
ABSTRACT
Tyrosine kinase inhibitors are novel therapies targeting specific cellular signalling pathways. Sunitinib and sorafenib primarily block tyrosine kinase receptors involved in the progression of many tumours, including clear cell renal cell cancer (ccRCC). Although developed to target selected receptors, it is becoming apparent that they inhibit other kinases; this may result in the development of unexpected side effects. This is potentially dangerous as kinases on noncancerous cells are also inhibited. TKI off-target effects contributing to cardiotoxicity, hypothyroidism, hypertension, fatigue, hair depigmentation, hand–foot syndrome and gastrointestinal perforation have been described. We report three patients (3/412) treated with sunitinib and sorafenib who developed chronic myeloid leukaemia (CML) during treatment for ccRCC, proposing a molecular mechanism of tyrosine kinase inhibitors action on bone marrow cells that might be co-responsible for CML development.
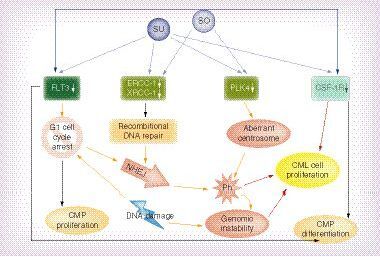
TKI (So and Su) and kinases inhibited by TKIs (Flt-3, ERCC-1, XRCC-1, PLK4 and CSF-1R). Blue arrows indicate inhibitory activity of TKI on target genes. Yellow arrows indicate promotion of cellular events: cell cycle arrest in G1 phase; DNA repair; NHEJ; and Ph developement. Black arrows indicate inhibition of cellular processes mediated by inhibited TKI: CMP proliferation and differentiation. Red arrows indicate processes leading to CML cells proliferation.
CML: Chronic myeloid leukaemia; CMP: Common myeloid progenitor; NHEJ: Non-homologous end-joining pathway; Ph: Philadelphia chromosome; So: Sorafenib; Su: Sunitinib; TKI: Tyrosine kinase inhibitor.
Background
A targeted therapy represents a major breakthrough in the treatment of clear cell renal cell cancer (ccRCC). Nevertheless TKI therapy is associated with multiple toxicities related to off-target inhibition [Citation1,Citation2,Citation3,Citation4,Citation5]. Sunitinib inhibits 73 and sorafenib inhibits 40 additional kinases resulting in a heterogeneous activity profile, which is poorly predictable at this point of time [Citation6,Citation7]. Toxicities induced by TKIs may be detrimental to patient quality of life. Common adverse events (AEs) with newer drugs include hypertension, skin reactions, anemia, neutropenia, gastrointestinal disturbances, thyroid dysfunction and fatigue [Citation8–11]. Despite good responses and treatments that are tolerable in ccRCC patients, little is known concerning the long-term toxicity of these agents [Citation11,Citation12]. Little is known about the mechanisms leading to these side effects, which makes prediction or causal treatment of side effects impossible. Their exploration remains a priority to improve ccRCC management [Citation13].
Secondary hematological malignancies that develop after oncological treatment are increasingly reported as an important late complication in cancer patients. Hematologic malignancies may arise after chemotherapy and/or radiotherapy, as well as to immunosuppressive treatment following allogenic organ transplantation [Citation14]. It was shown that CML non-related to cancer treatment (non-treatment-related chronic myeloid leukemia NTR CML), as a second malignancy, is a rare entity associated with elderly age (median 78 years of age). At the same time secondary CML – related to cancer treatment – develops at a younger age (median age 53 years) [Citation14]. Moreover in a retrospective analysis of patients with renal cell carcinoma, lymphoma, leukemia, or myeloma treated in the University of California in Los Angeles (1 July 1993–30 June 1995), it was reported that renal cell cancer (RCC) was diagnosed in 186 patients, whereas lymphoid malignancy was diagnosed in 405 patients and eight patients developed both disorders – RCC and lymphoma or leukemia. In four of eight patients, RCC was diagnosed prior to hematologic malignancy, whereas in the remaining four patients, the hematologic malignancy was diagnosed first. In this study none of the reported hematologic malignancies was CML. This analysis enables the understanding that that the probability of RCC-hematologic malignancy association arising by chance is unlikely given the incidence of these tumors in the general population. Treatment-related secondary malignancy development mechanism was shown as unlikely in five of eight patients, due to the timing or nature of the previous treatments, but was shown as possible in three of eight patients. For all first-RCC patients, treatment-related hematologic malignancy development was excluded at that time, but RCC patients were treated with nephrectomy only. Other possible explanations for the RCC–hematologic malignancy relationship suggested were: genetic mutation common to both malignancies; environmental exposure; immunomodulatory effect of the first tumor causing a predisposition to development of the second [Citation15]. Multiple cancers developing after chemotherapy or radiotherapy have been described, but in this report we presented peculiar cases of subsequent RCC and CML development and simultaneous treatment of a solid tumor – RCC and hematologic malignancy – CML. Until now, chronic myeloid leukemia was only reported as detected on fluorodeoxyglucose (18F) (FDG) PET/CT imaging in a patient with RCC after nephrectomy, but developed before TKI treatment [Citation15,Citation16]. Only two patients were treated simultaneously for CML and metastatic ccRCC with novel therapies (bevacizumab, everolimus) [Citation16,Citation17]. The first patient progressed on multiple lines of CML/metastatic renal cell carcinoma (mRCC) therapy, while experiencing significant dose-limiting side-effects of combined treatment of dasatinib and anti-RCC treatment – bevacizumab/everolimus [Citation16]. The second patient was treated with combined therapy of bevacizumab and imatinib and did not experience any serious AEs. The co-treatment was initialized after 4 years of imatinib therapy. In this patient, disease was reported as stable after 6 months of co-treatment [Citation17]. No patient was reported before to be treated with sunitinib or sorafenib and develop CML, while on TKI RCC treatment. The optimal treatment strategy for subjects with concurrent metastatic ccRCC and CML is unknown [Citation16,Citation17].
Cases presentation
Treatment setting
412 subsequent patients with mRCC eligible for the treatment [Citation18–20] received sunitinib 50 mg orally once daily in six-week cycles according to a four-week on/two-week off treatment schedule or sorafenib in 400 mg b.i.d. schedule in the Military Institute of Medicine in Warsaw. 148 mRCC patients were treated with sorafenib [Citation21] and 264 were treated with sunitinib [Citation20,Citation22] between March 2004 and March 2013, and monitored as reported before [Citation20–23]. Hematological and other toxicities were monitored as per standard treatment protocol [Citation18,Citation19].
Case No 1
At the age of 54, male Patient X was diagnosed with ccRCC in September 2004, due to spinal cord metastases, and underwent nephrectomy. After a laminectomy was performed due to spinal cord tumour compression, he was referred for radiotherapy of 3000cGy in November 2004. In March 2005, lung metastases were diagnosed via CT scan, and in April 2005 he started IFN treatment with a standard dose and schedule. In December 2005, disease progression was diagnosed and Patient X was referred for sunitinib 50 mg (4/2) treatment. Until May 2008, he received 22 cycles of sunitinib with good treatment tolerance and SD as best response, and continued the treatment. In February 2010 – after 4 years, 1 month of sunitinib treatment - he was diagnosed with CML bcr-abl(+), 46XY, t(9,22)(q34;q11) and received hydroxycarbamide 3 g/d. In April 2010, began imatinib treatment and a 37th cycle of sunitinib. In June 2010, during the 38th cycle, sunitinib was reduced to 37.5 mg due to fatigue grade 3. In July, he presented an optimal response to imatinib treatment, but by September had developed NSTEMI with EF 60%. The patient continued sunitinib (38 cycles total) and imatinib treatment until August 2011, and died due to multiorgan failure as a result of the progression of the CML disease after 5 years, 8 months since sunitinib treatment induction.
Case No 2
In December 1994, male at the age of 44, Patient Y was diagnosed with ccRCC and underwent radical nephrectomy that confirmed localized T3 ccRCC Fuhrman grade 3. In March 2003, lung metastases were diagnosed and he began chemoimmunotherapy of the Atzpodien regimen. After two cycles of treatment, a cerebral metastasis in the occipital lobe was found and a metastasectomy was performed. In January 2004, IFN immunotherapy was introduced. In September 2004, her condition deteriorated and treatment was suspended. In November 2004, he qualified for sorafenib treatment at a standard dose and schedule, and he continued the treatment with best response of SD with slow progression from February 2007. Sorafenib treatments continued, while in May 2008 a bcr-abl positive CML was diagnosed after 3 years and 5 months of sorafenib therapy (44 cycles). Hydroxycarbamide was administered at a dose of 3 g/d. Continuous sorafenib and recurring hydroxycarbamide treatments continued until the patient’s death in October 2010 – 15 years and 10 months after nephrectomy, and 5 years and 11 months after sorafenib induction (over 70 cycles).
Case No 3
Male at the age of 76, Patient Z, was diagnosed with ccRCC with synchronic lung metastases and underwent nephrectomy in April 2005. In July 2005, IFN therapy was started, but due to poor tolerance it was terminated after the seventh cycle. In December 2005, sunitinib treatment at the standard dose and schedule was started, and PR was achieved on the treatment. In August 2010, during the 39th sunitinib cycle, CML with bcr-abl(+),46XY, t(9,22)(q34;q11.2) was diagnosed. Hydroxycarbamide at a dose of 3 g/d was used for treatment. Sunitinib treatments continued, with slow progression beginning in September 2010. In January 2011, imatinib treatment for CML was introduced and continued until January 2012 with a poor response. Simultaneously, in February 2011, Patient Z’s sunitinib dose was reduced to 37.5 due to renal failure. In March 2012, dasatinib CML treatment was started, but in June 2012 a serious gastric bleeding event was noticed and dasatinib and sunitinib treatments were halted. In August 2012, the patient was rechallenged with sunitinib at a 25 mg dose and, in September, dasatinib treatment resumed. Due to hematological toxicity, dasatinib therapy was finished and, as a result of further renal abnormalities, the sunitinib dose was again decreased to 12.5 mg. The patient died from the progression of RCC disease in December 2012.
Conclusion & discussion
The introduction of targeted therapies represents a major advance in the treatment of tumour progression. Sunitinib and sorafenib are targeted TKIs designed to inhibit in primary VEGF receptor (VEGFR) types 1 (FLT1), 2 (KDR), and 3 (FLT4); platelet-derived growth factor receptors A and B (PDGFRA, PDGFRB); the stem cell factor receptor (cKIT); FMS-like tyrosine kinase 3 (FLT3); colony-stimulating factor 1 receptor (CSF-1R); and glial cell line-derived neurotrophic factor receptor (RET) with different affinity constants for each. The inhibition of these RTKs is primarily to decrease tumour growth, tumour progression and angiogenesis. Nevertheless, sunitinib inhibits 73 and sorafenib 40 additional kinases resulting in a heterogeneous activity profile, which is poorly predictable [Citation6,Citation7]. Despite good responses and treatments that are tolerable in ccRCC patients, little is known concerning the long-term toxicity of these agents [Citation1,Citation3,Citation12,Citation13].
At the same time an increased incidence of secondary malignancies has been well documented in a number of different neoplasms treatments. Secondary cancer development was reported in long term survivors of head and neck tumors, hairy cell leukemia Hodgkin’s lymphoma, Non-Hodgkin’s lymphoma, CML, multiple myeloma, testicular carcinoma and multiple pediatric tumors. Before now secondary cancers were not reported in ccRCC in large number [Citation14,Citation15,Citation24]. This lack of reports may result in part from relatively short time since RCC therapies - TKIs - have been introduced in routine clinical practice and therefore it is not enough long time for observation of secondary cancers development [Citation18,Citation19,Citation25]. Moreover relatively short OS of majority RCC patients [Citation26,Citation27] and low number of long-term responders [Citation28,Citation29] would not enable to observe secondary cancers. It is reasonable to postulate that in the long-term perspective, ccRCC treatment may also be correlated with increased risk of development of secondary malignancy.
At the same time it should be pointed that a preclinical 2-year study of rats receiving imatinib revealed neoplastic changes in kidneys, urinary bladder, urethra, preputial and clitoral glands, small intestine, parathyroid glands, adrenal glands and stomach. Renal carcinoma, urinary bladder, small intestine adenocarcinoma, parathyroid glands adenoma, malignant medullary tumors of the adrenal glands and stomach carcinomas were reported after prolonged exposition for this TKI [Citation30]. Nevertheless, recent analysis of malignancies developed during therapy with TKIs (imatinib) for CML has been conducted. In this study after a median follow-up of 107 months (range: 13–362) after CML diagnosis 4.6% patients developed secondary cancers, including of which: 31% skin; 15% prostate; 10% digestive system; 4% kidney; 4% thyroid; 3% breast or 3% hepatobiliary cancers; and 3% CLL or 13% melanoma. In the analysis, the risk of second cancer was lower than expected at observed-to-expected ratio of 0.6. Although the overall SIR of second cancers was 0.6, the incidences of melanoma, endocrine tumors, kidney cancers and CLL were higher than expected. The authors concluded that secondary cancers occur in a small percentage of patients receiving TKIs for CML [Citation30]. Another study on imatinib also confirmed higher incidence of secondary malignancies after prolonged treatment – particularly of prostate cancer. This trial however did not give answer whether the observed increased incidence of second malignancies is greater than expected in CML patients treated with standard chemotherapy or stem cell transplantation. The authors postulated, however, that reports from the literature did not support CML standard chemotherapy as significantly genotoxic [Citation31]. It is reasonable to postulate that in a long-term perspective, TKI treatment may also be correlated with increased risk of development of secondary malignancy.
A second cancer may develop coincidentally with the first in the same individual, or may be dependent on the treatment of first malignancy, which is most often genotoxic. The significant difference in median age of appearance of the first malignancy and CML diagnosis between CML non-related to cancer treatment, and cancer treatment-related CML was found, and it is in agreement with the age of our patients who are younger than the median age of 78 years reported for CML as second malignancy population [Citation14]. At the same time, epidemiological data seem to confirm that coincidence of RCC and CML is highly unlikely in the general population, including the Polish population. According to central national registry CML is diagnosed in Poland in approximately 800 cases and RCC in approximately 3800 cases annually, in the population of 36 million, which results in statistical RCC-CML coincidence at the level of 2x10–9. Possible mechanisms linking RCC, its treatment and CML development are unknown. We propose a multi-step model that may link prolonged TKI exposure and bone marrow abnormalities.
The first TKI activity that may be CML predisposing is to be assigned to FLT3 kinases inhibition. Sunitinib and sorafenib inhibit phosphorylation of FLT3, and subsequently inactivation of its signalling pathway. Decreased FLT3 levels results in increments of the time that hematopoietic progenitors stay in the G1-phase of the cell cycle and the time needed for each hematopoietic progenitor to divide. P65 and NF-κB proteins are mediators in this process [Citation32]. Prolonged G1 phase promote DNA ‘misrepair’ in nonhomologous end joining mechanism (NHEJ) [Citation33]. At the same time it is known that CML is associated with a specific chromosome abnormality – the t(9;22) reciprocal translocation forming the Philadelphia (Ph) chromosome [Citation34], which arises as a result of two NHEJ ‘misrepair’ [Citation35]. Taken together this data suggest that TKI induced prolonged G1 phase of the cell cycle and usage of NHEJ for DNA repair may increase the chance of BCR/ABL1 rearrangement and Ph arise in bone marrow cells [Citation35]. Moreover if BCR/ABL rearrangements already arise in pre-CML cells, those cells again utilize NHEJ to repair DNA and this further promotes genomic instability and proliferation of aberrant cells. Such increased proliferation may further contribute to the accumulation of BCR/ABL1 mutant cells and CML development [Citation35,Citation36]. Acquired BCR-ABL mutation initiates chronic phase CML and results in aberrant stem cell differentiation and survival [Citation37]. Those CML stem cells are characterized by increased NHEJ, enhanced susceptibility to repeated cycles of chromosome damage, through a breakage-fusion-bridge mechanism and presence of unstable DNA aberrations [Citation38]. Together, this suggests that in the CML development process, Bcr-Abl protein expression is already sufficient to 1) increase the number of multilineage progenitors, 2) increase the number of myeloid progenitors, 3) change the balance of lineage white blood cells development and 4) promote CML development. Acquisition of Bcr-Abl by bone marrow cells is being the sole genetic change needed for establishment of the chronic phase of CML [Citation36,Citation39–41]. It has also been experimentally proven that CML cells have increased susceptibility to anaphase bridge formation as a direct result of dysregulation of NHEJ repair, which clearly stimulates hyperproliferation in the myeloid compartment leading to the phenotype of CML and its blast crisis in consequence [Citation42]. Moreover BCR/ABL-mediates stimulation of WRN (mutated in Werner syndrome) protein activity that result in low fidelity of major DNA double-strand break repairs and further protects CML cells from apoptosis and facilitate increasing genomic instability [Citation43]. After all, CML may arise as a consequence of NHEJ in bone morrow cells, including those influenced by FLT3 inhibition [Citation44].
The second TKI activity that may be CML predisposing is to be assigned to ERK1/2 kinases inhibition. DNA instability in CML progenitor cells may be promoted by inhibiton of ERK1/2 phosphorylation. Inhibiton of ERK1/2 down-regulates the expression of DNA repair proteins – excision repair cross-complementing group 1 (ERCC-1) and x-ray cross-complementing group 1 (XRCC-1) – in a dose-dependent manner. Under-expression of nucleotide excision repair pathway genes may further enhance cell dependence on NHEJ and enhance the chance of DNA damage following exposure to genotoxic agents [Citation45], so TKI, by inhibiting ERCC-1 and XRCC-1, may promote the rise of the BCR/ABL fusion gene. Homologous mechanism of CML promotion may be applied to phosphatase and tensin homologs deleted on the chromosome ten (PTEN) gene down-regulation. De-activation of the PTEN gene locus is often silenced by methylation of its promoter on long-term sunitinib exposure [Citation46], but at the same time, PTEN down-regulation is known to cause acceleration of CML development [Citation47]. Therefore PTEN silenced by sunitinib may no longer act as a tumor suppressor, preventing CML development [Citation47].
A third TKI activity that may promote CML is induction of centrosomal defects [Citation12]. This abnormality seems to be mediated by Polo-like kinase 4 (PLK4), which is inhibited by both sunitinib and sorafenib, and regulates centriole duplication during the cell cycle, regulate mitosis, cytokinesis and cell cycle checkpoints in response to genotoxic stress. It has been proposed as a tumour suppressor [Citation6]. Down-regulated PLK4 was shown to cause cytokinesis failure, centrosome amplification and gross chromosomal abnormalities [Citation48]. It was proved that centrosome defects common and early detectable feature of CML and that may contribute to acquisition of chromosomal aberrations and aneuploidy [Citation49].
The fourth abnormality of bone marrow cells that may be attributed to inhibition of kinases is also dependent on FLT-3 blockage. FL ligand functions synergistically with various hematopoietic cytokines to promote the expansion of ‘short-term’ hematopoietic stem cells, multipotential progenitor (MPP) and common lymphoid progenitors (CLPs) [Citation50]. If the FLT3 receptor is activated, it promotes proliferation of normal progenitors (Philadelphia chromosome-negative cells) [Citation51], but if FLT3 activity is decreased in CML patients, proliferation of lymphoma cells is promoted, accelerating the manifestation of the disease.
A fifth mechanism influencing the bone marrow (BM) homeostasis is deregulation of the macrophage-colony stimulating factor receptor (CSF-1R, M-CSFR, CD115). Sorafenib showed inhibition against different phosphorylation states of CSF-1R, while sunitinib showed potent inhibition against dephosphorylated CSF-1R, preventing the activation of CSF-1R [Citation52]. If CSF-1R is active, its signalling through the receptor promotes the differentiation of myeloid progenitors into heterogeneous populations of monocytes, macrophages, dendritic cells and bone-resorbing osteoclasts [Citation53]. The activity of CSF-1R should be down-regulated during granulopoiesis. CSF1R inhibition promotes granulopoiesis and inhibits myeloid progenitor differentiation, which is a typical BM state in CML.
Similar activity may also apply to inhibition of the c-KIT receptor (CD115). This receptor is expressed on healthy hematopoietic cells, including CD34+ cells, but not on mature blood cells. At the same time CD34+ CML cells are considered as CML stem/progenitor cells [Citation54]. The cKIT receptor was found to activate JAK1–3 that further mediate STATs phosphorylation and dimerization and finally regulate CD34+ cell proliferation, differentiation and apoptosis [Citation55]. Downregulated Jak3 expression results is dysregulated myelopoiesis [Citation56]. This is due to fact that JAK3 expression must be up-regulated during granulocytic and monocytic differentiation. Deficiency in JAK3 expression results in increased levels of immature neutrophils and monocytes in the peripheral blood, which stays in concordance with some hematopoiesis perturbances found in CML [Citation57]. It may therefore be concluded that insufficient c-KIT signaling in hematopoietic stem cells, which is a result of TKI blockage, is partially responsible for poor differentiation of those cells. The importance of c-KIT signaling for hematopoietic cells is further supported by the phenomenon of c-KIT-inhibition-dependent macrocytosis resulting from sunitinib treatment [Citation58,Citation59].
Among the known RTKs, expression of Tie2, Flt-3, CSF1R, VEGFR2, VEGFR1 and FGF-receptors have been implicated in modulating the homeostasis of hematopoietic stem cells during the lifetime. However, none of these RTKs have before been fully analyzed in hematology. None of the above mentioned proteins were deemed satisfactory in basic research and were shown to play a major role in disrupting BM homeostasis during CML development. HSC self-renewal is dependent upon extrinsic signals, such as those provided by the niche environment, but also by intrinsic signals, such as anti-apoptotic proteins, polycombgroup proteins, transcription factors, adaptor proteins and cell cycle regulators that are expressed by the HSC itself and BM endothelial cells. CML development in TKI-treated patients proves that more basic research needs to be undertaken in order to clarify the effects of RTK inhibition in noncancer cells. Cell–cell interactions need to be analyzed and in vivo studies need to supplement in vitro experiments. The field is open for new hypotheses and molecular research, and long-term TKI treatment survivors need to be the subject of special in-clinic focus. All together it is becoming clearer that TKIs may target various pathways associated with the proliferation of aberrant clones in CML (). All unexpected off-target RTK inhibition activities may be important especially in the population of patients undergoing long-term treatment.
Future persepective
It is clear from this analysis of case series that the TKI-inhibited pathways may be defined as perturbed in hematopoietic disorders. There is also growing evidence that inhibition of multiple tyrosine kinase receptors (TKRs) plays an important role in transformation of primary bone marrow CD34+ stem cells into BCR/ABL(+) cells. Furthermore, the importance of defective TKR activation in CML progenitor cells and the extent of required TKR activation in the bone marrow niche cells needs to be established. However, the significance of particular kinase inhibition and the interaction or sequence of these molecular events remains less clear. It may be expected that in each case, the judicious expression of dominant-negative or constitutive inhibition (or siRNA) of kinase pathway components in either cell lines or mouse models should enable the relative contribution of altered signaling to the CML phenotype to be established. In addition, the availability of numerous mouse strains either deficient or transgenic for specific tyrosine kinase pathways components may provide additional opportunity for assessing their importance in vivo. It may also open the new way for a better understanding of TKI specificity at the molecular level and increase the knowledge required for designing novel compounds for ccRCC treatment. It may finally be anticipated that future developments will further facilitate the translation of the basic science of ccRCC signaling pathway into the onco-hematology clinics.
Executive Summary
Background
Secondary hematological malignancies may develop after oncological treatment and are increasingly reported as an important late complication in cancer patients.
Cancer treatment not-related chronic myeloid leukaemia (CML) is a second malignancy associated with elderly age (median78 years of age).
Suggested renal cell cancer (RCC)-hematologic malignancy relationship include: genetic mutation common to both malignancies; environmental exposure; immunomodulatory effect of the first cancer causing a predisposition to development of the second.
TKI therapy is associated with multiple toxicities related to off-target inhibition.
The effects of long-term exposure for TKI are not well defined until now.
Case No 1
Diagnosed with RCC at the age of 54, underwent nephrectomy and radiotherapy. Was treated with IFN (8 months) and with sunitinib 50 mg.
At 36th sunitinib cycle, diagnosed with CML. Treated with imatinib with optimal response. Died due to multiorgan failure.
Case No 2
Diagnosed with RCC at the age of 44, underwent radical nephrectomy. Was treated with chemoimmunotherapy (Atzpodien regimen – two cycles), IFN (9 months) and sorafenib.
Diagnosed with CML at 44th sorafenib cycle. Treated with hydroxycarbamide until the patient’s death.
Case No 3
Diagnosed with RCC at the age of 76, underwent nephrectomy. Was treated with IFN (seven cycles) and sunitinib.
Diagnosed with CML at 39th sunitinib cycle. Treated with hydroxycarbamide, imatinib and dasatinib subsequently. Died from RCC disease’s progression.
Conclusion
Possible mechanisms linking development and progression of clear cell renal cell cancer (ccRCC), ccRCC treatment with TKI and CML development are unknown.
Multi step model linking prolonged TKIs exposure and bone marrow abnormalities may be proposed.
The first TKI activity of interest in CML development may assigned to FLT3 kinases inhibition and nonhomologous end joining mechanism (NHEJ) and accumulation of BCR/ABL1 mutant cells.
The second TKI activity of interest in CML development may be assigned to ERK1/2 kinases inhibition and down-regulation of DNA repair proteins ERCC-1 and XRCC-1.
A third TKI activity of interest in CML development may be assigned to Polo-like kinase 4 inhibition and centrosomal defects.
CML development in TKI-treated patients proves that more basic research needs to be undertaken in order to clarify the effects of RTK inhibition in noncancer cells.
Ethical & informed consent disclosure
The manuscript has been prepared after obtaining OS from all patients. All patients have died prior to manuscript preparation. No sensitive personal data of the patient has been used in the manuscript. The authors state that they have obtained verbal and written informed consent from the patient/patients’ relatives for the inclusion of their medical and treatment history within this case report.
Financial & competing interests disclosure
C Szczylik received consulting and lecture honoraria from Pfizer, Bayer HealthCare, Astellas, GlaxoSmithKline and Novartis. AM Czarnecka received lecture honoraria from Pfizer, GlaxoSmithKline, Novartis, Merck, Roche, Vipharm and Hospira. S Oborska and P Rzepecki indicate no potential conflict of interest. All authors declare no other potential conflict of interests. This work was supported by the Military Institute of Medicine statutory founding. Treatment cost was covered by Polish National Health Fund. In development of molecular model CS and AMC have been supported by the Foundation for Polish Science (FNP) TEAM grant no. TEAM/2010–6/8. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- Di Lorenzo G , PortaC, BellmuntJet al. Toxicities of targeted therapy and their management in kidney cancer. Eur. Urol.59(4), 526–540 (2011).
- Guevremont C , AlaskerA, KarakiewiczPI. Management of sorafenib, sunitinib, and temsirolimus toxicity in metastatic renal cell carcinoma. Curr. Opin. Support. Palliat. Care3(3), 170–179 (2009).
- Hutson TE , FiglinRA, KuhnJG, MotzerRJ. Targeted therapies for metastatic renal cell carcinoma: an overview of toxicity and dosing strategies. The Oncologist13(10), 1084–1096 (2008).
- Schmidinger M , BellmuntJ. Plethora of agents, plethora of targets, plethora of side effects in metastatic renal cell carcinoma. Cancer Treat. Rev.36(5), 416–424 (2010).
- Aparicio-Gallego G , BlancoM, FigueroaAet al. New insights into molecular mechanisms of sunitinib-associated side effects. Mol. Cancer Ther.10(12), 2215–2223 (2011).
- Fabian MA , BiggsWH3rd, TreiberDKet al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol.23(3), 329–336 (2005).
- Stein MN , FlahertyKT. CCR drug updates: sorafenib and sunitinib in renal cell carcinoma. Clin. Cancer Res.13(13), 3765–3770 (2007).
- Cohen RB , OudardS. Antiangiogenic therapy for advanced renal cell carcinoma: management of treatment-related toxicities. Invest. New Drugs.30(5), 2066–2079 (2012).
- Oh WK , McdermottD, PortaCet al. Angiogenesis inhibitor therapies for advanced renal cell carcinoma: toxicity and treatment patterns in clinical practice from a global medical chart review. Int. J. Oncol.44(1), 5–16 (2014).
- Michel MS , VervenneW, De SantisMet al. SWITCH: A randomized sequential open-label study to evaluate efficacy and safety of sorafenib (SO)/sunitinib (SU) versus SU/SO in the treatment of metastatic renal cell cancer (mRCC). J. Clin. Oncol.32(Suppl.4), 393 abstr. (2014).
- Gore ME , SzczylikC, PortaCet al. Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: an expanded-access trial. Lancet Oncol.10(8), 757–763 (2009).
- Giehl M , LeitnerA, HaferlachCet al. Detection of centrosome aberrations in disease-unrelated cells from patients with tumor treated with tyrosine kinase inhibitors. Eur. J. Haematol.85(2), 139–148 (2010).
- Kollmannsberger C , BjarnasonG, BurnettPet al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist16(5), 543–553 (2011).
- Specchia G , BuquicchioC, AlbanoFet al. Non-treatment-related chronic myeloid leukemia as a second malignancy. Leuk. Res.28(2), 115–119 (2004).
- Nishikubo CY , KunkelLA, FiglinRet al. An association between renal cell carcinoma and lymphoid malignancies. A case series of eight patients. Cancer78(11), 2421–2426 (1996).
- Al-Najjar F , JarkowskiA3rd. Treatment of concurrent metastatic renal cell carcinoma and chronic myelogenous leukemia--easier said than done? A case report. J. Oncol. Pharm. Pract.17(4), 436–439 (2011).
- Pal SK , GuptaRK, DosikG, FiglinRA. Concomitant renal cell carcinoma and chronic myelogenous leukemia: use of a targeted approach. Curr. Oncol.16(2), 44–47 (2009).
- Escudier B , EisenT, StadlerWMet al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med.356(2), 125–134 (2007).
- Motzer RJ , HutsonTE, TomczakPet al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med.356(2), 115–124 (2007).
- Czarnecka A , SobczukP, BoguszK, SpychalskaM, SzczylikC. Survival, safety and treatment response duration in “real world” patients with metastatic clear cell renal cancer – an update from clinical practice. BJU Int.112(s3), 1–17 (2013).
- Szmit S , ZaborowskaM, Wasko-GrabowskaAet al. Cardiovascular comorbidities for prediction of progression-free survival in patients with metastatic renal cell carcinoma treated with sorafenib. Kidney Blood Press Res.35(6), 468–476 (2012).
- Czarnecka AM , OborskaS, RzepeckiP, SzczylikC. Development of CML in the course of ccRCC treatment with tyrosine kinase inhibitors – clinical report with molecular model. BJU Int.112(s3), 1–17 (2013).
- Szmit S , LangiewiczP, ZlnierekJet al. Hypertension as a predictive factor for survival outcomes in patients with metastatic renal cell carcinoma treated with sunitinib after progression on cytokines. Kidney Blood Press Res.35(1), 18–25 (2012).
- Jimenez VH . Coexistence between renal cell cancer and Hodgkin’s lymphoma: a rare coincidence. BMC urology6(10), (2006).
- Sternberg CN , HawkinsRE, WagstaffJet al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: final overall survival results and safety update. Eur. J. Cancer.49(6), 1287–1296 (2013).
- Motzer RJ , EscudierB, OudardSet al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer116(18), 4256–4265 (2010).
- Motzer RJ , HutsonTE, TomczakPet al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J. Clin. Oncol.27(22), 3584–3590 (2009).
- Molina AM , JiaX, FeldmanDRet al. Long-term response to sunitinib therapy for metastatic renal cell carcinoma. Clin. Genitourin. Cancer11(3), 297–302 (2013).
- Molina AM , LinX, KorytowskyBet al. Sunitinib objective response in metastatic renal cell carcinoma: analysis of 1059 patients treated on clinical trials. Eur. J. Cancer50(2), 351–358 (2014).
- Verma D , KantarjianH, StromSSet al. Malignancies occurring during therapy with tyrosine kinase inhibitors (TKIs) for chronic myeloid leukemia (CML) and other hematologic malignancies. Blood118(16), 4353–4358 (2011).
- Roy L , GuilhotJ, MartineauG, LarcheeR, GuilhotF. Unexpected occurrence of second malignancies in patients treated with interferon followed by imatinib mesylate for chronic myelogenous leukemia. Leukemia19(9), 1689–1692 (2005).
- Ohishi K , KatayamaN, ItohRet al. Accelerated cell-cycling of hematopoietic progenitors by the flt3 ligand that is modulated by transforming growth factor-beta. Blood87(5), 1718–1727 (1996).
- Sonoda E , HocheggerH, SaberiA, TaniguchiY, TakedaS. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA repair5(9–10), 1021–1029 (2006).
- Pasternak G , HochhausA, SchultheisB, HehlmannR. Chronic myelogenous leukemia: molecular and cellular aspects. J. Cancer Res. Clin. Oncol.124(12), 643–660 (1998).
- Gollin SM . Mechanisms leading to nonrandom, nonhomologous chromosomal translocations in leukemia. Semin. Cancer Biol.17(1), 74–79 (2007).
- Salles D , MencalhaAL, IrenoIC, WiesmullerL, AbdelhayE. BCR-ABL stimulates mutagenic homologous DNA double-strand break repair via the DNA-end-processing factor CtIP. Carcinogenesis32(1), 27–34 (2011).
- Jamieson CH . Chronic myeloid leukemia stem cells. Hematology/the Education Program of the American Society of Hematology. American Society of Hematology. Education Prog.436–442 (2008).
- Chakraborty S , StarkJM, SunCLet al. Chronic myelogenous leukemia stem and progenitor cells demonstrate chromosomal instability related to repeated breakage-fusion-bridge cycles mediated by increased nonhomologous end joining. Blood119(26), 6187–6197 (2012).
- Skorski T , SzczylikC, MalaguarneraL, CalabrettaB. Gene-targeted specific inhibition of chronic myeloid leukemia cell growth by BCR-ABL antisense oligodeoxynucleotides. Folia Histochem. Cytobiol.29(3), 85–89 (1991).
- Szczylik C , SkorskiT, NicolaidesNCet al. Selective inhibition of leukemia cell proliferation by BCR-ABL antisense oligodeoxynucleotides. Science253(5019), 562–565 (1991).
- Era T , WitteON. Regulated expression of P210 Bcr-Abl during embryonic stem cell differentiation stimulates multipotential progenitor expansion and myeloid cell fate. Proc. Natl Acad. Sci. USA97(4), 1737–1742 (2000).
- Carroll M . BCR/ABL and chromosomal instability: debate resolved. Blood119(26), 6180–6181 (2012).
- Slupianek A , PoplawskiT, JozwiakowskiSKet al. BCR/ABL stimulates WRN to promote survival and genomic instability. Cancer Res.71(3), 842–851 (2011).
- Era T . Bcr-Abl is a “molecular switch” for the decision for growth and differentiation in hematopoietic stem cells. Int. J. Hematol.76(1), 35–43 (2002).
- Yadav A , KumarB, TeknosTN, KumarP. Sorafenib enhances the antitumor effects of chemoradiation treatment by downregulating ERCC-1 and XRCC-1 DNA repair proteins. Mol. Cancer Ther.10(7), 1241–1251 (2011).
- Yang J , IkezoeT, NishiokaCet al. Long-term exposure of gastrointestinal stromal tumor cells to sunitinib induces epigenetic silencing of the PTEN gene. Int. J. Cancer.130(4), 959–966 (2012).
- Peng C , ChenY, YangZet al. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood115(3), 626–635 (2010).
- Holland AJ , FachinettiD, Da CruzSet al. Polo-like kinase 4 controls centriole duplication but does not directly regulate cytokinesis. Mol. Biol. Cell.23(10), 1838–1845 (2012).
- Giehl M , FabariusA, FrankOet al. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia19(7), 1192–1197 (2005).
- Dolence JJ , GwinK, FrankE, MedinaKL. Threshold levels of Flt3-ligand are required for the generation and survival of lymphoid progenitors and B-cell precursors. Eur. J. Immunol.41(2), 324–334 (2011).
- Lyman SD , JacobsenSE. c-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood91(4), 1101–1134 (1998).
- Kitagawa D , GoudaM, KiriiYet al. Characterization of kinase inhibitors using different phosphorylation states of colony stimulating factor-1 receptor tyrosine kinase. J. Biochem.151(1), 47–55 (2012).
- Hume DA , MacdonaldKP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood119(8), 1810–1820 (2012).
- Jiang X , ForrestD, NicoliniFet al. Properties of CD34+ CML stem/progenitor cells that correlate with different clinical responses to imatinib mesylate. Blood116(12), 2112–2121 (2010).
- Liang J , WuYL, ChenBJ, ZhangW, TanakaY, SugiyamaH. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci.9(5), 435–443 (2013).
- Ward AC , TouwI, YoshimuraA. The Jak–Stat pathway in normal and perturbed hematopoiesis. Blood95(1), 19–29 (2000).
- Miranda MB , JohnsonDE. Signal transduction pathways that contribute to myeloid differentiation. Leukemia21(7), 1363–1377 (2007).
- Schallier D , TrullemansF, FontaineC, DecosterL, De GreveJ. Tyrosine kinase inhibitor-induced macrocytosis. Anticancer Res.29(12), 5225–5228 (2009).
- Rini BI , ChoueiriTK, ElsonPet al. Sunitinib-induced macrocytosis in patients with metastatic renal cell carcinoma. Cancer113(6), 1309–1314 (2008).