and the infancy of the field of post-transcriptional gene regulation, particularly in cancer. It is exciting to see how post-transcriptional gene regulation by microRNAs and RNA-binding proteins will help predict and tailor drug therapy in the near future, as well as improve our understanding of the tumorigenesis process. We believe that HuR holds great potential to play a major role in the cancer cell response to the stress elicited by chemotherapeutic drugs and should be exploited accordingly.
Study of the ‘RNA-binding ome’ may help to uncover successful new therapies and therapeutic approaches that may ultimately reduce side effects and increase overall drug effectiveness. Future drug treatments may rely on not only information about a patient’s tumor genetic material, but also on the tumor’s RNA-binding protein distribution and microRNA expression profiles. Thus, we believe that this underappreciated level of gene regulation will provide important knowledge as we advance towards a modern era of personalized therapy in medical oncology.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
Bibliography
- Costantino CL , WitkiewiczAK, KuwanoYet al.: The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase.Cancer Res.69(11), 4567–4572 (2009).
- Hostetter C , LicataLA, WitkiewiczAet al.: Cytoplasmic accumulation of the RNA binding protein HuR is central to tamoxifen resistance in estrogen receptor positive breast cancer cells.Cancer Biol. Ther.7(9), 1496–1506 (2008).
- Galbán S , KuwanoY, PullmannRJret al.: RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1 α.Mol. Cell. Biol.28(1), 93–107 (2007).
- Kim HH , KuwanoY, SrikantanS, LeeEK, MartindaleJL, GorospeM: HuR recruits let-7/RISC to repress c-Myc expression.Genes Dev.23(15), 1743–1748 (2009).
- Masuda K , AbdelmohsenK, GorospeM: RNA-binding proteins implicated in the hypoxic response.J. Cell. Mol. Med. DOI: 10.1111/j.1582-4934.2009.00842.x (2009) (Epub ahead of print).
- Zou T , Mazan-MamczarzK, RaoJNet al.: Polyamine depletion increases cytoplasmic levels of RNA-binding protein HuR leading to stabilization of nucleophosmin and p53 mRNAs.J. Biol. Chem.281(28), 19387–19394 (2006).
- Scott GK , MarxC, BergerCEet al.: Destabilization of ERBB2 transcripts by targeting 3´ untranslated region messenger RNA associated HuR and histone deacetylase. 10.1158/1541-7786.MCR-09-0546 Mol. Cancer Res.6(7), 1250–1258 (2008).