
Abstract
Aim: Bioinformatic analysis of mutation sets in receptor-binding domain (RBD) of currently and previously circulating SARS-CoV-2 variants of concern (VOCs) and interest (VOIs) to assess their ability to bind the ACE2 receptor. Methods:In silico sequence and structure-oriented approaches were used to evaluate the impact of single and multiple mutations. Results: Mutations detected in VOCs and VOIs led to the reduction of binding free energy of the RBD–ACE2 complex, forming additional chemical bonds with ACE2, and to an increase of RBD–ACE2 complex stability. Conclusion: Mutation sets characteristic of SARS-CoV-2 variants have complex effects on the ACE2 receptor-binding affinity associated with amino acid interactions at mutation sites, as well as on the acquisition of other viral adaptive advantages.
Plain language summary
The increase in the infectious potential of SARS-CoV-2 variants (Alpha, Beta, Gamma, Delta, Omicron, etc.) that causes COVID-19 is mainly caused by virus mutations. Particularly important for the development of the disease is the interaction of the coronavirus spike protein with a receptor on the surface of human cell, as a result of which the virus penetrates the cell. Angiotensin-binding enzyme (ACE2) is such a receptor in humans, and there is a receptor-binding domain (RBD) in the coronavirus spike protein. In this study, using bioinformatic methods, an analysis of mutations in the RBD of the virus was carried out to find out their influence on the functionality and ability of the virus to interact with the ACE2 receptor with high stability, which ultimately leads to infection. A number of mutations increase the infectious potential of the virus. More recent variants of the virus have more than one mutation in the RBD, so their effects are complex. It is important that the coronavirus is constantly evolving, increasing the ability to bind to the ACE2 receptor, as well as avoiding the immune response. The Omicron variant, which has at least 15 mutations in the RBD, is the most successful in these directions.
Tweetable abstract
Mutation sets in the RBD of SARS-CoV-2 variants have complex effects on the ACE2 receptor-binding affinity.
At the end of 2019, the first acute respiratory disease cases caused by a new member of the coronavirus family were registered in Wuhan (Quebec Province, China). In the first World Health Organization (WHO) situation reports [Citation1], the pathogen was named 2019-nCoV. In February 2020, the official names of the pathogen – SARS-CoV-2 and the disease, COVID-19 – were approved [Citation2]. The disease spread rapidly in China and abroad, so, in March 2020, the COVID-19 pandemic was declared [Citation3].
For several years since the first reported cases, SARS-CoV-2 has shown a significant ability to mutate. A large number of virus variants have been identified, and various nomenclatures are now used to classify them, including GISAID [Citation4], Nextstrain [Citation5] and PANGO [Citation6]. Depending on the danger that SARS-CoV-2 variants may cause to society, it is customary to distinguish such categories as variants of concern (VOCs), variants of interest (VOIs) and variants under monitoring (VUMs) [Citation7]. Among them, VOCs cause the greatest danger. For the convenience of naming variants related to VOCs and VOIs in the non-scientific field, the WHO suggested using the Greek alphabet letters (for the Omicron variant, this nomenclature was not used) [Citation8]. At the time of writing, the WHO has classified as VOCs various sublineages of the Omicron (B.1.1.529) variant (here and further, the nomenclature according to the PANGO system is given in parentheses). Previously circulated VOCs include Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1) and Delta (B.1.617.2); previously circulated VOIs include Epsilon (B.1.427, B.1.429), Zeta (P.2), Eta (B.1.525), Theta (P.3), Iota (B.1.526), Kappa (B.1.617.1), Lambda (C.37) and Mu (B. 1.621) [Citation7].
Each SARS-CoV-2 variant is characterized by a specific mutation set that distinguishes it from the original wild-type (WT) virus. Mutations can affect the properties of the variant and cause an increase in its transmissibility, virulence and immune escape ability. Mutations in the spike glycoprotein are of particular importance because spike glycoprotein is required for SARS-CoV-2 to penetrate the host cell and is a target for antiviral vaccines. Spike glycoprotein is trimeric; each spike monomer contains 1273 amino acid residues and consists of two subunits: S1 (amino acid residues (aa) 14–685) and S2 (aa 686–1273), preceded by a short signal peptide (aa 1–13). The S1 subunit includes the receptor-binding domain (RBD), amino acid residues 319–541) and is responsible for binding to the angiotensin-converting enzyme 2 (ACE2), which acts as a receptor for SARS-CoV-2 on the surface of the host cell. The S2 subunit ensures the fusion of the virus membrane with the cytoplasmic membrane [Citation9].
Analysis of the 94,079 full-length SARS-CoV-2 nucleotide sequences from the GISAID database, conducted with a focus on the RBD [Citation10] in the first year of the pandemic, indicated that mutations were observed in 87.1 % of all amino acids in the RBD (169 out of 194 residues, with 216 mutational events), which points that the virus initially had a high variability. A similar rate of mutations was observed in S1 (88.7%, 597 out of 673 residues) and S2 (89.0%, 470 out of 528 residues) subunits. Most of the mutations had a low frequency of occurrence and were not fixed in viral populations at that time. Similar results were obtained in the second year of the pandemic when analyzing 1,036,030 GISAID genomic assemblies: 852 distinct mutations (646 recurrent) were detected in the RBD and only 5 (N439K, L452R, S477N, E484K, N501Y) were common to more than 1% of the mutated assembly sequences [Citation11]. Thus, SARS-CoV-2 spike protein (in particular, the RBD) is variable in most amino acid residues, and different substitutions are possible in one position. The phenomenon's nature lies in the frequent spontaneous replication errors and other important processes such as recombination, intra-host RNA editing, chronic infections and recombination [Citation12]. At the same time, only some mutations are fixed in the most common VOCs and VOIs and are characteristic of them, indicating the different advantages of mutations in evolutionary terms and, accordingly, their different fixability in virus lineages.
Each variant differs from the others in a specific set of mutations and simultaneously has different infectious potential, indicating a connection between these two concepts. Significantly more mutations were detected in the Omicron variant sublineages than in other previously circulated VOCs and VOIs [Citation7,Citation13–15], which created an evolutionary gap between the Omicron and previous variants. An important area of work is the characterization of the properties of SARS-CoV-2 variants in terms of the mutations established in them [Citation16]. Many mutations in spike protein were associated with increased SARS-CoV-2 infectious potential and resistance to neutralizing antibodies [Citation17,Citation18]. Because mutations in the RBD of spike protein have the most significant impact on the ACE2 binding ability of the virus variants, as well as the stability of the RBD–ACE2 complex, their study is a priority for understanding the threats caused by new SARS-CoV-2 variants in epidemiological, clinical, and therapeutic aspects.
This study proposes a bioinformatic analysis of missense mutations in the RBDs of currently and previously circulated SARS-CoV-2 VOCs and VOIs to assess their binding ability to the ACE2 receptor. The study aimed to characterize the effect of not only single mutations but their sets in VOCs and VOIs, to identify potential complex effects of mutations and determine mutational and infectious profiles of each of the variants.
Materials & methods
In silico methods were used to assess the impact of missense mutations in the RBD on the stability of the RBD–ACE2 complex.
Sequence-oriented approach
Methods based on the amino acid sequence of the spike protein were used. The surface glycoprotein SARS-CoV-2 sequence (GenBank: QHD43416.1) was used as a reference. This one corresponded to the Wuhan-Hu-1 genome (GenBank: MN908947.3) sampled on 26 December 2019 [Citation19] and was considered as the WT.
Mutations in the SARS-CoV-2 variants located in the RBD were identified according to existing databases and other sources [Citation7,Citation13–15].
Grantham's distances [Citation20,Citation21] (that take into account such properties as composition, polarity and molecular volume) were used for each of the amino acid pairs to assess the similarity of the properties of amino acid residues at positions in which mutations took place.
In silico mutation predictive tools SIFT [Citation22], PROVEAN [Citation23] and SNAP2 [Citation24] were used to assess the effect of mutations on the functions of the spike protein. SIFT and PROVEAN algorithms are based on the comparison of the query sequence with homologous sequences; SNAP2 utilize various features of the substitution in protein. Sequences of the spike protein (1273 aa) and the S1 subunit (672 aa) were used as queries. In the SIFT tool, homologous sequences from the non-redundant UniRef90 database [Citation25] were used as related sequences for comparison.
Structure-oriented approach
The three-dimensional structures of the RBD–ACE2 complex for different variants of SARS-CoV-2 were analyzed. The x-ray crystallography structure of the novel coronavirus spike receptor-binding domain complexed with its ACE2 receptor was used as a reference (Protein Data Bank [PDB ID]: 6LZG [Citation26]). Mutations were introduced into the reference structure using the Swiss-Pdb Viewer [Citation27] to obtain computer-simulated RBD–ACE2 complexes for other SARS-CoV-2 variants. For comparison, previously established x-ray crystallography structures for some VOCs were included in the analysis: Alpha (RDB ID: 7EKF [Citation28]), Beta (PDB ID: 7EKG [Citation28]), Gamma (PDB ID: 7EKC [Citation28]). Delta (PDB ID: 7WBQ [Citation29]), Omicron (PDB ID: 7WBP [Citation29]). All structures (both x-ray and computer-simulated) were prepared for further analysis (solvent was deleted and H atoms were added), followed by minimization in the GROMOS96 force field [Citation30]. All procedures were performed using the Swiss-Pdb Viewer [Citation27].
To assess the impact of mutations on the stability of the RBD–ACE2 complex, changes in binding free energy were assessed under the influence of each mutation separately (ΔΔGsingle) and a mutation set specific to a particular variant (ΔΔGmultiple). ΔΔGsingle and ΔΔGmultiple calculations were performed using bioinformatic tools whose algorithms are based on different methods: BeAtMuSiC (predicts ΔΔGsingle, is based on the use of a set of statistical potentials) [Citation31]; BindProfX (predicts ΔΔGsingle and ΔΔGmultiple; takes into account statistical and physics-based potentials) [Citation32]; Mutabind2 (predicts ΔΔGsingle and ΔΔGmultiple; is based on an accounting of changes in physicochemical features and evolutionary conservation) [Citation33]; SAAMBE-3D (predicts ΔΔGsingle; a machine-learning method based on features of the physical environment of the mutation site) [Citation34]; mmCSM-PPI (predicts ΔΔGsingle and ΔΔGmultiple; the machine-learning method which uses graph-based signatures integrated with other indicators of structure stability) [Citation35]. BindProfX, Mutabind2 and mmCSM-PPI were used to evaluate both ΔΔGsingle and ΔΔGmultiple.
Calculating the total level of binding free energy (ΔG) of complexes was performed for x-ray crystallography reference structure, computer-simulated structures of all VOCs and VOIs, as well as for x-ray crystallography structures of VOCs available in PDB. The calculations were performed using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method [Citation36] on the HawkDock server [Citation37].
Visualization of three-dimensional structures of the RBD–ACE2 complexes was performed using open-source PyMOL software [Citation38].
Results
Mutation sets in RBDs of SARS-CoV-2 variants
SARS-CoV-2 variants are characterized by different numbers and distributions of mutations in the RBD ( & ). Thus, previously circulating VOC Alpha has only one N501Y mutation in the RBD. In comparison, Beta and Gamma variants have two additional mutations — one common mutation E484K and one characteristic mutation in each of the variants (K417N in Beta, K417T in Gamma). The Delta variant was classified as VOC later than Alpha, Beta and Gamma and became the most prevalent variant in the world in the second half of 2021. The Delta variant doesn't have the N501Y mutation in the RBD, but has L452R (also characteristic of Epsilon and Kappa variants, as well as BA.4 and BA.5 sublineages of the Omicron variant) and T478K (identified in the Omicron sublineages) mutations.
Grantham’s distances [Citation16] for the corresponding pairs of amino acids are given in parentheses. Distances 0–50 are considered as a conservative substitution (green), 51-100 as moderately conservative (yellow), 101-150 as moderately radical (orange), or ≥151 as radical (red) according to the described classification [Citation17].
VOC: Variant of concern; WT: Wild-type.
![Figure 1. Mutations in the receptor-binding domains of the variants of concern.Grantham’s distances [Citation16] for the corresponding pairs of amino acids are given in parentheses. Distances 0–50 are considered as a conservative substitution (green), 51-100 as moderately conservative (yellow), 101-150 as moderately radical (orange), or ≥151 as radical (red) according to the described classification [Citation17].VOC: Variant of concern; WT: Wild-type.](/cms/asset/76bf06d6-5e73-45af-8774-25dd6ae66528/ifvl_a_12336087_f0001.jpg)
VOI: Variant of interest; WT: Wild-type.
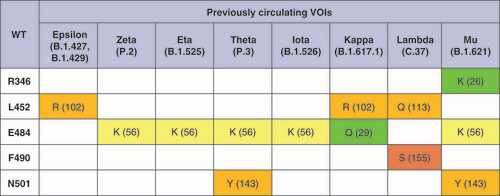
The Omicron variant was the last of these variants to be identified and quickly assigned to the group of VOCs [Citation7]. The Omicron variant includes sublineages BA.1, BA.1.1, BA.2, BA.3, BA.4 and BA.5. All Omicron sublineages contain about 30 mutations in the spike protein, of which at least 15 mutations in the RBD, which is much more than in other variants. The Omicron sublineages have the N501Y mutation (as most other variants), the K417N (as Beta) and the T478K (as Delta). The BA.1.1 sublineage also has the R346K mutation common to the Mu variant; BA.4 and BA.5 sublineages also have the L452R mutation, making them the only Omicron sublineages that have both RBD mutations of the Delta variant. Thus, the mutagenesis of the latter Omicron sublineages occurred precisely in the direction of incorporating the Delta RBD mutational profile into its own. Also worth noting is that other mutations in the RBD are specific to the Omicron sublineages and aren't found in other VOCs and VOIs.
Different approaches were used in the study to quantify the effect of mutations in the RBD. For the primary analysis, Grantham's distances were used to assess the evolutionary similarity between two amino acids at the mutation site based on their physicochemical properties ( & ). The distribution of all mutations into four groups (conservative, moderately conservative, moderately radical and radical) depending on the value of Grantham's distances made it possible to judge the nature of the evolutionary process that led to the emergence of VOCs and VOIs. According to the results obtained, moderately radical and radical amino acid substitutions are common among mutations in VOCs and VOIs, i.e., amino acid residues before and after mutations differ significantly in physicochemical properties. According to the existing classification, radical substitutions include S371F and S375F (Omicron sublineages), as well as F490S (Lambda). Moderately radical substitutions are L452R (Delta, Epsilon, Kappa, BA.4 and BA.5 Omicron sublineages); L452Q (Lambda); S371L, R408S and E484A (Omicron sublineages); N501Y (Alpha, Beta, Gamma, Omicron, Theta and Mu). At the same time, all VOCs and most VOIs (with the exception of Zeta, Eta, Iota) have at least one mutation in their RBD that is classified as radical or moderately radical amino acid substitution.
Predicted effects of mutations in the RBD on the functional properties of the spike protein
After characterizing the distribution of conservative and radical mutations in the RBDs of different SARS-CoV-2 variants, this study assessed the effect of these mutations on spike protein functions. Several bioinformatic tools (SIFT, PROVEAN and SNAP2) with different algorithms were used for more accuracy in such evaluation. Each of these tools gives mutations a numerical score on a scale specific to it; when the criterion value is reached, the mutation is classified as deleterious/effect or tolerated/neutral ().
Evaluation of the functional impact of mutations is presented according to the criteria of the developers of each of the methods: 1) SIFT: score <0.05 - ‘deleterious’; score ≥0.05 - ‘tolerated’ [Citation18]; 2) PROVEAN: score ≤-2.5 - ‘deleterious’, score >2.5 - ‘neutral’ [Citation19]; 3) SNAP2: score ≤0 - ‘neutral’, score >0 - ‘effect’ [Citation20]. Green indicates tolerated/neutral mutations; red indicates deleterious/effect mutations.
WT: Wild-type.
![Figure 3. Prediction of the functional effects of mutations in the RBD of the SARS-CoV-2.Evaluation of the functional impact of mutations is presented according to the criteria of the developers of each of the methods: 1) SIFT: score <0.05 - ‘deleterious’; score ≥0.05 - ‘tolerated’ [Citation18]; 2) PROVEAN: score ≤-2.5 - ‘deleterious’, score >2.5 - ‘neutral’ [Citation19]; 3) SNAP2: score ≤0 - ‘neutral’, score >0 - ‘effect’ [Citation20]. Green indicates tolerated/neutral mutations; red indicates deleterious/effect mutations.WT: Wild-type.](/cms/asset/40bc3370-ed04-4f69-ab5a-b58e56e181b0/ifvl_a_12336087_f0003.jpg)
presents the results of the functional impact assessment for each of the single mutations in the RBD that occurred in SARS-CoV-2 VOCs and VOIs. According to PROVEAN estimates, all mutations in the RBDs of different SARS-CoV-2 variants are neutral, i.e., do not have a significant effect on spike protein functions (). According to SIFT and SNAP2, functional changes can occur with the N501Y mutation, which is present in many variants, and the Y505H mutation, which was detected in the Omicron sublineages. In addition, SNAP2 prediction suggests that the N440K mutation, present only in the Omicron variant sublineages, may also have a potential effect on spike protein functions of SARS-CoV-2. The PROVEAN score for the N440K (-1,576) is also close to the criterion value, indicating the functional impact of this mutation.
The complex effect of RBD mutations on the ACE2 receptor-binding affinity of SARS-CoV-2
The penetration of SARS-CoV-2 into the cell is largely determined by the success of the interaction between the spike protein (in particular, RBD) and the ACE2 receptor. The ligand–receptor interactions are based on physicochemical processes: the formation of stable chemical contacts between interface amino acid residues and the formation of the RBD–ACE2 complex with a stable conformation are especially important.
One of the promising indicators to assess the stability of the RBD–ACE2 complex is the Gibbs binding free energy (ΔG) [Citation39]. Quantitative determination of the Gibbs binding free energy values for the RBD–ACE2 complexes of different SARS-CoV-2 variants makes it possible to conduct a comparative analysis between them and identify variants with the most stable complexes and, therefore, with the highest potential to infect cells.
In addition to the calculations of Gibbs binding free energy of the entire RBD–ACE2 complex, important indicators are also the calculations of energy changes under the influence of single or multiple mutations (ΔΔGsingle and ΔΔGmultiple), which allows us to conclude the significance of certain mutations for the stabilization of the RBD–ACE2 complex. Thus, three indicators are considered in this study: changes in Gibbs binding free energy under the influence of single mutations in the RBD (ΔΔGsingle), changes in binding energy under the influence of mutation sets specific to each of the variants (ΔΔGmultiple), and calculation of the binding energy for entire RBD–ACE2 complexes (ΔG).
The results of the calculations of ΔΔGsingle are presented in . Worth mentioning that in this study, it is assumed that mutations leading to a decrease (ΔΔG<0) and an increase (ΔΔG>0) in the binding free energy have a stabilizing and destabilizing effect, respectively. To conduct a more comprehensive analysis of ΔΔGsingle (as in the case of the analysis of the functional impact of mutations) several bioinformatic tools (BeAtMuSiC, BindProfX, MutaBind2, SAAMBE-3D, mmCSM-PPI) were used at once. Since these computational tools use different algorithms to predict the effects of mutations on the stability of the protein complexes, their estimates are partially different.
ΔΔGsingle calculation with BindProfX was performed only for interface mutations due to service limitations (fields for which calculations were not performed are marked with a dash). For mmCSM-PPI, all ΔΔGsingle values are written with the opposite sign to match them with other tools. Stabilizing mutations are marked in light green, and destabilizing mutations in light yellow.
*Mutations in the interface of the RBD–ACE2 complex.
WT: Wild-type.
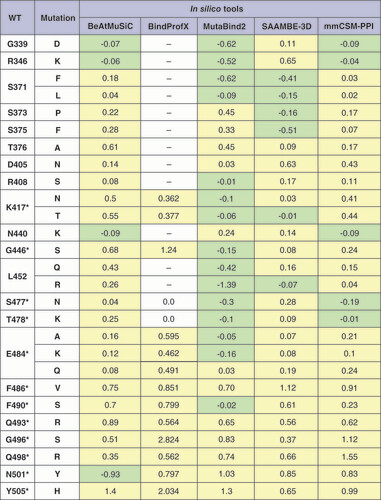
Among the single mutations in the RBDs of the VOCs and VOIs for which a stabilizing effect was shown by at least one of the computational tools are following: R346K (Mu variant, BA.1.1 Omicron BA.1.1 sublineage), K417N (Beta, Omicron. Zeta, Eta, Theta, Iota, Mu variants), K417T (Gamma variant), L452R (Delta, Epsilon, Kappa variants, BA.4 and BA.5 Omicron sublineages), L452Q and F490S (Lambda variant), E484K (Beta, Gamma and Mu variants), N501Y (Alpha, Beta, Gamma, Omicron, Theta, Mu variants); as well as mutations G339D, S371L/F, S373P, S375F, R408S, N440K, G446S, S477N, T478K, E484A, which are available only in the sublineages of the Omicron variant. Among all studied mutations K417N/T, G446S, S477N, T478K, E484A/K, F486V, F490S and N501Y are in the interface of the RBD–ACE2 complex, therefore, may have the greatest impact on the stability of the complex.
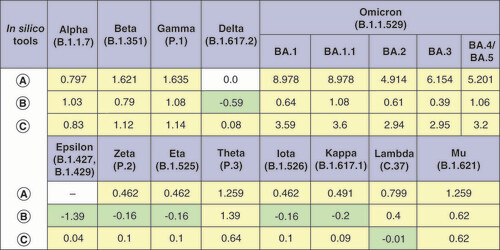
Evaluation of ΔΔGmultiple was performed using BindProfX, MutaBind2 and mmCSM-PPI tools (). The results suggest a possible stabilization effect of mutation sets in the Delta variant (MutaBind2 estimation) and some of the VOIs. For other VOCs, no ΔΔGmultiple values, indicating a stabilizing effect of mutation sets on the RBD–ACE2 complex, were found, which may, nevertheless, be related to the algorithms of used bioinformatic tools.
In silico tools used for ΔΔGmultiple calculations are labeled as follows: (A) BindProfX, (B) MutaBind2 and (C) mmCSM-PPI. Only interface mutations were taken into account when calculating ΔΔGmultiple with BindProfX due to service limitations (field for which calculation was not performed is marked with a dash). For mmCSM-PPI, all ΔΔGmultiple values are written with the opposite sign to match them with other tools. Stabilizing mutations are marked in light green, and destabilizing mutations in light yellow.
Estimation of the Gibbs binding free energy (ΔG) of the entire RBD–ACE2 complex performed using the MM/GBSA method is the most accurate and efficient approach [Citation36,Citation40] presented in this study for evaluation of the stability of RBD–ACE2 complexes. Obtained results showed higher ACE2 receptor-binding affinity of VOC and VOI RBDs and greater stability of their complexes compared with the WT (). It is important to note that more negative values of the ΔG here correspond to more stability of the RBD–ACE2 complexes. Comparison of the x-ray crystallography structures of RBD–ACE2 complex for WT variant with similar complexes for VOCs showed that ACE2 affinity of Beta, Delta and especially Omicron variants is much higher than in the WT variant because the ΔG is lower for them. At the same time, the ΔG of complexes for the Alpha and Gamma variants was higher than for the WT which did not reflect the increase in the ACE2 binding affinity of these variants.
The arrows indicate that all computer-simulated structures were built based on the x-ray WT structure. SARS-CoV-2 variants that have more stable RBD–ACE2 complexes compared with WT are marked in light green, and variants that have less stable complexes are marked in light yellow. X-ray structures were not considered for the Omicron lineages BA.1.1, BA.2, BA.3, BA.4/BA.5; the corresponding fields are marked with a dash.
WT: Wild-type.
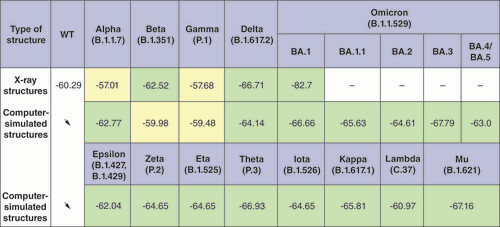
Although the x-ray crystallography structures obtained in various experiments reasonably accurately reflect the proper arrangement of atoms in space, such structures have differences due to the different resolutions of the practical means used, which limits the possibility of comparing them. Another approach used in this study was to introduce mutations into the reference structure of the RBD–ACE2 complex for the WT variant using computer methods and compare the obtained computer-simulated structures with the reference structure. Thus, all structures were derived from the reference, allowing compare them better. The analysis of the ΔG performed by this approach showed lower values of the ΔG and, accordingly, greater stability of the complexes for all VOCs and VOIs except Beta and Gamma ().
The following are the contributions of individual amino acids at mutation sites to the ΔG of the RBD–ACE2 complex. More negative values here also indicate a greater stabilization effect. According to the results of the ΔG calculation, it was found that greater stability of the RBD–ACE2 complex for the Alpha variant was provided by the N501Y mutation (ΔGN501 = -2.82 kcal/mol, ΔGY501 = -7.6 kcal/mol; hereinafter, in parentheses, the contribution of amino acids at mutation sites to the ΔG of the RBD–ACE2 complex is indicated); for Zeta, Eta, Iota – mutation E484K (ΔGE484 = 1.95 kcal/mol, ΔGK484 = -0.57 kcal/mol). Complexes with Beta and Gamma variants also contain N501Y and E484K mutations but significantly lose their stability due to K417N and K417T mutations, respectively, because lysine at position 417 (ΔGK417 = -3.91 kcal/mol) provides relatively greater stability than asparagine (ΔGN417 = -0.08 kcal/mol) and threonine (ΔGT417 = 0.0 kcal/mol).
The stability of one of the previously circulating VOCs Delta is provided by mutations L452R (ΔGL452 = 0.01 kcal/mol, ΔGR452 = -1.04 kcal/mol) and T478K (ΔGT478 = 0.16 kcal/mol, ΔGK478 = -0.86 kcal/mol), both of which help reduce the ΔG of the RBD–ACE2 complex of the Delta variant. Similarly, the L452R mutation has a stabilizing effect on the complex of the Epsilon variant.
As for the Lambda variant, radical substitution F490S (ΔGF490 = -0.08 kcal/mol, ΔGS490 = -0.60 kcal/mol) has a moderate stabilizing effect on the RBD–ACE2 complex, while substitution L452Q (ΔGQ452 = 0.06 kcal/mol) has a much smaller destabilizing effect. In general, the ΔG of the complex with the Lambda variant is slightly lower than the corresponding value for the WT variant.
Among the sublineages of the Omicron variant, the RBD–ACE2 complexes with BA.3 (ΔG = -67.79 kcal/mol) and BA.1 (ΔG = -66.66 kcal/mol) were the most stable (). The stability of complexes with all Omicron sublineages is provided by mutations N440K (ΔGN440 = 0,.03 kcal/mol, ΔGK440 = -1.3 kcal/mol), T478K (ΔGK478 = -0.86 kcal/mol), E484A (ΔGA484 = 0.06 kcal/mol), G496S (ΔGG496 = -0.65 kcal/mol, ΔGS496 = -3.92 kcal/mol), N501Y (ΔGY501 = -7.06 kcal/mol), as well as D405N in BA.2, BA. 3, BA.4/BA.5 sublineages (ΔGD405 = 2.56 kcal/mol, ΔGN405 = 0.06 kcal/mol), and L452R in BA.4/BA.5 sublineages (ΔGR452 = -1.03 kcal/mol). Other mutations have a neutral or destabilizing effect on the complex. The presence of additional D405N mutations in sublineage BA.3 in the absence of destabilizing mutation R408S (ΔGR408 = -1.16 kcal/mol, ΔGS408 = 0.05 kcal/mol) gives this sublineage advantages over both BA.1 and BA. 2. As for the latter sublineages BA.4 and BA.5, the L452R mutation, similar to that in Delta, gives an additional stabilizing effect. However, the presence of a new F486V (ΔGF486 = -3.3 kcal/mol, ΔGV486 = -1.94 kcal/mol) mutation, the presence of R408S and the absence of G496S make these complexes less stable than for other sublineages.
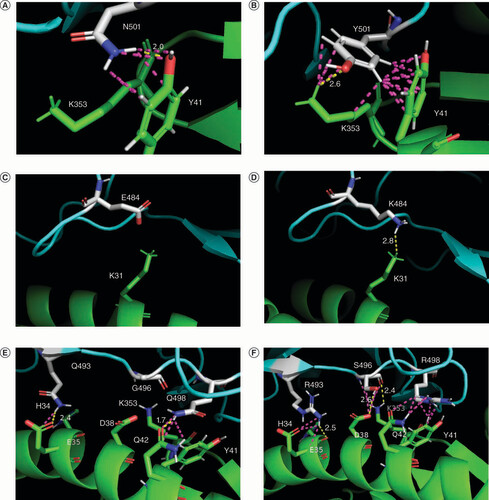
Mutations in the RBDs of SARS-CoV-2 variants provide greater stability of the RBD–ACE2 complexes by providing more energy-efficient conformation (such as Delta variant mutations) and by forming additional chemical bonds with ACE2 amino acids in the interface of the complex. The results of computer visualization revealed that, in particular, mutations N501Y, E484K and G496S provide the formation of H-bonds and other interchain contacts (). Thus, the presence of an aromatic ring in tyrosine (N501Y) in Alpha and latter variants makes it possible to create a greater number of contacts with ACE2 interface amino acids K353 and Y43. Mutation E484K in Beta, Gamma and some VOIs creates a new H-bond with K31 in ACE2. As for the G496S mutation in the Omicron, it acts in a complex way together with Q493R, Q498R and N501Y (the last is not shown on the E–F) determining the rearrangement of interface contacts in the RBD–ACE2 complex; while S496 forms new H-bonds with ACE2 interface amino acids D38 and K353.
(A & B) Mutation N501Y: Y501 in Alpha variant (B) in comparison with N501 in the WT (A) forms additional contacts with ACE2 K353. (C & D) Mutation E484K: K484 in Beta, Gamma, Eta, Zeta, Theta, Iota and Mu variants creates a new H-bond with ACE2 K31, absent in the RBD–ACE2 complex of the WT. (E & F) Mutations Q493R, G496S, Q498R: Omicron (sublineage BA.1) mutations (F) in comparison with the WT (E) determine the rearrangement of interface contacts in the RBD–ACE2 complex; S496 forms new favorable H-bonds with ACE2 D38 and K353.
ACE2 is shown in green; RBD is shown in cyan; SARS-CoV-2 amino acids involved in mutations are shown in white. H-bonds are shown in yellow; other interchain contacts with a distance of fewer than 3.0 Å (ångström) are shown in light mangenta. The distances (Å) between the atoms are indicated only for H-bonds.
Based on the data in & , was generated, which shows the RBD mutational profiles of SARS-CoV-2 VOCs. illustrates the quantitative distribution of mutations in VOCs and shows the ACE2 binding affinity gradation line based on the ΔG calculations. Thus, Alpha, Delta and Omicron variants show higher ACE2 affinity compared to the WT, while Gamma and Beta variants are inferior to the WT. It should be noted that the ability to ACE2 binding is one of the key, but not the only, factor of adaptive success for the virus. For this reason, in the immune escape gradation line is also presented, built based on data obtained from [Citation16]. Thus, it worth paying attention that the lower stability of the RBD–ACE2 complexes in the case of Beta and Gamma variants may be compensated by other evolutionary advantages, such as immune escape, which is shown in more detail in the Discussion section.
Characteristic mutation sets are indicated for five variants of concern (Alpha, Beta, Gamma, Delta and Omicron [BA.1 sublineage]). The red arrow indicates the ACE2 binding affinity of VOCs compared with the WT (based on the ΔG calculated values). The blue arrow indicates the gradation of immune escape ability (based on [Citation16] data).
WT: Wild-type.
![Figure 8. Mutational profiles of the SARS-CoV-2 variants.Characteristic mutation sets are indicated for five variants of concern (Alpha, Beta, Gamma, Delta and Omicron [BA.1 sublineage]). The red arrow indicates the ACE2 binding affinity of VOCs compared with the WT (based on the ΔG calculated values). The blue arrow indicates the gradation of immune escape ability (based on [Citation16] data).WT: Wild-type.](/cms/asset/381da350-ec2c-47e6-8cbb-f5f49a8fa97b/ifvl_a_12336087_f0008.jpg)
Discussion
This study showed the distribution of mutations in the RBDs of SARS-CoV-2 VOCs and VOIs and indicated which mutations are conservative and which are radical. The first thing that draws attention in the analysis is that the Omicron sublineages carry many more mutations than the previous variants. To some extent, the phenomenal increase in the number of mutations in the RBD of the Omicron compared with previous VOCs could be fixed by evolution in a stable lineage, provided that the complex effect of the mutation set on the characteristics of the virus gives it certain adaptive advantages. Other studies showed that the Omicron is characterized by greater infectivity, increased risk of reinfections, immune escape, and low vaccine efficacy [Citation41–44]. The Omicron has quickly become the most common variant while achieving a balance between high transmissibility and low mortality [Citation16]. It can be noted that this process played a major role in crowding out other SARS-CoV-2 variants and, ultimately, led to their reclassification as previous VOCs and VOIs. The acquisition of both RBD mutations of the Delta variant (L452R, T478K) by the Omicron BA.4 and BA.5 sublineages is important for further monitoring, as this may affect the infectious characteristics of the virus and its further evolution.
Since a significant number of mutations in the RBDs of SARS-CoV-2 variants are characterized in this study as radical or moderately radical according to Grantham's distances, and most of the variants have at least one such mutation in their mutational profile, this allows us to conclude that a radical change in the physicochemical properties of amino acids in the RBD is important for achieving evolutionary progress.
The significance of the moderately radical N501Y mutation presented in most VOCs was described in many publications [Citation45–49]. Thus, it was shown that tyrosine (Y), which can exhibit both polar and non-polar properties, and interact with aromatic amino acids and non-carbon atoms, is likely to be more conducive to the formation of a stable RBD–ACE2 complex [Citation49]. The extreme importance of this mutation for the adaptive potential of the virus is even evidenced by the fact that it was definitely often found in the mutational profiles of VOCs.
Another L452R mutation that was classified in this study as moderate radical allows the virus to evade cellular immunity, provide stronger attachment to the ACE2, and increase its infectivity [Citation50,Citation51] (which in particular takes place for the Omicron sublineages carrying this mutation [Citation52]).
Also noteworthy are the radical S→F substitutions (S371F and S375F) in Omicron sublineages and F→S substitution (F490S) in Lambda. Aromatic non-polar phenylalanine (F) contrasts with polar hydroxyl serine (S), which may underlie significant changes in the functions of the RBD and the spike protein in these SARS-CoV-2 variants. The recent study found [Citation53] that mutation F371S in the BA.2 sublineage of the Omicron led to avoidance of the immune system. Notable that for the F490S mutation, it was shown [Citation54] that it may underlie the decrease in the effectiveness of vaccination. Thus, both phenylalanine and serine may be conducive for SARS-CoV-2, depending on the position in the RBD.
In the evolution of the SARS-CoV-2 RBD structure, both radical mutations and an abrupt increase in their number (in the case of the Omicron) take place, which, of course, affects the properties of the spike protein. Evolution favors such changes if they confer certain benefits on the virus, as seen in SARS-CoV-2 VOCs and VOIs, which supplanted the WT and each other due to the increasing transmissibility, infectious potential, and greater immune escape capability provided by characteristic mutation sets.
The assessment of the impact of mutations on the RBD functions made using SIFT, PROVEAN and SNAP2 bioinformatic tools indicated the presence of the effect from mutations N501Y, Y505H and, to a certain extent, N440K. As for the N501Y mutation, its functional significance was confirmed not only in this but also in many other studies, as already mentioned above. At the same time, the functional effect of the Y505H mutation is less frequently discussed in scientific studies. It is known that Y505H is associated with escape from the monoclonal antibody casirivimab, and together with Q493R, Q498R and N501Y takes part in the creation of additional bonding pairs, which enhances the strength of interaction with the ACE2 receptor [Citation55,Citation56]. As for the N440K mutation, it should be noted that the SARS-CoV-2 variant with the N440K mutation has previously been shown to avoid antibody neutralization. However, the RBD–ACE2 complex of this variant was less stable than of the WT [Citation57].
In this study, the evaluations of the Gibbs binding free energy level (ΔG) and its change under the influence of single and multiple mutations (ΔΔGsingle and ΔΔGmultiple) serves as key indicators for assessing the change in the stability of RBD–ACE2 complexes formed by virus variants, and thus for assessing their potential to interact with the ACE2 receptor and infect host cells. In particular, the results of ΔG calculations obtained using the accurate MM/GBSA method indicate that most of VOCs and VOIs (with the exception of Beta and Gamma) have a mutational profile that leads to the reduction of the ΔG and strengthening interactions with the ACE2 receptor. The obtained results are generally consistent with the data of many other studies, a comparison with some of which is given. In the study [Citation58], it was predicted using the MM/GBSA method that the Alpha variant with additional mutation E484K, as well as the Delta variant, have a higher binding free energy in complex with ACE2 and correspondingly increased ACE2 binding affinity. Several studies showed using Molecular Dynamics simulations and Monte Carlo sampling that the N501Y mutation and two mutations in the RBD of the Delta variant enhanced electrostatic interactions with ACE2 [Citation45,Citation59]. In the study [Citation60], it was determined using molecular dynamics simulations and binding free energy calculations with the MM/PBSA method that mutations in different SARS-CoV-2 variants (Delta plus, Iota, Kappa, Mu, Lambda and C.1.2) helped reduce the binding free energy of the RBD–ACE2 complex, forming more H-bonds and accordingly increasing the contagiousness of these SARS-CoV-2 variants.
The calculation results for Beta and Gamma variants performed in this study indicated that the RBD–ACE2 complexes formed by these variants have increased ΔG levels compared with WT and, accordingly, are less stable. This is due to the presence of destabilizing K417N/T mutations. It should be noted that previous studies indicated similar patterns [Citation18,Citation61,Citation62].
As for Omicron, all sublineages of this variant have a confident advantage over the WT variant; they are characterized by a more negative level of the ΔG for RBD–ACE2 complexes formed, and as a result, their greater stability. The binding affinity of the Omicron variant was characterized previously in several studies. For comparison in the study [Citation63], complexes of ACE2 with the WT, Delta and Omicron variants were investigated via density functional theory (DFT) simulations to obtain binding energies. The results indicated that the binding of the Omicron variant to the ACE2 was much more favorable than the binding of the Delta variant and, especially, the WT of the virus. In the study [Citation64], MD simulation and the MM/PBSA method were used to obtain the binding free energy of the WT virus and the Omicron variant. Authors pointed to the greater affinity of the Omicron for ACE2 and suggested that the emergence of a new variant with a higher infection rate compared with that of the Omicron is still possible if new combination of mutations take place in the interface region.
The results obtained in this study for Omicron showed that its greater ACE2 binding affinity is provided by several mutations (N440K, T478K, E484A, G496S, N501Y) in BA.1 and BA.1.1 sublineages, as well as additionally D405N and L452R mutations in latter sublineages). These mutations have a stabilizing effect on the RBD–ACE2 complex; on the contrary, the remaining mutations in the Omicron RBD are neutral or destabilizing. At the same time, the entire mutation sets in the Omicron sublineages have complex effect, strongly stabilizing interactions with the ACE2 receptor. For comparsion, it was shown in various studies that Omicron mutations may both increase and decrease the ACE2 receptor affinity, but the total effect of all Omicron variant mutations increases the stability of RBD–ACE2 [Citation65–67]. This, in turn, confirms that mutation sets characteristics of the virus variants have complex effects. Due to this reason, a large predictive power is obtained specifically for the analysis of mutation sets in the RBD than of single mutations of SARS-CoV-2 variants, which is especially evident for the Omicron.
The effect of mutations in spike protein is not limited to changes in the ACE2 receptor affinity. The complex effect of mutation set in each of the SARS-CoV-2 variants has a profound impact on the spike protein itself, creating the basis for the virus to acquire the necessary adaptive advantages.
Although the organization of spike as a protein molecule at all levels, from the primary sequence of amino acid residues to the three-dimensional structure, is of great importance for SARS-CoV-2 infectivity, post-translational modifications also have a significant influence [Citation68]. Glycosylation is the most common protein post-translational modification in viruses that modulates interactions with receptors and the following innate and adaptive immune response [Citation69]. Among all the sites of N-glycosylation of asparagine residues in spike protein, it is necessary to pay attention to those localized in the RBD (N331 and N343, as well as N334). Mutations leading to the substitution of asparagine with another amino acid prevent glycosylation, which in the case of N331 and N343, results in a decrease in the infectivity of the virus [Citation69]. The N343A mutation leads to a 56% decrease in ACE2 binding [Citation70]. By comparing the glycosylation sites in the RBD of SARS-CoV-2 with the mutational profiles in VOCs and VOIs, it can be noted that none of these most common variants distributed mutations in the glycosylation sites in the RBD, which indicates the importance of the conservatism of glycosylated asparagines for the preservation infectious potential of the virus. At the same time, the Omicron lineages are characterized by the presence of mutations G339D, S373P and S375F (common to all lineages), as well as mutations R346K (lineage BA.1.1), S371L (BA.1 and BA.1.1) and S371F (BA.2, BA.3, BA.4/BA.5) near N-glycosylation sites. These mutations are absent in most other SARS-CoV-2 variants and, accordingly, may affect the effect of glycosylation at the N331 and N343 sites. In the study [Citation71] it was indicated that the change in the environment of N343 in the Omicron variant could probably provoke reduced recognition by the neutralizing antibodies and, therefore, avoidance of the immune response by the virus.
Another way (except for the increased ACE2 receptor affinity) in which mutations give SARS-CoV-2 variants evolutionary advantage is the ability to immune escape. In this aspect, the mutations in the RBD are important, given that it is the main target of neutralizing antibodies after infection or vaccination [Citation72]. Among the individual mutations presented in the RBDs of VOCs and VOIs, E484K, K417N, L452R, as well as S477N are important for immune escape [Citation61,Citation73]. Considering the virus variants with specific RBD mutational profiles, it is worth noting that before the appearance of the Omicron variant, the Beta had the greatest ability to escape from both COVID-19 convalescent plasma and vaccine-induced antibodies, that is conditioned by the presence of RBD mutations K417N, E484K, as well as N501Y [Citation16,Citation72]. The Gamma variant has a similar mutational profile (E484K, K417T, N501Y) and the ability to immune escape is also significant, although less than in the case of the Beta [Citation16]. This is well consistent with the results obtained in this study: the K417N and K417T mutations and ΔG levels of these variants indicate relatively less stability of the RBD–ACE2 complexes compared with the WT; however, the K417N and K417T mutations are fixed in these previously circulating VOCs, which is probably because of their role for immune escape (as also mentioned in [Citation61,Citation62]).
The latter of the considered Omicron variant had the highest ability to avoid immune response in studies using both plasma of COVID-19 convalescent patients and plasma from vaccinated patients. It was determined that the neutralizing activity of various vaccines reduced many times, and in some cases, efficiency was absent. The escape from therapeutic antibodies is also explained by many mutations in the Omicron variant, which was significantly increased compared with other variants, thus forming a certain evolutionary gap [Citation16,Citation72].
Increased ACE2 receptor affinity and stability of the RBD–ACE2 complex (which enhances the infectious potential of the virus) and the ability to evade immunity are two key adaptive benefits that the virus gains through mutations and one of the main directions of virus evolution over time. schematically shows the mutational profiles of VOCs, as well as the gradation of ACE2 affinity (based on the ΔG calculations carried out in this study) and immune escape capacity (based on data in[Citation16]) among the WT and VOCs. It is worth noting that variants such as Beta and Gamma are partially less successful in terms of ACE2 affinity than other variants. However, they benefit from a greater ability of immune escape, which indicates a different direction of mutagenesis in SARS-CoV-2. At the same time, the Omicron has absolute advantages in all respects.
Given the high ability of SARS-CoV-2 not only to mutagenesis but also to quickly acquire the most favorable mutations, monitoring its evolution is necessary on an ongoing basis. In addition, given the presence of mutations in the SARS-CoV-2 variants, the risks of the virus overcoming new interspecies barriers and the emergence of animal reservoirs of COVID-19 are high [Citation74,Citation75]. Timely identification of new variants and assessment of their infectious potential and ability to immune escape (which can be carried out using bioinformatic approaches) will help prevent all possible adverse outcomes associated with the COVID-19 pandemic.
Conclusion
Using databases and bioinformatic methods, an analysis of mutation sets in the RBD of spike protein was carried out for SARS-CoV-2 VOCs and VOIs, from Alpha to the latest Omicron sublineages. Even though the RBD of spike protein is quite variable, only a part of mutations were fixed in virus lineages of epidemiological significance. Some mutations such as K417N, L452R, E484K and N501Y are present in more than one variant of SARS-CoV-2, which indicates their role in virus evolution. Omicron acquired significantly more mutations than other variants, including those that were previously absent among VOCs and VOIs, and received the greatest adaptive advantages. At the same time, mutagenesis of the latest Omicron BA.4 and BA.5 sublineages was associated with incorporating the Delta RBD mutational profile (L452R and T478K) into its own.
To obtain the most comprehensive characterization of the RBD mutations and assess their impact on the ability of SARS-CoV-2 to interact with the ACE2 receptor, we consistently used bioinformatic tools that predict the impact of mutations on the functions of spike protein and stability of the RBD–ACE2 complex. The most significant among these estimates were calculations of the Gibbs binding free energy obtained using the MM/GBSA method. This accurate approach revealed a complex effect of the RBD mutations in each of the variants associated with amino acid interactions at the RBD mutation sites both with ACE2 and within the RBD. The stability of the RBD–ACE2 complexes is provided by mutations N440K, L452R, T478K, E484K, G496S, N501Y, as well as F490S, D405N; mutations E484K, G496S, N501Y, in particular, provide the formation of additional H-bonds with ACE2 and other interchain contacts. The study results indicate that one of the directions of the SARS-CoV-2 evolution and mutagenesis in the RBD is increasing the ACE2 receptor affinity. At the same time, mutations that do not enhance such interactions (K417N/T) can provide other adaptive benefits, such as immune escape. In the case of the Omicron, this is the variant with the most developed mutational profile, which allowed it to obtain an absolute advantage over the other variants. This, in turn, confirms the fact that evolution consolidates the most favorable mutational changes.
Both single mutations and mutation sets in the RBD of the SARS-CoV-2 spike protein of currently and previously circulating VOCs and VOIs (from Alpha to the latest Omicron sublineages) were evaluated using bioinformatic methods in terms of their impact on the ACE2 receptor affinity and the stability of the RBD–ACE2 complex.
In silico sequence and structure-oriented approaches were used to assess the impact of mutations: Grantham's distances; predictive mutation tools; creation of three-dimensional structures of the RBD–ACE2 complexes, their visualization; estimation of the Gibbs binding free energy.
Mutations N440K, L452R, T478K, E484K, G496S, N501Y, as well as F490S and D405N in the RBD of SARS-CoV-2 spike protein have a great impact on the ability of the virus variants to bind to the ACE2 receptor stabilizing the RBD–ACE2 complex.
Mutation sets characteristic of the SARS-CoV-2 variants lead to the reduction of binding free energy of the RBD–ACE2 complex, forming additional chemical bonds with the ACE2 receptor and, consequently, to an increase of the RBD–ACE2 complex stability.
Analysis of mutation sets of the SARS-CoV-2 variants has greater predictive power than the analysis of single mutations.
One of the main directions of the SARS-CoV-2 evolution and mutagenesis in the RBD is increasing the ACE2 receptor-binding affinity. In addition, the RBD mutations allow the virus to achieve other adaptive benefits, such as immune escape.
Omicron is the variant with the most developed mutational profile, which allowed it to obtain an absolute advantage over the other variants. This confirms the fact that evolution consolidates the most favorable mutational changes.
Author contributions
M Peka and V Balatsky were responsible for study conception and design; M Peka was responsible for conducting in silico experiments; M Peka and V Balatsky were responsible for data analysis, drafting and revision of the manuscript.
Financial & competing interests disclosure
This work was supported by the National Academy of Agrarian Sciences of Ukraine. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- World Health Organization . Novel Coronavirus (2019-nCoV). Situation report - 1 (2020). www.who.int/docs/default-source/coronaviruse/situation-reports/20200121-sitrep-1-2019-ncov.pdf?sfvrsn=20a99c10_4 (Accessed 20December2021).
- World Health Organization . Novel Coronavirus (2019-nCoV). Situation report - 22 (2020). www.who.int/docs/default-source/coronaviruse/situation-reports/20200211-sitrep-22-ncov.pdf?sfvrsn=fb6d49b1_2 (Accessed 20December2021).
- World Health Organization . WHO Director-General's opening remarks at the media briefing on COVID-19 – 11 March 2020 (2020). www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (Accessed 20December, 2021).
- Shu Y , McCauleyJ. GISAID: global initiative on sharing all influenza data – from vision to reality. Euro Surveill.22(13), 30494 (2017).
- Hadfield J , MegillC , BellSMet al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics.34(23), 4121–4123 (2018).
- Rambaut A , HolmesEC , O'TooleÁet al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol.5(11), 1403–1407 (2020).
- World Health Organization . Tracking SARS-CoV-2 variants. (2022). www.who.int/activities/tracking-SARS-CoV-2-variants/tracking-SARS-CoV-2-variants (Accessed 30July2022).
- World Health Organization . WHO announces simple, easy-to-say labels for SARS-CoV-2 Variants of Interest and Concern (2021). www.who.int/news/item/31-05-2021-who-announces-simple-easy-to-say-labels-for-sars-cov-2-variants-of-interest-and-concern (Accessed 20December2021).
- Huang Y , YangC , XuXF , XuW , LiuSW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta. Pharmacol. Sin.41(9), 1141–1149 (2020).
- Li C , TianX , JiaXet al. The impact of receptor-binding domain natural mutations on antibody recognition of SARS-CoV-2. Sig. Transduct. Target Ther.6, 132 Doi:10.1038/s41392-021-00536-0 (2021).
- Schrörs B , Riesgo-FerreiroP , SornPet al. Large-scale analysis of SARS-CoV-2 spike-glycoprotein mutants demonstrates the need for continuous screening of virus isolates. PlOS ONE.16(9), e0249254 (2021).
- Safari I , ElahiE. Evolution of the SARS-CoV-2 genome and emergence of variants of concern. Arch. Virol.167(2), 293–305 (2022).
- SARS-CoV-2 Variants . Standford Coronavirus Antiviral & Resistance Database (CoVDB). (2023). https://covdb.stanford.edu/page/mutation-viewer/ (Accessed 10April2022).
- Desingu PA , NagarajanK , DhamaK. Emergence of Omicron third lineage BA.3 and its importance. J. Med. Virol.94(5), 1808–1810 (2022).
- European Centre for Disease Prevention and Control . Epidemiological update: SARS-CoV-2 Omicron sub-lineages BA.4 and BA.5 (2022). www.ecdc.europa.eu/en/news-events/epidemiological-update-sars-cov-2-omicron-sub-lineages-ba4-and-ba5 (Accessed 28May2022).
- Kudriavtsev AV , VakhrushevaAV , NovoseletskyVNet al. Immune escape associated with RBD Omicron mutations and SARS-CoV-2 evolution dynamics. Viruses.14(8), 1603 (2022).
- Gobeil SM , JanowskaK , McDowellSet al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science373(6555), eabi6226 (2021).
- Barton MI , MacGowanSA , KutuzovMA , DushekO , BartonGJ , vander Merwe PA. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife.10, e70658 Doi:10.7554/eLife.70658 (2021).
- Wu F , ZhaoS , YuBet al. A new coronavirus associated with human respiratory disease in China. Nature579(7798), 265–269 (2020).
- Grantham R . Amino acid difference formula to help explain protein evolution. Science185(4154), 862–864 (1974).
- Li WH , WuCI , LuoCC. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Mol. Evol.21(1), 58–71 (1984).
- Sim NL , KumarP , HuJ , HenikoffS , SchneiderG , NgPC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res.40(Web Server issue), W452–W457 (2012).
- Choi Y , SimsGE , MurphyS , MillerJR , ChanAP. Predicting the functional effect of amino acid substitutions and indels. PLOS ONE.7(10), e46688 (2012).
- Hecht M , BrombergY , RostB. Better prediction of functional effects for sequence variants. BMC Genomics.16(Suppl. 8), S1 (2015).
- Suzek BE , WangY , HuangH , McGarveyPB , WuCH. UniProt Consortium. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics.31(6), 926–932 (2015).
- Wang Q , ZhangY , WuLet al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell181(4), 894–904.e9 (2020).
- Guex N , PeitschMC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis18(15), 2714–2723 (1997).
- Han P , SuC , ZhangYet al. Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants. Nat. Commun.12(1), 6103 (2021).
- Han P , LiL , LiuSet al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell185(4), 630–640.e10 (2022).
- van Gunsteren WF , BilleterSR , EisingAAet al. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Biomos, Zürich, Federal Republic of Germany (1996).
- Dehouck Y , KwasigrochJM , RoomanM , GilisD. BeAtMuSiC: prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res.41(Web Server issue), W333–W339 (2013).
- Xiong P , ZhangC , ZhengW , ZhangY. BindProfX: Assessing Mutation-Induced Binding Affinity Change by Protein Interface Profiles with Pseudo-Counts. J. Mol. Biol.429(3), 426–434 (2017).
- Zhang N , ChenY , LuHet al. MutaBind2: Predicting the Impacts of Single and Multiple Mutations on Protein-Protein Interactions. iScience.23(3), 100939 (2020).
- Pahari S , LiG , MurthyAKet al. SAAMBE-3D: predicting effect of mutations on protein–protein interactions. Int. J. Mol. Sci.21(7), 2563 (2020).
- Rodrigues CHM , PiresDEV , AscherDB. mmCSM-PPI: predicting the effects of multiple point mutations on protein-protein interactions. Nucleic Acids Res.49(W1), W417–W424 (2021).
- Genheden S , RydeU. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert opinion on drug discovery.10(5), 449–461 (2015).
- Weng G , WangE , WangZet al. HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res.47(W1), W322–W330 (2019).
- Schrödinger L , DeLanoW. PyMOL (2020). http://www.pymol.org/pymol (Accessed 22March2022).
- Du X , LiY , XiaYLet al. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci.17(2), 144 (2016).
- Hou T , WangJ , LiY , WangW. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model.51(1), 69–82 (2011).
- Tian D , SunY , XuH , YeQ. The emergence and epidemic characteristics of the highly mutated SARS-CoV-2 Omicron variant. J. Med. Virol.94(6), 2376–2383 (2022).
- Chaguza C , CoppiA , EarnestRet al. Rapid emergence of SARS-CoV-2 Omicron variant is associated with an infection advantage over Delta in vaccinated persons. Med (N Y).3(5), 325–334.e4 (2022).
- Araf Y , AkterF , TangYDet al. Omicron variant of SARS-CoV-2: genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol.94(5), 1825–1832 (2022).
- World Health Organization . Enhancing response to Omicron SARS-CoV-2 variant: technical brief and priority actions for Member States World (2022). https://www.who.int/docs/default-source/coronaviruse/2022-01-21-global-technical-brief-and-priority-action-on-omicron-sars-cov-2-variant.pdf?sfvrsn=f3ac8bc3_9&download=true (Accessed 14February2022).
- Ali F , KasryA , AminM. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med Drug Discov.10, 100086 Doi:10.1016/j.medidd.2021.100086 (2021).
- Lu L , ChuAW , ZhangRRet al. The impact of spike N501Y mutation on neutralizing activity and RBD binding of SARS-CoV-2 convalescent serum. EBioMedicine.71, 103544 Doi:10.1016/j.ebiom.2021.103544 (2021).
- Tian F , TongB , SunLet al. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife.10, e69091 Doi:10.7554/eLife.69091 (2021).
- Liu Y , LiuJ , PlanteKSet al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature602(7896), 294–299 (2022).
- Nguyen KV . Problems associated with antiviral drugs and vaccines development for COVID-19: approach to intervention using expression vectors via GPI anchor. Nucleosides Nucleotides Nucleic Acids.40(6), 665–706 (2021).
- Motozono C , ToyodaM , ZahradnikJet al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe.29(7), 1124–1136.e11 (2021).
- Tchesnokova V , KulasekaraH , LarsonLet al. Acquisition of the L452R Mutation in the ACE2-Binding Interface of Spike Protein Triggers Recent Massive Expansion of SARS-CoV-2 Variants. J. Clin. Microbiol.59(11), e0092121 (2021).
- Zhang Y , ZhangT , FangY , LiuJ , YeQ , DingL. SARS-CoV-2 spike L452R mutation increases Omicron variant fusogenicity and infectivity as well as host glycolysis. Signal. Transduct. Target Ther.7(1), 76 (2022).
- Liu L , IketaniS , GuoYet al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature602(7898), 676–681 (2022).
- Novazzi F , BajA , PasciutaRet al. Acluster of SARS-CoV-2 Delta variant of concern additionally harboring F490S, Northern Lombardy, Italy. Int. J. Infect. Dis.116, 271–272 Doi:10.1016/j.ijid.2021.12.362 (2022).
- Cao Y , WangJ , JianFet al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature602(7898), 657–663 (2022).
- Alkhatib M , SalpiniR , CariotiLet al. Update on SARS-CoV-2 Omicron Variant of Concern and Its Peculiar Mutational Profile. Microbiol. Spectr.10(2), e0273221 (2022).
- Kullappan M , MaryU , AmbroseJM , VeeraraghavanVP , SurapaneniKM. Elucidating the role of N440K mutation in SARS-CoV-2 spike - ACE-2 binding affinity and COVID-19 severity by virtual screening, molecular docking and dynamics approach. J Biomol Struct Dyn.41(3), 912–929 (2021).
- Verma J , SubbaraoN. Insilico study on the effect of SARS-CoV-2 RBD hotspot mutants' interaction with ACE2 to understand the binding affinity and stability. Virology561, 107–116 Doi:10.1016/j.virol.2021.06.009 (2021).
- Goher SS , AliF , AminM. The Delta variant mutations in the receptor binding domain of SARS-CoV-2 show enhanced electrostatic interactions with the ACE2. Med. Drug Discov.13, 100114 Doi:10.1016/j.medidd.2021.100114 (2021).
- Celik I , KhanA , DwivanyFM , Fatimawali , WeiDQ , TalleiTE. Computational prediction of the effect of mutations in the receptor-binding domain on the interaction between SARS-CoV-2 and human ACE2. Mol Divers.26(6), 3309–3324 (2022).
- Harvey WT , CarabelliAM , JacksonBet al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol.19, 409–424 Doi:10.1038/s41579-021-00573-0 (2021).
- Rayati Damavandi A , DowranR , AlSharif Set al. Molecular variants of SARS-CoV-2: antigenic properties and current vaccine efficacy. Med. Microbiol. Immunol.211(2–3), 79–103 (2022).
- Yamacli S , AvciM. Computation of the Binding Energies between Human ACE2 and Spike RBDs of the Original Strain, Delta and Omicron Variants of the SARS-CoV-2: A DFT Simulation Approach. Adv. Theory Simul.5, 2200337 Doi:10.1002/adts.202200337 (2022).
- Nguyen HL , ThaiNQ , NguyenPH , LiMS. SARS-CoV-2 Omicron Variant Binds to Human Cells More Strongly than the Wild Type: Evidence from Molecular Dynamics Simulation. J. Phys. Chem. B. B.126(25), 4669–4678 (2022).
- Ortega JT , JastrzebskaB , RangelHR. Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: an In Silico Analysis. Pathogens.11(1), 45 (2021).
- Shah M , WooHG. Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies. Front Immunol.12, 830527 Doi:10.3389/fimmu.2021.830527 (2022).
- Lupala CS , YeY , ChenH , SuXD , LiuH. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun.590, 34–41 Doi:10.1016/j.bbrc.2021.12.079 (2022).
- Kapoor K , ChenT , TajkhorshidE. Posttranslational modifications optimize the ability of SARS-CoV-2 spike for effective interaction with host cell receptors. Proc. Natl Acad, Sci, U S A.119(28), e2119761119 (2022).
- Gong Y , QinS , DaiL , TianZ. The glycosylation in SARS-CoV-2 and its receptor ACE2. Sig. Transduct. Target Ther.6, 396 Doi:10.1038/s41392-021-00809-8 (2021).
- Sztain T , AhnSH , BogettiATet al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem.13, 963–968 Doi:10.1038/s41557-021-00758-3 (2021).
- López-Cortés GI , Palacios-PérezM , VeledíazHFet al. The Spike Protein of SARS-CoV-2 Is Adapting Because of Selective Pressures. Vaccines (Basel).10(6), 864 (2022).
- Cameroni E , BowenJE , RosenLEet al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature602, 664–670 Doi:10.1038/s41586-021-04386-2 (2022).
- Yi C , SunX , LinYet al. Comprehensive mapping of binding hot spots of SARS-CoV-2 RBD-specific neutralizing antibodies for tracking immune escape variants. Genome Med.13, 164 Doi:10.1186/s13073-021-00985-w (2021).
- Bashor L , GagneRB , Bosco-LauthAM , BowenRA , StengleinM , VandeWoudeS. SARS-CoV-2 evolution in animals suggests mechanisms for rapid variant selection. Proc. Natl Acad. Sci. U S A118(44), e2105253118 (2021).
- Meekins DA , GaudreaultNN , RichtJA. Natural and Experimental SARS-CoV-2 Infection in Domestic and Wild Animals. Viruses.13(10), 1993 (2021).